
Abstract
Diabetic kidney disease (DKD) is a common microvascular complication of diabetes mellitus, and oxidative stress and mitochondrial dysfunction play an important role in this process. It has been shown that aldose reductase (ALR2) catalyzes NADPH-dependent reduction of glucose to sorbitol, resulting in oxidative stress and mitochondrial dysfunction in diabetic patients. Astragalin (AG), a flavonoid extracted from Thesium chinense Turcz., shows an inhibitory activity on ALR2. In this study, we investigated the therapeutic effects of AG against renal injury in streptozocin (STZ)-induced diabetic mouse model. Diabetic mice were orally administered AG (5, 10 mg·kg−1·d−1) for 4 weeks. We showed that AG treatment greatly improved the proteinuria and ameliorated renal pathological damage without affecting the elevated blood glucose in diabetic mice. Furthermore, AG treatment significantly suppressed highly activated ALR2, and reduced oxidative stress in the kidney of diabetic mice and in high glucose and lipids-stimulated HK2 cells in vitro. We demonstrated that AG treatment modulated mitochondrial quality control and ameliorated apoptosis, boosting mitochondrial biogenesis, maintaining mitochondrial dynamic homeostasis, and improving energy metabolism disorder in vivo and in vitro. In high glucose and lipids-stimulated HK2 cells, we found that AG (20 μM) restored the phosphorylation level of AMPK, and upregulated the expression and transcriptional activity of PGC1α, whereas treatment with H2O2, blockade of AMPK with Compound C or knockdown of AMPKα with siRNA abolished the protective effect of AG on mitochondrial function, suggesting that antioxidant effects and activation of AMPK-dependent PGC1α pathway might be the molecular mechanisms underlying the protective effects of AG on mitochondrial quality control. We conclude that AG could be a promising drug candidate for the treatment of diabetic renal injury through activating AMPK.
Similar content being viewed by others
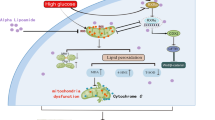


Introduction
Diabetic kidney disease (DKD), characterized by proteinuria and a progressive decline in glomerular filtration rate (GFR), is the main cause of chronic kidney disease (CKD) and end-stage renal disease (ESRD) worldwide [1]. However, the underlying pathobiology of DKD is complex and still not fully understood.
The proximal tubules are susceptible to energy deficiency due to the high energy demands for sustaining normal renal reabsorption. Proximal tubules are rich in mitochondrion, and the maintenance of mitochondrial quality control is responsible for renal function. Studies have found that mitochondrial damage is involved in the pathogenesis of kidney injury [2, 3]. Also, several studies have shown that renal injury can be attenuated by restoring the mitochondrial function of renal tubular cells in diabetes [4,5,6].
The mitochondrion is also the main source and primary target of intracellular reactive oxygen species (ROS) [3]. In diabetes, persistent hyperglycemia generates excessive ROS, representing a critical contributor to the impairment of mitochondrial quality control, including an imbalance between mitochondrial fission and fusion, less mitochondrial biogenesis, which can induce mitochondrial dysfunction and, in turn, cause cell death, tissue injury, and even organ failure. Therefore, it is conceivable that maintaining mitochondrial homeostasis and enhancing mitochondrial function may be viable therapeutic approaches for DKD.
Aldose reductase (ALR2), a key enzyme in the polyol pathway, is widely expressed in the kidney and activated in response to hyperglycemia [7]. ALR2 catalyzes nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reduction of glucose to sorbitol, which results in oxidative stress and mitochondrial dysfunction in diabetic patients [8]. ALR2 inhibitors have been shown to protect DKD via metabolic modulation [9]. Astragalin (AG) is a natural flavonoid extracted from Thesium chinense Turcz. with excellent antioxidant properties [10, 11]. Furthermore, AG can suppress ALR2 activity in vitro [12]. In this study, we hypothesized that AG has a protective impact on diabetic kidney injury. We evaluated the therapeutic effect of AG in a mouse model of DKD induced by streptozocin (STZ) and explored the potential molecular mechanisms through which AG improves mitochondrial dysfunction.
Materials and methods
Drug and regents
Astragalin (AG, HPLC ≥98%, the chemical structure was shown in Fig. 1a) was purchased from BioBioPha (Kunming, Yunnan, China). Epalrestat (Epa) was obtained from Yangtze River Pharmaceutical Group (Taizhou, Jiangsu, China). STZ was purchased from Sigma (St Louis, Colorado, USA). The blood glucose kit (F006-1-1), total triglyceride (TG, F001-1-1), total cholesterol (TCHO, A111-2-1), low-density lipoprotein cholesterol (LDL-C, A113-2-1), creatinine (C011-2-1), and superoxide dismutase (SOD, A001-1-2), malondialdehyde (MDA, A003-1-2), catalase (CAT, A007-1-1) assay kits were purchased from Nanjing JianCheng (Nanjing, Jiangsu, China). NADPH-Na4 (60302ES01), MitoSOX (40778ES50), and qPCR SYBR Green Master Mix (11198ES08) were purchased from YEASEN (Shanghai, China). Secondary antibody (1:20,000, A0208), MitoTracker Green (C1048), BCA protein (P0010), ATP (S0027), NADP+/NADPH (S0179), and GSH/GSSG (S0053) assay kits were purchased from Beyotime (Shanghai, China). JC1 (J8030) was obtained from Solarbio (Beijing, China). The primary antibodies including AMPKα (#5832S), p-AMPKα (Thr172) (#2535S), caspase3 (#9662), cleaved caspase3 (Asp175) (#9661), and β-actin (#3700S) were purchased from CST (Danvers, MA, USA); DRP1 (A2586), MFN2 (A19678) were from ABclonal (Shanghai, China); PGC1α (ab72230), Bax (ab32503) and Bcl2 (ab182858) were from Abcam (Cambridge, UK).
