
Abstract
Hypoxia is a key concern during the treatment of non-small cell lung cancer (NSCLC), and hypoxia-inducible factor 1 alpha (HIF-1α) has been associated with increased tumor resistance to therapeutic modalities such as cisplatin. Compensatory activation of nucleotide excision repair (NER) pathway is the major mechanism that accounts for cisplatin resistance. In the present study, we suggest a novel strategy to improve the treatment of NSCLC and overcome the hypoxia-induced cisplatin resistance by cotreatment with Oroxylin A, one of the main bioactive flavonoids of Scutellariae radix. Based on the preliminary screening, we found that xeroderma pigmentosum group C (XPC), an important DNA damage recognition protein involved in NER, dramatically increased in hypoxic condition and contributed to hypoxia-induced cisplatin resistance. Further data suggested that Oroxylin A significantly reversed the hypoxia-induced cisplatin resistance through directly binding to HIF-1α bHLH-PAS domain and blocking its binding to HRE3 transcription factor binding sites on XPC promoter which is important to hypoxia-induced XPC transcription. Taken together, our findings not only demonstrate a crucial role of XPC dependent NER in hypoxia-induced cisplatin resistance, but also suggest a previously unrecognized tumor suppressive mechanism of Oroxylin A in NSCLC which through sensitization of cisplatin-mediated growth inhibition and apoptosis under hypoxia.
Similar content being viewed by others


Introduction
Lung cancer is the most commonly diagnosed and the deadliest cancer worldwide [1]. As the most common type of lung cancer, non-small cell lung cancer (NSCLC) accounts for ~75% frequency of occurrence, compared to small cell lung cancer (SCLC) with only 25%. Although several new therapeutic strategies have been developed, cisplatin-based therapy remains the golden standard treatment for late stage non-small cell lung cancer [2]. Cisplatin (CDDP) is used either alone or combined with other drugs for patients who are not amenable to surgical resection or with no ‘identifiable’ mutation, or for maintenance therapies [3].
The effectiveness of cisplatin-based NSCLC chemotherapy, however, is still facing a great clinical challenge from the chemoresistance in the therapeutic strategies [4]. A growing body of experimental evidence suggest that the effects of cisplatin are markedly diminished under hypoxic condition, a distinctive property of solid tumors [5,6,7,8]. To adapt the hypoxic condition, cancer cells are known to upregulate a protein called hypoxia inducible factor (HIF) [9]. HIF-1 [10,11,12] and to a lesser extent HIF-2 [13], the oxygen-regulated HIF isoforms, have been associated with chemotherapy failure. The expression of HIF-1α is associated with disease progression in NSCLC tissues, and is expected as a potential therapeutic target for lung cancer [14, 15]. However, in lung cancer, therapeutic inhibition of HIF-2α may paradoxically promote tumor growth by reducing expression of tumor suppressor genes [16]. It is reported that moderate hypoxia can lead to replication stress and activation of the DNA Damage Repair (DDR) pathway proteins even in the absence of measurable DNA damage [17, 18]. Previous research reported an increased number of double-strand breaks (DSBs) in untreated as well as etoposide-treated HIF-1α-deficient murine embryonic fibroblasts (MEFs) [19]. Despite strong similarities between the two isoforms, HIF-2α fails to affect DDR due to its phosphorylation at Thr-324, thereby abrogating Sp1 binding [20]. Therefore, hypoxia plays a critical role in DNA damage repair.
Anti-cancer potency of cisplatin is due to formation of platinum-DNA adducts, mainly between adjacent purines generating intrastrand crosslinks (80–90%) or bases on opposite strands giving rise to interstrand crosslinks (ICLs). Cytotoxicity of these DNA lesions is considered to be derived from impediment of transcription and replication, which eventually causes cell cycle arrest or apoptosis [21]. Increased capacity of DNA damage repair, especially the nucleotide excision repair (NER) [22], is proposed to be one of the most crucial determinants [23, 24]. Cells deficient in NER pathway are sensitive to the drug [25, 26]. Indeed, according to various evidences, increased NER capacity has been proposed as an important mechanism of CDDP resistance [27,28,29,30]. Thus, the induction of DNA damage repair by hypoxia could be an important mechanism leading to cisplatin resistance. Here, we demonstrated an unexpected role for XPC in hypoxia-induced cisplatin resistance. XPC is upregulated by HIF-1α under hypoxic condition, which leads to the failure of cisplatin treatment.
Oroxylin A, an effective component of a Chinese traditional medicinal plant Scutellaria baicalensis Georgi, has various pharmacological activities, including anti-inflammation [31], anti-cancer [32], antiviral and anti-bacterial infections [33]. Previous studies have widely reported that Oroxylin A could reverse drug resistance [32, 34, 35]. Oroxylin A improves sensitivity of chronic myelogenous leukemia cells to Imatinib treatment in bone marrow microenvironment through regulating CXCL12/CXCR7 pathway [34], reverses multidrug-resistant by G2/M arrest and the underlying mechanism attributed to the suppression of P-gp expression via Chk2/P53/NF-κB pathway [36], and improves the sensitivity of HT-29 human colon cancer cells to 5-FU through modulation of the COX-2 pathway [37]. In our study, we investigated the efficacy of co-treatment with cisplatin and Oroxylin A on inhibiting NSCLC development under hypoxic condition. Oroxylin A enhanced the anticancer effect of cisplatin through suppressing XPC transcription, which was promoted by HIF-1α under hypoxia condition.
Results
Oroxylin A attenuates hypoxia-induced cisplatin resistance
To verify the hypoxia-induced resistance in cisplatin-induced cell death, H460 cells were treated with an increasing dose of cisplatin under normoxic (20% O2) and hypoxic (1% O2) condition respectively (Fig. 1a, b). Indeed, IC50 of cisplatin in H460 cells under hypoxia was two-time higher than normoxia (41.10 μM vs 22.71 μM, respectively). Furthermore, DMOG, which inhibits prolyl-4-hydroxylase to stabilize HIF-1α, was used to mimic hypoxic condition. DMOG treatment also notably decreased both the inhibition of cell growth and apoptosis induced by cisplatin in H460 cells (Supplementary Fig. S1A, B).