Animals and treatments
Six-week-old male C57BL/6J mice (21 ± 2 g) were purchased from Slaccas-Shanghai Lab Animal Ltd. (Shanghai, China). After 1 week of adaptation feeding, mice were given STZ (100 mg·kg−1·d−1, dissolved in citrate solution) intraperitoneally for two consecutive days to induce the diabetic model. Seven days after the injection, these mice with random blood glucose (RBG) ≥ 16.7 mmol/L were randomly divided into four groups (n = 6): model group, low-dose (5 mg/kg) and high-dose AG (10 mg/kg) groups, and Epalrestat group (40 mg/kg). The control group received an equal amount of citrate solution intraperitoneally. AG and Epalrestat were prepared by 0.5% CMC-Na and were given by gavage once a day for 4 weeks. The control and model group were administered an equal volume of 0.5% CMC-Na solution by gavage. After 4 weeks of treatment, mice were disposed according to following scheme. In the day before sacrificing the animals, blood and 5-h urine of mice were collected at a random time. Next day, all mice were euthanized after 6-h fasting (6:00 AM-12:00 PM), and blood sample and kidney tissues were collected.
All the animals were housed in an environment with a temperature of 22 ± 1 °C and a light/dark cycle of 12/12 h. All animal studies were done in compliance with the regulations and guidelines of Fudan University institutional animal care and approved by the Ethics Committee of School of Pharmacy Fudan (2021-11-YL-WHB-112) and conducted according to the IACUC guidelines.
Serum and urine biochemical parameters measurement
The blood samples were centrifuged (3500 rpm, 10 min) to collect the serum. Blood glucose, TG, TCHO, LDL-C, serum creatinine (SCr), and urine creatinine (UCr) levels were measured using the commercial kit by following the manufacturers’ instructions. ELISA was used to measure urine microalbumin. The urine albumin-to-creatinine ratio (UACR, mg/mmol) and 5-h urinary albumin excretion rate (UAER, mg/5 h) were calculated: UACR = urine microalbumin (μg/mL)/urine creatinine (mM), UAER = urine microalbumin (μg/mL) × urine volume (mL).
Histopathological analysis
The kidney tissues were fixed in 10% neutral formalin and cut in sections (3 μm thickness), after which Hematoxylin and Eosin stain (H&E), Periodic Acid-Schiff (PAS) and Masson’s Trichrome staining were performed. H&E staining was used to display the morphology of renal tissue; PAS used for staining structures containing a high proportion of carbohydrates such as glycogen, glycoproteins, and proteoglycans to estimate basement membrane lesion, extracellular matrix proliferation and exudation of various cells; and Masson’s staining to reveal the proliferation of collagen fibers in the kidney. All stained images were statistically analyzed using ImageJ software.
Renal NADP+/NADPH assay
The ratio of NADP+ to NADPH was assayed following the manufacturer’s instruction. Briefly, a 5% renal homogenate was prepared with lysis and centrifuged to collect the supernatant for analysis of total NADPH and NADP+. Another 100 μL sample was heated for 30 min at 60 °C to degrade NADP+ and then used to assay NADPH content. [NADP+]/[NADPH] = ([NADPtotal] − [NADPH])/[NADPH].
Renal GSH/GSSH assay
GSH and GSSH content were determined using the commercial kit. In brief, a sample mixture (10 µL) and glutathione assay solution (150 µL) were incubated for 5 min at 25 °C and then mixed with 50 µL NADPH solution. OD412nm was tested every 5 min for a total of 25 min using an enzyme marker. A sufficient sample was extracted and used to measure the GSSG concentration after eliminating GSH. A standard curve was obtained by calculating the amount of total glutathione or GSSG in the sample at the moment of measurement. GSH (µM) = total Glutathione (µM) − GSSG (µM) × 2.
Renal oxidative stress level examination
A 10% homogenization of kidney tissue was prepared with PBS and centrifuged at 4 °C (12,000 rpm, 10 min) to collect the supernatant for subsequent assays. The levels of SOD, CAT, and MDA in the kidney were assayed according to the commercial kit instructions.
Cell culture and treatment
HK2 cells were cultured in RPMI-1640 medium supplemented with 10% FBS in 5% CO2 at 37 °C. Cells were then exposed to high glucose and lipids (40 mM glucose + 0.35 mM palmitic acid, HG + PA) and simultaneously treated with AG (20 μM). To further explore the mechanism of AG, HK2 was treated with or without AG, MitoQ (1 μM, a mitochondrial antioxidant), H2O2 (0.1 μM, an oxidative agent), and Compound C (10 μM, an AMPK inhibitor). Cells cultured for 12 h and 24 h were analyzed using PCR and Western blotting, respectively.
Cell activity assay
HK2 cells were cultured with RPMI-1640 medium containing 10% FBS in 96 well plates. After reaching a 90% confluency, cells were exposed to different concentrations of AG for 24 h. Then, 200 μL of sterile MTT dye (0.5 mg/mL) was added to each well and incubated for another 2 h at 37 °C. After removal of the medium, DMSO was added to each well and properly mixed for another 10 min. The absorbance at 570 nm was determined using a microplate reader.
Aldose reductase activity assay
Renal aldose reductase was obtained, as previously reported [13]. The protein was extracted, and concentration was measured. ALR2 activity was assessed by the absorption of NADPH at 340 nm at 37 °C for 5 min.
Mitochondrial ROS (mtROS) and mitochondrial mass detection
The mtROS levels of renal tissues and HK2 cells were measured by MitoSOX red mitochondrial superoxide indicator, while MitoTracker Green was used to measure mitochondrial mass. Renal tissues and cells were incubated for 30 min at 25 °C with MitoSOX (5 μM) or MitoTracker Green solution (10 nM). After washing with PBS, the stained sections were observed on a fluorescent microscope. Sodium gradient-dependent glucose transporter protein (SGLT2) was used as a special marker for tubulicular epithelial cells and the location of mtROS was identified by co-staining with the SGLT2 antibody.
Mitochondrial membrane potential (MMP) measurement
The MMP levels of renal tissues and HK2 were analyzed using JC1 staining. Kidney-frozen sections and cells were stained with JC1 (10 μg/mL) for 30 min at 25 °C and were viewed on a fluorescent microscope.
ATP content measurement
ATP content was determined using the commercial kit. Briefly, 20% homogenate of kidney tissue was prepared by lysis. Tissues were then centrifuged at 4 °C (12,000 rpm, 5 min), and the supernatant was collected for subsequent assays, following the manufacturer’s instructions.
siRNA transfection
The siRNA was incubated with Lipo3000 in 500 μL serum-free media without penicillin and streptomycin for 10 min at room temperature and then added to the HK2 cells. Six hours after transfection, the cells were treated with HG + PA or/and AG and incubated again for 24 h at 37 °C.
Western blotting
Total protein was extracted from renal tissues and cells using RIPA lysis buffer. The protein concentration was determined using a BCA assay. The protein was separated by SDS-PAGE and then electrophoretically blotted onto PVDF membranes. After blocking with 5% skim milk, the membranes were incubated with the primary antibodies [AMPKα (1:1000), pAMPKα (1:1000), PGC1α (1:1000), DRP1 (1:1000), MFN2 (1:1000), Bax (1:1000), Bcl2 (1:1000), caspase3 (1:1000), cleaved caspase3 (1:500), and β-actin (1:1000)] at 4 °C overnight, and then with the secondary antibody (1:20,000) for 2 h at 25 °C. An enhanced ECL kit detected protein bands, and visualized images were captured with a camera-based imaging system.
Immunofluorescence
Paraffin sections of kidney tissues were dewaxed, hydrated, and antigenically repaired with citrate buffer (pH 6.0). After permeabilizing the cell membrane by 0.1% TrionX-100, the sections were blocked with 10% goat serum for 1 h at 37 °C. Samples were then incubated with the primary antibody at 4 °C overnight and then with the secondary antibody for 90 min at 25 °C. Cell nuclei were stained with DAPI for 10 min, and the sections were observed using inverted fluorescence microscopy.
qRT-PCR
Total RNA was isolated from kidney tissue and HK2 cells with TRIzol and then reverse transcribed to cDNA using a DNA Reverse Transcription SuperMix for qPCR kit. Then real-time qPCR was performed utilizing a qPCR SYBR Green Master Mix kit. The gene expression was normalized by the β-actin gene, and the 2-ΔΔCT method was applied to analyze the relative expression levels of genes. The sequences of all primers are shown in Supplementary Table S1.
Statistical analysis
Data were presented as mean ± SD. One-way ANOVA, followed by Bonferroni’s post hoc test, was used to analyze the differences between multiple groups. P value < 0.05 was considered statistically significant.
Results
Astragalin improves renal function in diabetic mice
The protective effects of AG on renal injury in STZ-induced diabetic mice were investigated. The diabetic mice showed significantly decreased body weight and elevated blood glucose compared with the control group, while treatment with astragalin and epalrestat did not affect the changes (Fig. 1b–d). Moreover, diabetic mice developed hyperlipidemia; serum TG and LDL-C levels in diabetic mice were significantly higher in the model group compared to the control group, while there was no significant increase in serum TCHO. After AG treatment, serum TG levels decreased, but LDL-C levels did not improve (Fig. 1e–g).