a H460 cells were treated with cisplatin under normoxic (20% O2) or hypoxic (1% O2) condition for 36 h. The cell viability was analyzed by CCK8 assay. [mean ± S.D. (error bars), n = 3; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, compared with normoxia group, Student’s t test]. b H460 cells were treated with cisplatin under normoxic or hypoxic condition for 36 h. Apoptotic cells were detected by Annexin V and PI staining. One representative experiment out of three is shown. c H460 cells were treated with cisplatin in the presence or absence of 50 μmol/L Oroxylin A under normoxic or hypoxic condition for 36 h. The cell viability was analyzed by CCK8 assay. [mean ± S.D. (error bars), n = 3; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, compared with normoxia group; #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001, compared with hypoxia group, two-way analysis of variance]. d H460 cells were treated with cisplatin in the presence or absence of 50 μmol/L Oroxylin A under hypoxic condition for 36 h. Apoptotic cells were detected by Annexin V and PI staining. One representative experiment out of three is shown. e A549 cells, 95D cells, PC9 cells, HCC827 cells, and H1975 cells were treated with cisplatin in the presence or absence of 50 μmol/L of Oroxylin A under normoxic or hypoxic condition for 36 h. The cell viability was analyzed by CCK8 assay. [mean ± S.D. (error bars), n = 3. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, compared with normoxia group; #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001, compared with hypoxia group, two-way analysis of variance]. f A549 cells, 95D cells, PC9 cells, HCC827 cells, and H1975 cells treated with cisplatin were loaded with 0.5 mmol/L of DMOG in combination with or without 50 μmol/L of Oroxylin A for 36 h. The cell viability was analyzed by CCK8 assay. [mean ± S.D. (error bars), n = 3. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, compared with control group; #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001, compared with DMOG group, two-way analysis of variance]. The data shown are representative of three independent experiments.
To evaluate the potential reversal effect of Oroxylin A on hypoxic-induced cisplatin resistance in H460 cells, proper doses should be chosen primarily. We first assessed the influence of Oroxylin A on cell viability of H460 cells. The 50 μM dose of Oroxylin A was selected because of no severe proliferation inhibition (Supplementary Fig. S1C, D). Both flow cytometric and CCK8 assays showed that Oroxylin A reversed hypoxia-induced cisplatin resistance in H460 cells (Fig. 1c, d). Then we analyzed the chemosensitivity of other non-small cell lung cancer cells (A549, 95D, PC9, HCC827 and H1975 cells) to cisplatin with or without Oroxylin A under hypoxic condition. All these cells treated with Oroxylin A under hypoxic condition showed an enhanced susceptibility towards cisplatin (Fig. 1e). The reverse effect of Oroxylin A was further confirmed with DMOG treatment (Fig. 1a). However, Oroxylin A at 50 μM did not significantly alter the sensitivity of cisplatin in A549/CDDP cell lines compared to their parental A549 cell lines under normoxic condition (Supplementary Fig. S1E). Taken together, Oroxylin A sensitizes cisplatin-induced cell death in NSCLC cells under hypoxia.
Oroxylin A inhibits nucleotide excision repair through downregulating XPC expression
To investigate whether nucleotide excision repair (NER) is a crucial factor in hypoxia-mediated suppression in tumor cells pre-treated with cisplatin, we tested the production of Pt-DNA adducts pre-treated with cisplatin for 4 h (Supplementary Fig. S2A). Hypoxia significantly increased Pt-DNA adducts production than normoxia (Fig. 2a, b). In addition, hypoxia increased XPC and other NER-associated proteins expression levels in H460 cells (Fig. 2c and Supplementary Fig. S2B). Most NER-associated proteins were significantly increased in cisplatin-resistant NSCLC cell line (A549/CDDP) as compared to the parental cell line (A549), which led to more efficient DNA repair (Supplementary Fig. S2C, D). TCGA data analysis showed that XPC expression was lower in human non-small lung cancer tumor tissues compared with the normal lung tissues (Supplementary Fig. S2E). These results suggested that in the initial step of tumor, downregulation of XPC may be involved in promoting carcinogenesis via impairment of DNA repair. Consistent with our findings, XPC expression was found to be significantly associated with poor outcomes of NSCLC patients treated with cisplatin, including decreased overall survival, which indicates the role of XPC in cisplatin resistance (Fig. 2d).

a Representative slot blot analysis of the levels of Pt-DNA adducts in H460 cells at 4, 12, 24 h after cisplatin treatment under hypoxic or normoxic condition. b Quantification of percentage (%) of Pt-DNA adducts repair from A. Data represent the mean ± S.D. of three independent experiments. *p ≤ 0.05, compared with normoxia group, Student’s t test. c Representative immunoblot analysis of NER-related proteins and GAPDH in H460 cells under hypoxic or normoxic condition. d Kaplan–Meier analysis and log-rank test for overall survival of LUSC patients treated cisplatin according to XPC expression level. e Representative slot blot analysis of the levels of Pt-DNA adducts in H460 cells. Cells were collected for analysis at 4, 12 and 24 h after cisplatin treatment in the presence or absence of 50 μmol/L Oroxylin A under normoxic or hypoxic condition. f Quantification of percentage (%) of Pt-DNA adducts repair from E. Data represent the mean ± S.D. of three independent experiments. *p ≤ 0.05, compared with normoxia group, #p ≤ 0.05; ##p ≤ 0.01, compared with hypoxia group, two-way analysis of variance. g Representative immunoblot analysis of XPC, HIF-1α and GAPDH in H460 cells in the presence or absence of 50 μmol/L Oroxylin A under normoxic or hypoxic condition. h Representative immunoblot analysis of XPC and GAPDH in H460 cells stably transfected with control shRNA or 2 independent shRNAs targeting XPC (XPC shRNA #1 or XPC shRNA #2). i Real-time reverse transcriptase-PCR analysis of XPC and GAPDH in H460 cells stably transfected with control shRNA or 2 independent shRNAs targeting XPC (XPC shRNA #1 or XPC shRNA #2). [mean ± S.D. (error bars), n = 3. ***p ≤ 0.001, compared with shCON group, one-way analysis of variance]. j H460 cells stably transfected with shCON or shXPC were treated with cisplatin in the presence or absence of 50 μmol/L Oroxylin A under hypoxic condition for 36 h. The cell viability was analyzed by CCK8 assay. [mean ± S.D. (error bars), n = 3. **p ≤ 0.01; ***p ≤ 0.001, compared with shCON group, two-way analysis of variance]. The data shown are representative of three independent experiments.