a Chemical structure of AG. b–k Physiological parameters of mice were measured, including body weight (b), fasting blood glucose (FBG, c), random blood glucose (RBG, d), serum total triglycerides (TG, e), total cholesterol (TCHO, f), low-density lipoprotein cholesterol (LDL-C, g), serum creatinine (SCr, h), kidney weight/body weight [kidney index (%), i], urine albumin-to-creatinine ratio (UACR, j), and 5-h urinary albumin excretion rate (UAER, k). l Relative levels of NGAL mRNA were determined in the kidney. Data represent the mean ± SD, n = 6; *P < 0.05; ***P < 0.001 vs. control group; #P < 0.05; ##P < 0.01; ###P < 0.001 vs. model group (one-way ANOVA, with Bonferroni’s post hoc test).
To assess renal function in diabetic mice, we measured SCr level, kidney index (KI), UACR, and UAER. The results showed that the SCr, KI, UACR, and UAER were significantly higher in the model group compared to the control group (Fig. 1h–k); the PCR results showed that the level of NGAL mRNA in the kidney of the model group was also markedly increased (Fig. 1l), indicating renal dysfunction. Notably, the parameters could be significantly improved by AG administration, confirming the therapeutic effect of AG on renal injury in diabetic mice.
Astragalin ameliorates pathological renal damage in diabetic mice
The kidney dysfunction in the diabetic mice was accompanied by tubular swelling and structural derangements of glomerular vascular tufts and interstitial space. PAS staining revealed an expanded tubulointerstitial space and a thicker glomerular basement membrane in the model group. In addition, Masson staining showed an increase in the area of fibrosis in the diabetic kidney (Fig. 2a). AG treatment effectively inhibited glomerular proliferation and improved tubule-glomerular morphology (Fig. 2b). Furthermore, the kidney’s fibrosis levels, particularly in the tubular interstitium, were remarkably improved after AG treatment (Fig. 2c, d).

a Representative images of H&E (400×), PAS (400×), and Masson (200×) staining in kidneys. b–d H&E, PAS, and Masson staining were analyzed using the ImageJ, respectively, glomerular cell count per unit area (b), mesangial index (%, c) and area of fibrosis (%, d). Data represent the mean ± SD, n = 4; **P < 0.01; ***P < 0.001 vs. control group; #P < 0.05; ##P < 0.01; ###P < 0.001 vs. model group (one-way ANOVA, with Bonferroni’s post hoc test).
Astragalin inhibits aldose reductase activity and ameliorates oxidative stress in the kidney of diabetic mice
The diabetic mice showed high ALR2 activity in the kidney (Fig. 3a), suggesting activation of the polyol pathway. Increased polyol bypass flux is the major contributor to oxidative stress in diabetic mice’s kidneys, leading to diabetic kidney injury [14]. Thus, we examined the oxidative stress-related indicators in the kidney of diabetic mice. NADPH/NADP+ (Fig. 3b), GSH/GSSG (Fig. 3c), and CAT activities (Fig. 3d) were significantly lower in diabetic mice kidneys compared to control, while MDA contents were remarkably increased (Fig. 3e), with no significant change in SOD activity (Fig. 3f). The results indicated that kidneys of diabetic mice were under severe oxidative stress. Contrary, AG administration could inhibit ALR2 activity and thus reduce oxidative stress, relieving diabetic kidney injury.

a Aldose reductase (ALR2) activity was measured in the kidney. b–f Oxidative stress-related indicators were analyzed in the kidney, including NADP+/NADPH (b), GSH/GSSG (c), catalase (CAT, d), malondialdehyde (MDA, e), and superoxide dismutase (SOD, f). Data represent the mean ± SD, n = 6; **P < 0.01; ***P < 0.001 vs. control group; #P < 0.05; ##P < 0.01; ###P < 0.001 vs. model group (one-way ANOVA, with Bonferroni’s post hoc test).
Astragalin alleviates mitochondrial dysfunction in the kidney of diabetic mice
Mitochondria are the major source of intracellular ROS and the key damage target of oxidative stress. Based on the potent antioxidant effect of AG, we explored whether it could improve mitochondrial dysfunction in diabetic mice kidneys. Increased mitochondrial superoxide anion generation, decreased mitochondrial abundance, and depolarized mitochondrial membrane potential (MMP) were seen in the kidney of diabetic mice (Fig. 4a–c), indicating depressed mitochondrial quality. AG treatment significantly reduced mitochondrial oxidative stress, increased mitochondrial mass, and elevated MMP.

a Representative images of MitoTracker staining to exhibiting mitochondrial mass in the kidney. b SGLT2 and MitoSOX co-staining to exhibiting mitochondrial oxidative stress in the proximal tubule. c Representative images of staining with JC1 demonstrating intrarenal mitochondrial membrane potential. d–f Relative mRNA expression of gene involved in mitochondrial biogenesis (d), mitochondrial oxidative phosphorylation (e), and fatty acid metabolism (f) in the kidney. g ATP contents were measured in the kidney. h Relative mRNA expression of genes involved in mitochondrial fission and fusion in the kidney. Data represent the mean ± SD, n = 3; *P < 0.05; **P < 0.01; ***P < 0.001 vs. control group; #P < 0.05; ##P < 0.01; ###P < 0.001 vs. model group (one-way ANOVA, with Bonferroni’s post hoc test).