Subsequently, pre-treatment of Oroxylin A for 6 h inhibited Pt-DNA adducts repair and XPC expression in hypoxic condition to a level similar to normoxic condition (Fig. 2e–g). However, Oroxylin A did not affect the expression of XPC in A549/CDDP cells (Supplementary Fig. S2F). The cell growth inhibition and apoptosis induced by cisplatin had no difference between normoxic and hypoxic condition in XPC stably knockdown cells (Fig. 2h, i, Supplementary Fig. S2G, H). The removal of Pt-DNA adducts under hypoxic condition was significantly reduced when knocking down XPC (Supplementary Fig. S2I). Oroxylin A had no effect on cisplatin-induced cell growth inhibition of XPC stably knockdown H460 cells under hypoxic condition (Fig. 2j). Taken together, Oroxylin A inhibits hypoxic-induced cisplatin resistance through downregulation of nucleotide excision repair-related protein XPC.
Oroxylin A inhibits hypoxia-induced XPC transcription
To determine the mechanism how Oroxylin A downregulates XPC, the mRNA level of XPC and the transcriptional activity of the XPC promoter in H460 cells were analyzed. Oroxylin A treatment decreased the XPC mRNA level and the transcription activity of XPC under hypoxic condition (Fig. 3a, b). Stable overexpression of XPC in H460 cells (Fig. 3c, d) suppressed the cisplatin associated inhibition of cell growth (Supplementary Fig. S3A) and apoptosis (Supplementary Fig. S3B) and accelerated the removal of Pt-DNA adducts (Supplementary Fig. S3C, D), while the treatment of Oroxylin A did not significantly affect cisplatin-induced inhibition of H460 cells stably transfected with pLenti XPC under hypoxia (Fig. 3e). It is due to the fact that the expression of exogenous XPC was driven by the constitutive CMV promoter, which could not be affected by Oroxylin A (Fig. 3f). These data suggest that Oroxylin A inhibits hypoxic-induced cisplatin resistance through downregulation of XPC transcription.

a Real-time reverse transcriptase-PCR analysis of XPC mRNA levels in H460 cells in the presence or absence of 50 μmol/L Oroxylin A under normoxic or hypoxic condition. [mean ± S.D. (error bars), n = 3. ***p ≤ 0.001, compared with normoxia group, ###p ≤ 0.001, compared with hypoxia group, two-way analysis of variance]. b Luciferase reporter assay of the XPC promoter in H460 cells in the presence or absence of 50 μmol/L Oroxylin A under normoxic or hypoxic condition. [mean ± S.D. (error bars), n = 3. **p ≤ 0.01, compared with normoxia group, ###p ≤ 0.001, compared with hypoxia group, two-way analysis of variance]. c Representative immunoblot analysis of XPC and GAPDH in H460 cells stably transfected with pLenti CON or pLenti XPC. d RT-PCR assay of XPC and GAPDH in H460 cells stably transfected with pLenti CON or pLenti XPC. [mean ± S.D. (error bars), n = 3. ***p ≤ 0.001, compared with pLenti CON group, Student’s t test]. e H460 cells stably transfected with pLenti CON or pLenti XPC were treated with cisplatin in the presence or absence of 50 μmol/L Oroxylin A under hypoxic condition for 36 h. The cell viability was analyzed by CCK8 assay. [mean ± S.D. (error bars), n = 3. **p ≤ 0.01; ***p ≤ 0.001, compared with pLenti CON group, two-way analysis of variance]. f Representative immunoblot analysis of XPC and GAPDH in H460 cells stably transfected with pLenti CON or pLenti XPC in the presence or absence of 50 μmol/L Oroxylin A. The data shown are representative of three independent experiments.
Hypoxia promotes XPC transcription through binding of HIF-1α to HRE3 site on XPC promoter
HIF-1α is a master regulator of cellular adaptation to hypoxia [38], and DMOG, which inhibits prolyl-4-hydroxylase to stabilize HIF-1α, notably decreased the inhibition of cell growth and apoptosis induced by cisplatin (Supplementary Fig. S1A, B). We tested whether HIF-1α plays a role in hypoxia-induced cisplatin resistance which was mediated by enhanced NER activity in H460 cells. Knockdown of HIF-1α in H460 cells (Fig. 4a, b) abolished the effect of hypoxia-induced NER (Supplementary Fig. S4A, B). Furthermore, the protein and mRNA levels of XPC were reduced (Fig. 4c, d). Hypoxia promoted the transcription of XPC, which was inhibited by the knockdown of HIF-1α (Fig. 4e). It suggests that hypoxia promotes nucleotide excision repair and XPC transcription via activating HIF-1α.