In the present study, we determined the number of mitochondria-related genes and intrarenal ATP content in the kidney. We found that the model group showed an obvious decrease in the expression of genes encoding PGC1α, TFAM, and ERRα, indicating suppression of mitochondrial biogenesis in the kidney during the progression of diabetes (Fig. 4d). In addition, the gene expression levels of ATP5F1e, NDUFV1, Cox7a, and PPARα were greatly downregulated in the model group (Fig. 4e, f), and the intrarenal ATP content was decreased (Fig. 4g), suggesting a severe impairment of mitochondrial energy metabolism in the diabetic kidney. The elevated mRNA levels of DRP1 and FIS1 in the model group indicated excessive mitochondrial fission in the diabetic kidney, whereas the levels of genes encoding mitochondrial fusion proteins (OPA1, MFN1, MFN2) did not significantly differ between the model and control group (Fig. 4h). AG administration elevated diabetes-induced inhibition of mitochondrial biogenesis and energy metabolism, inhibited excessive mitochondrial fission, and promoted mitochondrial fusion-related gene and protein expression, displaying the effect of regulating mitochondrial dynamics homeostasis (Fig. 4h and Supplementary Fig. S1).
Astragalin facilitates AMPK/PGC1α activation and inhibits mitochondria-dependent apoptosis in the kidney of diabetic mice
AMPK has been identified as a metabolism guardian and a central integrator of mitochondrial homeostasis [15]; inactivation of AMPK induced by oxidative stress may be a major cause of mitochondrial dysfunction in diabetic kidney injury. Moreover, PGC1α is crucial in regulating mitochondrial metabolism and biogenesis [16]. Therefore, we hypothesized that AG regulates mitochondrial quality control via the AMPK/PGC1α pathway. Western blotting analysis showed that the phosphorylation level of AMPK and the expression of PGC1α were significantly decreased in the model group, which were reversed by the administration of AG or Epa (Fig. 5a). Consistently, immunofluorescence staining results showed lower PGC1α expression in the kidney of the model group, especially in the renal tubules, compared with the control group. Also, AG treatment promoted PGC1α protein expression in diabetic mice kidneys (Fig. 5b). These findings suggested that inhibited AMPK/PGC1α signaling in diabetic mice could be activated by AG, contributing to restoring energy deficiency.

a Western blotting analysis of the expression of p-AMPKα, AMPKα, PGC1α and apoptosis-related proteins (Bcl2, Bax, caspase3, cleaved caspase3) in the kidney. b Immunofluorescence analysis of PGC1α expression in the kidney. Data represent the mean ± SD, n = 4; **P < 0.01; ***P < 0.001 vs. control group; #P < 0.05; ##P < 0.01; ###P < 0.001 vs. model group (one-way ANOVA, with Bonferroni’s post hoc test).
Recent studies have identified the involvement of apoptosis in the pathogenesis of DKD [17]. Mitochondria have a central role in the regulation of apoptosis, and Bcl2 family proteins are the primary regulators of mitochondria-associated apoptotic factor release [18]. We found that Bcl2 protein content was significantly lower in the kidney of diabetic mice compared with the control group, while the expressions of Bax and cleaved caspase3 were significantly increased, suggesting that the mitochondria-mediated apoptotic pathway is activated in the diabetic kidney. Furthermore, our data demonstrated that AG reduced mitochondria-dependent apoptosis in the kidney of diabetic mice.
Astragalin modulates mitochondrial quality control via antioxidant effect and AMPK activation in the HK2 cells
To further validate the protective mechanism of AG against diabetic kidney injury, we assessed the effect of AG on HK2 cell damage induced by high glucose and lipids. Firstly, HK2 cell viability was determined to identify the non-toxic dose of AG within 24 h. AG (0–80 µM) had no impact on HK2 cell proliferation (Fig. 6a). Next, we measured ALR2 activity in HK2 cells. The results showed that AG of 5–40 µM abolished the elevated ALR2 activity, indicating the suppressive effect of AG on ALR2 activity (Fig. 6b). Thereby, 20 μM AG was chosen for further in vitro studies.

a Viability analysis of AG on HK2 cells. b ALR2 activity assay. c, d Relative mRNA levels of NGAL (c) and KIM1 (d) were determined. e Representative images of MitoTracker and MitoSOX staining. f Representative images of JC1 staining. g Mitochondria-related gene mRNA expression levels were tested, including TFAM, ERRα, NDUFB8, ATP5F1A, PPARα, CPT1A, DRP1 and MFN2. h ATP content was analyzed. Data represent the mean ± SD of three independent experiments; *P < 0.05; **P < 0.01; ***P < 0.001 vs. control group; #P < 0.05; ##P < 0.01; ###P < 0.001 vs. HG + PA group; &P < 0.05; &&P < 0.01; &&&P < 0.001 vs. AG group (one-way ANOVA, with Bonferroni’s post hoc test).
Next, we examined whether the role of AG in regulating mitochondrial quality control was dependent on antioxidant effects and AMPK activation. In the set of experiments, HK2 cells were treated with or without AG, MitoQ, H2O2, and Compound C. We also examined the mRNA levels of NGAL and KIM1, markers of renal tubular injury. The obtained results showed that H2O2 and Compound C abolished the inhibition of AG on NGAL and KIM1 expression (Fig. 6c, d). Then, we tested the roles of H2O2 and Compound C in AG-improving mitochondrial function. Staining results demonstrated that AG significantly inhibited mtROS production, increased mitochondrial mass, and elevated MMP, while H2O2 reversed the effects and Compound C (Fig. 6e, f). Consistently, mRNA gene expression analysis showed that AG significantly upregulated TFAM, ERRα, NDUFB8, ATP5F1A, PPARα, and increased protein and mRNA levels of MFN2, downregulated DRP1, whereas AG combined with H2O2 or Compound C exhibited the opposite effects when compared to AG alone. For CPT1A mRNA expression, elevated action of AG was enhanced by Compound C (Fig. 6g and Supplementary Fig. S2). Moreover, increased intracellular ATP contents by AG treatment were also diminished by H2O2 and Compound C (Fig. 6h), suggesting that H2O2 and Compound C counteracted the protective effect of AG on mitochondrial function. These results suggested that the antioxidant effects and AMPK activation are key approaches for AG to regulate mitochondrial homeostasis and protect renal function.
Astragalin upregulated AMPK-mediated PGC1α expression contributing to the restoration of mitochondrial biogenesis and suppression of apoptosis
A growing number of studies have demonstrated that functional activation of PGC1α can be induced by AMPK activation and that it promotes mitochondrial biogenesis and energy metabolism [19]. Our in vivo data showed that AG increased the expressions of phosphorylated AMPK and PGC1α in the kidney of diabetic mice. Therefore, we sought to determine whether AG’s AMPK activation was responsible for the increase in PGC1α expression. Firstly, we examined cellular protein expressions by Western blotting to assess the role of AG in HK2 cells stimulated by high glucose and lipids. The damaged cells exhibited an increase in AMPK phosphorylation, PGC1α, and Bcl2 expression and a decrease in Bax and cleaved caspase3 expression following administration of AG and MitoQ (Fig. 7a), which is consistent with in vivo results.

a, b WB detection of protein expression in HK2 cells. c Immunofluorescence staining was performed to observe the expression and distribution of PGC1α protein in HK2 cells. Relative mRNA levels of KIM1 (d) and NGAL (e) were determined. f WB detection of protein expression in HK2 cells transfected with siAMPK. Data represent the mean ± SD of three independent experiments. a–c, *P < 0.05; **P < 0.01; ***P < 0.001 vs. control group; #P < 0.05; ##P < 0.01; ###P < 0.001 vs. model group; &P < 0.05; &&P < 0.01; &&&P < 0.001 vs. AG group; d–f, *P < 0.05; **P < 0.01; ***P < 0.001 vs. control + siNC group; #P < 0.05; ##P < 0.01 vs. HG + PA + siNC group; &P < 0.05; &&P < 0.01; &&&P < 0.001 vs. control+siAMPK group (one-way ANOVA, with Bonferroni’s post-hoc test).
Next, Compound C or H2O2 was used to block AMPK signaling or trigger oxidative stress. The data showed that Compound C greatly inhibited the AG-mediated increase of AMPK phosphorylation and PGC1α (Fig. 7b); the result was further confirmed by immunofluorescence staining of PGC1α in protein expression and nuclear translocation (Fig. 7c). Moreover, a similar reversal was presented in apoptosis-related protein regulation, including elevated Bcl2 and decreased Bax and cleaved caspase3 expression. In agreement with this, H2O2 also abrogated the beneficial influence of AG.
To further determine the therapeutic approach of AG, we knocked down AMPKα expression with specific small, interfering RNAs. The results exhibited that silencing of AMPKα increased the level of DRP1, Bax and cleaved caspase-3 in normal conditions, indicating that AMPKα knockdown sensitized HK2 cells to apoptosis (Fig. 7f). Then, we observed the effect of AG on the injury of HK2 cells induced by high glucose and lipids. The data showed that reduced mRNA levels of KIM1 and NGAL by AG were abolished in AMPKα silence cells (Fig. 7d, e). WB results also confirmed that AG could not increase expressions of PGC1α and Bcl2 and could not inhibit the expression of Bax and cleaved caspase3 in HK2 cells after AMPKα silencing. It was concluded that the knockdown of AMPK diminished protective role of AG in the damaged HK2 cells (Fig. 7f). The data strongly suggested that AMPK activation by AG is required for PGC1α upregulation.
Then, we treated HK2 cells with the PGC1α inhibitor SR18292 to further verify whether AG exerts the protective effect via activating PCG1α. Consistent with AMPKα silence, inhibition of PCG1α also abolished the ameliorative effect of AG on the injury of HK2 cells, observing the reverses for expressions of KIM1 and NGAL, DRP1 and MFN2, and apoptosis-related factors such as Bcl2, Bax and cleaved caspase3 (Supplementary Fig. S3). Therefore, we believe that activating AMPK/PGC1α might be the molecular mechanism of AG to alleviate mitochondrial dysfunction and suppress apoptosis in diabetic-induced renal injury.