a Representative immunoblot analysis of HIF-1α and GAPDH in H460 cells stably transfected with control shRNA or 5 independent shRNAs targeting HIF-1α (HIF-1α shRNA #1, HIF-1α shRNA #2, HIF-1α shRNA #3, HIF-1α shRNA #4 and HIF-1α shRNA #5) under hypoxic condition. b RT-PCR assay of HIF-1α and GAPDH in H460 cells stably transfected with control shRNA or 5 independent shRNAs targeting HIF-1α (HIF-1α shRNA #1, HIF-1α shRNA #2, HIF-1α shRNA #3, HIF-1α shRNA #4 and HIF-1α shRNA #5) under hypoxic condition. [mean ± S.D. (error bars), n = 3. *p ≤ 0.05; ***p ≤ 0.001, compared with shCON group, one-way analysis of variance]. c Representative immunoblot analysis of XPC, HIF-1α and GAPDH in H460 cells stably transfected with shCON or shHIF-1α under normoxic or hypoxic condition. d RT-PCR assay of XPC mRNA levels in H460 cells stably transfected with shCON or shHIF-1α under normoxic or hypoxic condition. [mean ± S.D. (error bars), n = 3. ***p ≤ 0.001, compared with shCON normoxia group, ##p ≤ 0.01, compared with shCON hypoxia group, two-way analysis of variance]. e Luciferase reporter assay of the XPC promoter in H460 cells stably transfected with shCON or shHIF-1α. [mean ± S.D. (error bars), n = 3. *p ≤ 0.05, compared with shCON normoxia group, #p ≤ 0.05, compared with shCON hypoxia group, two-way analysis of variance]. f Schematic representation of the HRE mutation sites of the XPC promoter. Red nucleotides indicate mutations made in human XPC promoter. g Luciferase reporter assay of the XPC promoter with an intact XPC or mutation of three HRE sites in H460 cells under normoxic or hypoxic condition for 36 h. [mean ± S.D. (error bars), n = 3. ***p ≤ 0.001, compared with normoxia group, Student’s t test]. h Chromatin immunoprecipitation assays of HIF-1α interaction with the XPC promoter in H460 cells transfected with pCMV-FLAG-HIF-1α under hypoxic condition for 36 h. After ChIP with anti-FLAG, the results were tested by quantitative PCR amplifications. [mean ± S.D. (error bars), n = 3. ***p ≤ 0.001, compared with IgG group, Student’s t test]. The data shown are representative of three independent experiments.
Hypoxia response elements (HREs) are composite regulatory elements comprising the conserved HIF-binding site with an (A/G) CGTG core sequence and highly variable flanking sequences [39]. By analyzing JASPAR database, we discovered three predicted binding sites of HIF-1α in the promoter region of XPC gene (Supplementary Fig. S4C). To determine whether HRE elements are involved in hypoxia-mediated upregulation of XPC transcription, we mutated the putative HRE sites individually or in combination (Fig. 4f). The mutation of HRE3 site but not HRE1 or HRE2 sites reversed the upregulated XPC transcription induced by hypoxia (Fig. 4g). Chromatin immunoprecipitation assay (CHIP) was performed and the results suggested that the representative HIF-1α specifically bound to the HRE3 site of XPC promoter (Fig. 4h). These results indicate that hypoxia promotes NER and XPC transcription through the binding of HIF-1α to HRE3 site on XPC promoter.
Oroxylin A inhibits the HIF-1α DNA binding property through directly binding to the bHLH-PAS domain of HIF-1α
HIF-1α is essential for hypoxia-induced XPC transcription. However, our data suggested that Oroxylin A had no effect on hypoxia-induced HIF-1α expression and nuclear translocation (Figs. 2g and 5a, b). Chromatin immunoprecipitation combined with quantitative polymerase chain reaction (ChIP-qPCR) revealed that the treatment of Oroxylin A abolished the binding of HIF-1α to HRE2/3 site on XPC promoter (Fig. 5c). To test whether Oroxylin A binds to HIF-1α, H460 cell lysates were incubated with biotin-Oroxylin A and Oroxylin A was pulled down by streptavidin-coupled agarose beads (Supplementary Fig. S5A). Western blot analysis identified HIF-1α in the pulldown, indicating that HIF-1α physically associates with Oroxylin A (Fig. 5d, e and Supplementary S5D). To determine whether Oroxylin A interacts directly with HIF-1α, GST tagged full length HIF-1α was expressed in bacteria and purified (Fig. 5f and Supplementary Fig. S5B). Microscale Thermophoresis (MST) experiment was performed, and Oroxylin A showed binding activity to HIF-1α, yielding a Kd value of ~39 μM (Fig. 5g). These data demonstrate that Oroxylin A interacts directly with HIF-1α protein, which is responsible for the activation of XPC transcription.

a Representative immunoblot analysis of HIF-1α, HDAC1 and GAPDH in cytosolic and nuclear fraction from H460 cells after Oroxylin A treatment under hypoxic condition. b Representative immunofluorescence assay of HIF-1α in H460 cells in the presence or absence of 50 μmol/L Oroxylin A under hypoxic condition. Scale bar, 100 μm. c Chromatin immunoprecipitation assays of HIF-1α interaction with the XPC promoter in H460 cells transfected with pCMV-FLAG-HIF-1α in the presence or absence of 50 μmol/L Oroxylin A under hypoxic condition for 36 h. After ChIP with anti-FLAG, the results were tested by quantitative PCR amplifications. [mean ± S.D. (error bars), n = 3. ***p ≤ 0.001, compared with IgG group, ###p ≤ 0.001, compared with pCMV-FLAG-HIF-1α group, two-way analysis of variance]. d H460 cell lysates were incubated with Biotin or Bio-Oroxylin A at 1 mM with or without Oroxylin A at 10 mM overnight, and were subjected to immunoprecipitation using streptavidin agarose and western blot using indicated antibodies. The data shown are representative of three independent experiments. e Protein levels of HIF-1α were quantified using the ImageJ software. Biotin group was used as control. Data represent the mean ± S.D. of three independent experiments. **p ≤ 0.01, compared with Biotin group, ##p ≤ 0.01, compared with Biotin-OA group, two-way analysis of variance. f Schematic of HIF-1α protein and the truncations used in the present study. g Representative microscale Thermophoresis binding analysis of Oroxylin A to the GST-HIF-1α. h Representative microscale Thermophoresis binding analysis of Oroxylin A to the indicated GST-HIF-1α truncations. The data shown are representative of three independent experiments.