Discussion
The present study demonstrates that AG alleviates renal injury in the kidney of STZ-induced diabetic mice, as evidenced by a decrease in UACR and the expression of NGAL, as well as an improvement in histopathological damage, irrespective of glucose-lowering. Furthermore, AG exerted potent antioxidant effects by inhibiting ALR2 activity, and improved mitochondrial dysfunction by activating the AMPK/PGC1α pathway, promoting mitochondrial biogenesis, enhancing energy metabolism, maintaining dynamic mitochondrial homeostasis, and ultimately reducing renal apoptosis. Therefore, AG represents a potential therapeutic option for diabetic-induced renal injury.
Our results demonstrated the excellent inhibitory effect of AG on ALR2 activity in vitro and in vivo. Aldose reductase, an NADPH-dependent enzyme, has been implicated as a causative agent and potential therapeutic target in DKD [20]. Activation of ALR2 leads to disruption of glucolipid metabolism and oxidative stress due to NADPH deficiency, culminating in tubular, glomerular, and microvascular damage in the diabetic kidney [21]. Our findings showed that AG decreases NADP+/NADPH and MDA contents while increasing GSH/GSSH and CAT activities in diabetic mice, indicating that AG reduces intracellular oxidative stress in the kidney of diabetic mice, which is an important approach to protect the renal injury.
Excessive activation of ALR2 destroys intracellular redox equilibrium, while mitochondria, the main source of intracellular free radicals, are particularly vulnerable to ROS damage, eventually forming a vicious cycle in the kidney of DKD [22]. Mitochondrial dysfunction, manifested as less ATP production and loss of MMP, accompanied by an increase in mtROS [23], has been affirmed to precede renal damage and further contribute to the development of DKD [24]. Mitochondrial dynamics and biogenesis are quality control mechanisms for maintaining a healthy mitochondrial population and function. Our data indicated that AG could reduce mtROS production and regulate mitochondrial quality control, such as less mitochondrial biogenesis and dynamic imbalances, promoting energy metabolism. Furthermore, the protective effect of AG on mitochondria was abolished by H2O2, suggesting that anti-mitochondrial oxidative stress may be crucial for regulating mitochondrial dysfunction by AG.
Furthermore, we found that AG reduces renal tubular epithelial cell apoptosis in DKD. The apoptotic pathway is involved in the onset and progression of DKD, and mitochondria have a crucial role in the regulation of apoptosis mediated by cytochrome c and apoptosis-induced factor [25]. Excessive production of mtROS resulting from hyperglycemia triggers apoptosis signaling within mitochondria, such as Bcl2 and caspase family, leading to cell death. Mitochondrial dysfunction is an early manifestation of renal histopathological injury in DKD, as many apoptotic renal tubular epithelial cells appear in early DKD. Therefore, AG might have a therapeutic role in the early stages of DKD, thereby retarding the progression of diabetic kidney injury.
The molecular mechanisms through which oxidative stress causes mitochondrial dysfunction are unclear. Several studies have reported that AMPK has a central protective role in limiting oxidative damage and regulating mitochondrial function [26, 27]. PGC1α is the major metabolic regulator, and mitochondria are highly sensitive to intracellular PGC1α levels [27, 28]. Previous studies have reported lower PGC1α protein levels in the kidneys of diabetic patients [29]. AMPK activation promotes PGC1α expression, which interacts with various transcription factors to coordinate essential mitochondrial processes, including biogenesis, oxidative phosphorylation, fatty acid oxidation, and mitochondrial dynamics [19, 30]. Our study found that AG could activate AMPK and boost PGC1α expression and transcriptional activity, thereby modulating mitochondrial quality control in the diabetic kidney. However, Compound C, an AMPK inhibitor, eradicated these effects. This indicates that AG regulates mitochondrial function via AMPK agonism, suggesting that AG could be used as an AMPK agonist in DKD treatment, which is subject to further research.
Conclusion
This study demonstrates that AG exerts its renoprotective effects by inhibiting oxidative stress induced by aldose reductase activation in the kidney of diabetic mice and modulating mitochondrial quality control by activating the AMPK-dependent PGC1α pathway. These findings point to AG’s potential role and mechanism in treating diabetic kidney injury.
References
Chinese Diabetes Society. Guideline for the prevention and treatment of type 2 diabetes mellitus in China (2020 edition). Int J Endocrinol Metab. 2021;41:482–548.
Forbes JM, Thorburn DR. Mitochondrial dysfunction in diabetic kidney disease. Nat Rev Nephrol. 2018;14:291–312.
Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017;13:629–46.
Qin X, Jiang M, Zhao Y, Gong J, Su H, Yuan F, et al. Berberine protects against diabetic kidney disease via promoting PGC-1α-regulated mitochondrial energy homeostasis. Br J Pharmacol. 2020;177:3646–61.
Qin X, Zhao Y, Gong J, Huang W, Su H, Yuan F, et al. Berberine protects glomerular podocytes via inhibiting Drp1-mediated mitochondrial fission and dysfunction. Theranostics. 2019;9:1698–713.
Yuan Y, Yuan L, Li L, Liu F, Liu J, Chen Y, et al. Mitochondrial transfer from mesenchymal stem cells to macrophages restricts inflammation and alleviates kidney injury in diabetic nephropathy mice via PGC-1α activation. Stem Cells. 2021;39:913–28.