To define the domain of HIF-1α that interacts with Oroxylin A, three truncations of HIF-1α were generated (Fig. 5f and Supplementary Fig. S5C). We found that the bHLH-PAS motif of HIF-1α showed binding activity to Oroxylin A, yielding a Kd value of ~106 μM. But either no binding, or no statistically significant binding, was detected when ODD domain or Inhibitor domain was titrated with Oroxylin A (Fig. 5h). Together, Oroxylin A interacts with the N-terminus of HIF-1α.
Oroxylin A enhances cisplatin efficacy in vivo
To determine the functional significance of Oroxylin A mediated sensitization of cisplatin in NSCLC, a xenografted tumor model derived from H460 cells was used (Fig. 6a). The data showed that sequential administration of cisplatin plus Oroxylin A tremendously reduced tumor growth compared with cisplatin alone, while Oroxylin A used alone had minor effects (Fig. 6b and Supplementary Fig. S6B). Immunofluorescence analysis showed a strong colocalization of XPC and HIF-1α in the tumor tissues especially in the internal tumor tissue (Supplementary Fig. S6C) and Oroxylin A obviously reduced XPC expression in hypoxic tumor tissues (the central part of tumors and normoxic tumor tissues referred to the outer part of tumors) (Supplementary Fig. S6D). Moreover, the cisplatin plus Oroxylin A combination administration reduced cell proliferation (Ki67, PCNA expression), affected cisplatin-induced DNA damage and increased apoptosis by TUNEL test (Fig. 6d, e).

a Schematic design of in vivo experiments with H460 cells stably transfected with shCON or shXPC xenografts testing the efficacy of suboptimal doses of cisplatin (4 mg/kg) and Oroxylin A (50 mg/kg) alone and in combination. b Analysis of tumor growth in each shCON experimental group described in a. [mean ± S.D. (error bars), n = 5. *p ≤ 0.05, compared with shCON Vehicle group, one-way analysis of variance]. c Analysis of tumor growth in each shXPC experimental group described in a. [mean ± S.D. (error bars), n = 5. *p ≤ 0.05; **p ≤ 0.01, compared with shXPC Vehicle group, one-way analysis of variance]. d Representative immunohistochemical analysis of Ki67, PCNA protein levels and Pt-DNA adducts (brown) in each experimental group described in a. Scale bar, 200 μm. f Representative immunofluorescence assay of TUNEL in each experimental group described in a. Scale bar, 100 μm. The data shown are representative of three independent experiments.
Since XPC is important to Oroxylin A mediated sensitization of cisplatin, XPC stable knockdown H460 cells were generated for xenografted tumor model (Fig. 6a and Supplementary Fig. S6A). The similar results were obtained between cisplatin-treated group and combination therapy group which suggested that Oroxylin A sensitizes cisplatin cytotoxicity through regulation of XPC (Fig. 6c–e, and Supplementary Fig. S6B).
Overall, these data demonstrate that Oroxylin A improves the efficacy of cisplatin in vitro and in vivo.
Discussion
During the past years, new therapeutic strategies have been developed for NSCLC, such as anti-PD-1/PD-L1 agents and AZD9291 which overcomes T790M-mediated resistance to EGFR inhibitors. Though, cisplatin-based therapy still remains the golden standard treatment of advanced non-small cell lung cancer [2]. Anti-cancer potency of cisplatin is due to formation of platinum-DNA adducts, which eventually causes cell cycle arrest or apoptosis [21]. Nucleotide excision repair, a versatile DNA repair pathway that eliminates a wide variety of helix-distorting base lesions such as platinum-DNA adducts, is proposed to be one of the most crucial determinants for cisplatin resistance [23, 24]. Hypoxia, which commonly exists in solid tumors, leads to cancer cell chemoresistance including cisplatin resistance via diverse mechanisms. Recent reports showed that HIF-1α-induced cisplatin resistance is mediated by regulating nucleotide excision repair-related protein XPA [40]. In the present study, the NER-related proteins were screened and the data showed that XPA slightly increased under hypoxia, while another NER-related protein XPC dramatically increased, which suggested that XPC may also play an important role in hypoxia-induced cisplatin resistance. Consistent with our hypothesis and other findings [41], TCGA data analysis showed that lower XPC level was associated with better OS in NSCLC patients treated with cisplatin, indicating the role of XPC in cisplatin resistance. HIF-1α as an important transcription factor, which is induced under hypoxia, was reported to prevent etoposide-induced DNA damage in cancer cells. In the present study, we firstly pinpointed that the binding of HIF-1α to HRE3 transcription site on XPC promoter activated XPC transcription, which eventually reduced the accumulation of cisplatin-induced DNA damage. Our findings reveal a previously unexpected role of XPC in hypoxia-induced cisplatin resistance, elucidate a crucial role of HIF-1α in NER and provide an important molecular mechanism linking hypoxia and genomic integrity in cancer.
In different cancer cell lines, combination of different drugs with Oroxylin A leads to significantly better antitumor effects than single-drug treatments because of cumulative or synergistic effects [32, 36, 37, 42, 43]. Here we demonstrated that, compared to cisplatin alone, co-treatment with the flavonoid Oroxylin A induced higher pro-apoptotic and anti-proliferative activity in NSCLC cells. Our in vitro and in vivo data indicated that co-treatment with Oroxylin A and cisplatin significantly reduced NSCLC cell viability compared to cisplatin alone under hypoxic condition (Figs. 1 and 6). Due to the dose-dependent toxicity and drug resistance of platinum-based compounds in clinic, the efficacy of cisplatin in non-small cell lung cancer treatment is very limited. Unfortunately, an increased drug dose often leads to increased toxicity. In this study, the in vivo animal results showed that the combination of Oroxylin A and cisplatin significantly decreased H460 tumor volumes and tumor weights compared to each drug treatment alone, which suggested that Oroxylin A combined with platinum-based compounds has potential therapeutic value in targeting non-small cell lung cancer. Both in vitro and in vivo study showed the low toxicity of Oroxylin A [44], indicating that a lower dose of cisplatin combined with Oroxylin A could be used in order to minimize the toxicity of cisplatin.
Oroxylin A inhibits HIF-1α/hedgehog pathway by promoting the degradation of HIF-1α on glioma cells [35]. However, our studies suggest that Oroxylin A did not affect the level of HIF-1α protein in NSCLC cells under hypoxia, whereas it markedly restrained its binding to XPC promotor (Fig. 5). In the present study, three truncations of HIF-1α were generated. In general, the bHLH-PAS motifs are indispensable to form the heterodimer of HIF-1α and HIF-1β subunits and for their binding to the HRE-DNA sequence on the target genes [45]. All HIF-1α subunits have an oxygen-dependent degradation domain (ODDD) overlapping N-TAD in their structures. This ODD domain is important in mediating O2 regulated stability [46]. An inhibitor domain (ID) is located in the C-terminal of HIF-1α, which could repress its transcriptional activity [47]. Oroxylin A specifically bound to bHLH-PAS domain, in accordance with its inhibition effect on HIF-1α DNA binding ability. Although our data clearly indicate the interaction between Oroxylin A and HIF-1α bHLH-PAS motif in H460 cells, we cannot exclude the possibility that Oroxylin A also affects the stability of HIF-1α in other cell lines.
HIF-1α is considered to be a promising target for the development of novel therapeutics against cancer metastasis [48, 49]. In recent years, a number of small-molecule inhibitors have been identified to reduce the expression of HIF-1α or to induce HIF-1α degradation [50,51,52,53]. However, the expression of genes that promote cell death, such as BNIP3 and NIX (BNIP3L), is also induced by hypoxia through HIF-1α [54]. In this sense, HIF-1α plays dual roles in the survival and death pathways of tumor cells [55]. Decreasing HIF-1α protein levels may affect the expression of most genes induced by hypoxia. A functional discrepancy of these targets of HIF-1α at the level of HIF-1α–DNA binding might have clinical relevance [55]. Therefore, Oroxylin A could be a potential drug that interferes with the HIF-1α–DNA interaction.
In view of a clinical application, we are aware that Oroxylin A has limitations in that it specifically increases the efficacy of cisplatin on NSCLC cells under hypoxic condition, while has no significant effect on cisplatin-resistance cell lines. Our results showed that overexpression of most NER-related proteins and accelerated removal of Pt-DNA adducts were observed in A549/CDDP cells (Supplementary Fig. S2C, D). The mechanisms of enhanced NER capacity are complicated. It is reported that overexpression of Rap1 interacting factor 1 (RIF1) enhances the NER activity by promoting the NER proteins in ovarian cancer cells [56], and that autophagy positively regulates NER through enhancing DNA damage recognition [57]. In our study, Oroxylin A impaired NER through inhibiting HIF-1α-induced XPC transcription, but did not play a role in the other pathways that regulates NER.
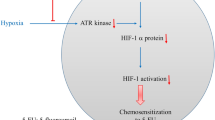
In summary, our findings demonstrate that hypoxia positively regulates cisplatin-DNA damage recognition by nucleotide excision repair via stimulating HIF-1α-mediated XPC transcription. Oroxylin A directly interacts with HIF-1α bHLH-PAS motif and suppresses XPC transcription through inhibiting the binding of HIF-1α to the XPC promoter (Fig. 7). Our findings provide an effective therapeutic approach to enhance cisplatin-induced apoptosis in NSCLC cells especially under hypoxic condition, providing theoretical evidence that Oroxylin A may be a promising anti-cancer agent for further clinical development.

Under normoxic condition, cisplatin generates intra-strand and inter-strand crosslinks (ICLs) that activates DNA damage response (DDR) and, subsequently, induces cell apoptosis. While under hypoxic condition, HIF-1α increases XPC mRNA expression by directly binding to a hypoxia response element (HRE) in the XPC promoter region. Increased XPC expression promotes NER dependent removal of Pt-DNA adducts and attenuates cisplatin-induced apoptosis and growth inhibition. Oroxylin A binds to HIF-1α bHLH motif and inhibits HIF-1α binding to the XPC promotor, thus downregulates XPC expression and reverse hypoxia-induced cisplatin resistance.
Materials and methods
Cell lines and reagents
Non-small cell lung cancer cells H460 cells, A549 cells, 95D cells, PC9 cells, HCC827 cells, and H1975 cells were purchased from Cell Bank of Shanghai Institute of Biochemistry & Cell Biology, Chinese Academy of Sciences (Shanghai, China). CDDP-resistant A549/CDDP cells [58] were kindly provided by Dr. Feihong Chen (Southeast University at Nanjing, China). Cells were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA, 31800-089) with 10% fetal bovine serum (Biological Industries Israel Beit Haemek Ltd., Kibbutz Beit Haemek, Israel, 04-001-1ACS), 100 units per mL penicillin and 100 μg per mL streptomycin (Biological Industries Israel Beit Haemek Ltd., Kibbutz Beit Haemek, Israel, 03-031-1B) at 37 °C with the CO2 concentration 5%. To maintain drug resistance, A549/CDDP cells were maintained in the presence of 2 μM cisplatin. These above cell lines were authenticated by short tandem repeats (STRs) DNA profiling. All cells were tested for mycoplasma contamination before use with the Myco-Blue Mycoplasma Detector (Vazyme Biotec, Nanjing, China, D101-02) and were not contaminated by mycoplasma. Oroxylin A (Oroxylin A purity>99%) and Biotin-Oroxylin A were prepared at China Pharmaceutical University (Nanjing, China). The compound was dissolved in dimethyl sulfoxide (DMSO, Sigma–Aldrich, St. Louis, USA, D2650) to a concentration of 100 mM and stored at −20 °C. Cisplatin was purchased from Selleck Chemicals (Shanghai, China, S1166) and dissolved in N,N-Dimethylformamide (DMF, Selleck Chemicals, Shanghai, China, S6192) to 5 mM. Dimethyloxalylglycine (DMOG, Selleck Chemicals, Shanghai, China, S7483) was dissolved in water to 0.1 M and was diluted with RPMI-1640 to its final concentration.