Terubayashi H, Sato S, Nishimura C, Kador PF, Kinoshita JH. Localization of aldose and aldehyde reductase in the kidney. Kidney Int. 1989;36:843–51.
Tang WH, Stitham J, Jin Y, Liu R, Lee SH, Du J, et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation. 2014;129:1598–609.
He J, Gao HX, Yang N, Zhu XD, Sun RB, Xie Y, et al. The aldose reductase inhibitor epalrestat exerts nephritic protection on diabetic nephropathy in db/db mice through metabolic modulation. Acta Pharmacol Sin. 2019;40:86–97.
Peng L, Gao X, Nie L, Xie J, Dai T, Shi C, et al. Astragalin attenuates dextran sulfate sodium (DSS)-induced acute experimental colitis by alleviating gut microbiota dysbiosis and inhibiting NF-κB activation in mice. Front Immunol. 2020;11:2058.
Kim YH, Kang MK, Lee EJ, Kim DY, Oh H, Kim SI, et al. Astragalin inhibits cigarette smoke-induced pulmonary thrombosis and alveolar inflammation and disrupts par activation and oxidative stress-responsive MAPK-signaling. Int J Mol Sci. 2021;22:3692.
Choung WJ, Hwang SH, Ko DS, Kim SB, Kim SH, Jeon SH, et al. Enzymatic synthesis of a novel kaempferol-3-o-β-d-glucopyranosyl-(1→4)-o-α-d-glucopyranoside using cyclodextrin glucanotransferase and its inhibitory effects on aldose reductase, inflammation, and oxidative stress. J Agric Food Chem. 2017;65:2760–7.
Liu CH, Chen HL, Zha XL, Zhu XX. Isolation and purification of aldose reductase and analysis of its properties. Acta Acad Med Shanghai. 1996;6:427–30.
Ighodaro OM. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed Pharmacother. 2018;108:656–62.
Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–35.
Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90.
Ricciardi CA, Gnudi L. Kidney disease in diabetes: from mechanisms to clinical presentation and treatment strategies. Metab Clin Exp. 2021;124:154890.
Schapira AH. Mitochondrial diseases. Lancet. 2012;379:1825–34.
Kou G, Li Z, Wu C, Liu Y, Hu Y, Guo L, et al. Citrus tangeretin improves skeletal muscle mitochondrial biogenesis via activating the AMPK-PGC1-α pathway in vitro and in vivo: a possible mechanism for its beneficial effect on physical performance. J Agric Food Chem. 2018;66:11917–25.
Kador PF, Wyman M, Oates PJ. Aldose reductase, ocular diabetic complications and the development of topical Kinostat®. Prog Retin Eye Res. 2016;54:1–29.
Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol. 2012;3:87.
Kowalczyk P, Sulejczak D, Kleczkowska P, Bukowska OI, Kucia M, Popiel M, et al. Mitochondrial oxidative stress-a causative factor and therapeutic target in many diseases. Int J Mol Sci. 2021;22:13384.
Wang J, Zhu P, Li R, Ren J, Zhang Y, Zhou H. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics. 2020;10:384–97.
Tirichen H, Yaigoub H, Xu W, Wu C, Li R, Li Y. Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front Physiol. 2021;12:627837.
Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100.
Wu SB, Wu YT, Wu TP, Wei YH. Role of AMPK-mediated adaptive responses in human cells with mitochondrial dysfunction to oxidative stress. Biochim Biophys Acta. 2014;1840:1331–44.
Han X, Tai H, Wang X, Wang Z, Zhou J, Wei X, et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD+ elevation. Aging Cell. 2016;15:416–27.
Haemmerle G, Moustafa T, Woelkart G, Büttner S, Schmidt A, van de Weijer T, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med. 2011;17:1076–85.
Li SY, Susztak K. The role of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) in kidney disease. Semin Nephrol. 2018;38:121–6.
Dong YZ, Li L, Espe M, Lu KL, Rahimnejad S. Hydroxytyrosol attenuates hepatic fat accumulation via activating mitochondrial biogenesis and autophagy through the AMPK pathway. J Agric Food Chem. 2020;68:9377–86.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number 82073859 and 81673658).
Author information
Authors and Affiliations
Contributions
MYS and HBW conceptualized the study. MYS and HJY contributed to the animal experiments. MYS and CZ contributed to the molecular biology experiments. MYS and ZJ contributed to the data collection and statistical analysis. YY contributed to the methodology. MYS and HBW drafted and finalized the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sun, My., Ye, Hj., Zheng, C. et al. Astragalin ameliorates renal injury in diabetic mice by modulating mitochondrial quality control via AMPK-dependent PGC1α pathway. Acta Pharmacol Sin 44, 1676–1686 (2023). https://doi.org/10.1038/s41401-023-01064-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-023-01064-z
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
ALCAT1-mediated abnormal cardiolipin remodelling promotes mitochondrial injury in podocytes in diabetic kidney disease
Cell Communication and Signaling (2024)
-
Exploring a new mechanism between lactate and VSMC calcification: PARP1/POLG/UCP2 signaling pathway and imbalance of mitochondrial homeostasis
Cell Death & Disease (2023)