DNA constructs
We cloned XPC from the pCMV vector to the pENTER vector. pENTER XPC were recombined into pLenti CMV Dest vector using Gateway LR Clonase Enzyme Mix (Invitrogen, Carlsbad, CA, 11791043) following the manufacturer’s instructions. The XPC promoters were cloned from genomic DNA purified from NHEK cells based on publicly available sequence data. Nested PCR was necessary to clone the promoters. The PCR product was digested and cloned into pGL3-Basic vectors. We cloned synthesized-FLAG to pCMV vector. The HIF-1α protein sequence was cloned from cDNA purified from 293T cells based on publicly available sequence data. Nested PCR was necessary to clone the protein sequence. The PCR product was digested and cloned into pCMV Flag vector and the pGEX-6P-1. We then cloned HIF-1α bHLH/PAS domain, ODD domain and Inhibitor domain into the pGEX-6P-1. DNA fragment encoding HIF-1α-shRNA were cloned into pLKO.1 vector. The primers are listed in Supplementary Table S1. All constructs were confirmed by sequencing.
Real-time PCR analysis
Total RNA was extracted using RNAiso Plus (Vazyme Biotech, Nanjing, China, R401-01). 1 μg of total RNA was used to transcribe the first-strand cDNA with HiScript II Q RT SuperMix (Vazyme Biotech, Nanjing, China, R223-01). Real-time PCR was completed on a LightCycler® 480 Instrument II (Roche, Basel, Switzerland). ChamQ SYBR qPCR Master Mix was purchased from Vazyme Biotech (Nanjing, China, Q331-02). Forward and reverse primers for targeted mRNA were purchased from Sangon Biotech (Shanghai, China). The threshold cycle number (CQ) for each sample was determined in triplicate and normalized against GAPDH as described previously [57]. Amplification primers are listed in Supplementary Table S2.
Statistical analysis
Sample size was determined upon availability, and chosen based on previous experience and previous studies. No exclusion criteria were pre-established. Samples or animals were randomly allocated among groups. Investigators were not blinded to group allocation during data collection and analyzes. No data were excluded. All statistical tests were performed with SigmaPlot 14.0. Data shown were presented by means ± Standard Deviation (S.D.) from triplicate experiments performed in a parallel manner unless otherwise indicated. Statistical significance was performed using an unpaired two-tailed Student’s t test for two data sets when the data met the normal distribution tested by F-test. The variance between comparison groups was not similar.
Detailed descriptions of all methods are provided in Supplementary Materials and Methods.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Deben C, Deschoolmeester V, Lardon F, Rolfo C, Pauwels P. TP53 and MDM2 genetic alterations in non-small cell lung cancer: evaluating their prognostic and predictive value. Crit Rev Oncol Hematol. 2016;99:63–73.
Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ Jr., Wu YL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299–311.
Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer. 2011;71:3–10.
Shannon AM, Bouchier-Hayes DJ, Condron CM, Toomey D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev. 2003;29:297–307.
Song X, Liu X, Chi W, Liu Y, Wei L, Wang X, et al. Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1alpha gene. Cancer Chemother Pharm. 2006;58:776–84.
Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225–39.
Wohlkoenig C, Leithner K, Deutsch A, Hrzenjak A, Olschewski A, Olschewski H. Hypoxia-induced cisplatin resistance is reversible and growth rate independent in lung cancer cells. Cancer Lett. 2011;308:134–43.
Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–34.
Brown LM, Cowen RL, Debray C, Eustace A, Erler JT, Sheppard FC, et al. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol Pharm. 2006;69:411–8.
Nardinocchi L, Puca R, Sacchi A, D’Orazi G. Inhibition of HIF-1alpha activity by homeodomain-interacting protein kinase-2 correlates with sensitization of chemoresistant cells to undergo apoptosis. Mol Cancer. 2009;8:1.
Sowa T, Menju T, Chen-Yoshikawa TF, Takahashi K, Nishikawa S, Nakanishi T, et al. Hypoxia-inducible factor 1 promotes chemoresistance of lung cancer by inducing carbonic anhydrase IX expression. Cancer Med. 2017;6:288–97.
Roberts AM, Watson IR, Evans AJ, Foster DA, Irwin MS, Ohh M. Suppression of hypoxia-inducible factor 2alpha restores p53 activity via Hdm2 and reverses chemoresistance of renal carcinoma cells. Cancer Res. 2009;69:9056–64.
Yang SL, Ren QG, Wen L, Hu JL. Clinicopathological and prognostic significance of hypoxia-inducible factor-1 alpha in lung cancer: a systematic review with meta-analysis. J Huazhong Univ Sci Technol Med Sci. 2016;36:321–7.
Yohena T, Yoshino I, Takenaka T, Kameyama T, Ohba T, Kuniyoshi Y, et al. Upregulation of hypoxia-inducible factor-1alpha mRNA and its clinical significance in non-small cell lung cancer. J Thorac Oncol. 2009;4:284–90.
Mazumdar J, Hickey MM, Pant DK, Durham AC, Sweet-Cordero A, Vachani A, et al. HIF-2alpha deletion promotes Kras-driven lung tumor development. Proc Natl Acad Sci USA. 2010;107:14182–7.
Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Mol Cell. 2010;40:509–20.
Olcina MM, Foskolou IP, Anbalagan S, Senra JM, Pires IM, Jiang Y, et al. Replication stress and chromatin context link ATM activation to a role in DNA replication. Mol Cell. 2013;52:758–66.
Wirthner R, Wrann S, Balamurugan K, Wenger RH, Stiehl DP. Impaired DNA double-strand break repair contributes to chemoresistance in HIF-1 alpha-deficient mouse embryonic fibroblasts. Carcinogenesis. 2008;29:2306–16.
To KK, Sedelnikova OA, Samons M, Bonner WM, Huang LE. The phosphorylation status of PAS-B distinguishes HIF-1alpha from HIF-2alpha in NBS1 repression. EMBO J. 2006;25:4784–94.
Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107:1387–407.
Wood RD, Araujo SJ, Ariza RR, Batty DP, Biggerstaff M, Evans E, et al. DNA damage recognition and nucleotide excision repair in mammalian cells. Cold Spring Harb Symp Quant Biol. 2000;65:173–82.
Liu RY, Dong Z, Liu J, Yin JY, Zhou L, Wu X, et al. Role of eIF3a in regulating cisplatin sensitivity and in translational control of nucleotide excision repair of nasopharyngeal carcinoma. Oncogene. 2011;30:4814–23.
Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. 2008;14:1291–5.
Furuta T, Ueda T, Aune G, Sarasin A, Kraemer KH, Pommier Y. Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res. 2002;62:4899–902.
Selvakumaran M, Pisarcik DA, Bao R, Yeung AT, Hamilton TC. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003;63:1311–6.
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–83.
Hu LB, Chen Y, Meng XD, Yu P, He X, Li J. Nucleotide excision repair factor XPC ameliorates prognosis by increasing the susceptibility of human colorectal cancer to chemotherapy and ionizing radiation. Front Oncol. 2018;8:290.
Slyskova J, Sabatella M, Ribeiro-Silva C, Stok C, Theil AF, Vermeulen W, et al. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018;46:9537–49.
Xie X, Huang N, Zhang Y, Wei X, Gao M, Li M, et al. MiR-192-5p reverses cisplatin resistance by targeting ERCC3 and ERCC4 in SGC7901/DDP cells. J Cancer. 2019;10:1039–51.
Lee JY, Park W. Anti-inflammatory effects of oroxylin A on RAW 264.7 mouse macrophages induced with polyinosinic-polycytidylic acid. Exp Ther Med. 2016;12:151–6.
Cao H, Li W, Zhou Y, Tan R, Yang Y, Zhou Y, et al. Oroxylin a inhibits the protection of bone marrow microenvironment on CML cells through CXCL12/CXCR4/P-gp signaling pathway. Front Oncol. 2019;9:188.
Jin J, Chen S, Wang D, Chen Y, Wang Y, Guo M, et al. Oroxylin A suppresses influenza A virus replication correlating with neuraminidase inhibition and induction of IFNs. Biomed Pharmacother. 2018;97:385–94.
Li W, Ding Q, Ding Y, Lu L, Wang X, Zhang Y, et al. Oroxylin A reverses the drug resistance of chronic myelogenous leukemia cells to imatinib through CXCL12/CXCR7 axis in bone marrow microenvironment. Mol Carcinog. 2017;56:863–76.
Wei M, Ma R, Huang S, Liao Y, Ding Y, Li Z, et al. Oroxylin A increases the sensitivity of temozolomide on glioma cells by hypoxia-inducible factor 1alpha/hedgehog pathway under hypoxia. J Cell Physiol. 2019;234:17392–404.
Zhu L, Zhao L, Wang H, Wang Y, Pan D, Yao J, et al. Oroxylin A reverses P-glycoprotein-mediated multidrug resistance of MCF7/ADR cells by G2/M arrest. Toxicol Lett. 2013;219:107–15.
Ha J, Zhao L, Zhao Q, Yao J, Zhu BB, Lu N, et al. Oroxylin A improves the sensitivity of HT-29 human colon cancer cells to 5-FU through modulation of the COX-2 signaling pathway. Biochem Cell Biol. 2012;90:521–31.
Akanji MA, Rotimi D, Adeyemi OS. Hypoxia-inducible factors as an alternative source of treatment strategy for cancer. Oxid Med Cell Longev. 2019;2019:8547846.
Kaluz S, Kaluzova M, Stanbridge EJ. Regulation of gene expression by hypoxia: integration of the HIF-transduced hypoxic signal at the hypoxia-responsive element. Clin Chim Acta. 2008;395:6–13.
Liu Y, Bernauer AM, Yingling CM, Belinsky SA. HIF1alpha regulated expression of XPA contributes to cisplatin resistance in lung cancer. Carcinogenesis. 2012;33:1187–92.
Lai TC, Chow KC, Fang HY, Cho HC, Chen CY, Lin TY, et al. Expression of xeroderma pigmentosum complementation group C protein predicts cisplatin resistance in lung adenocarcinoma patients. Oncol Rep. 2011;25:1243–51.
Yang HY, Zhao L, Yang Z, Zhao Q, Qiang L, Ha J, et al. Oroxylin A reverses multi-drug resistance of human hepatoma BEL7402/5-FU cells via downregulation of P-glycoprotein expression by inhibiting NF-kappaB signaling pathway. Mol Carcinog. 2012;51:185–95.
Zhu B, Zhao L, Zhu L, Wang H, Sha Y, Yao J, et al. Oroxylin A reverses CAM-DR of HepG2 cells by suppressing Integrinbeta1 and its related pathway. Toxicol Appl Pharm. 2012;259:387–94.
Lu L, Guo Q, Zhao L. Overview of Oroxylin A: a promising flavonoid compound. Phytother Res. 2016;30:1765–74.
Masoud GN, Li W. HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. 2015;5:378–89.
Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40.
Chun YS, Kim MS, Park JW. Oxygen-dependent and -independent regulation of HIF-1alpha. J Korean Med Sci. 2002;17:581–8.
Li Z, You Q, Zhang X. Small-molecule modulators of the hypoxia-inducible factor pathway: development and therapeutic applications. J Med Chem. 2019;62:5725–49.
Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharm Sci. 2012;33:207–14.
An H, Lee S, Lee JM, Jo DH, Kim J, Jeong YS, et al. Novel hypoxia-inducible factor 1alpha (HIF-1alpha) inhibitors for angiogenesis-related ocular diseases: discovery of a novel scaffold via ring-truncation strategy. J Med Chem. 2018;61:9266–86.
Kwon DY, Lee HE, Weitzel DH, Park K, Lee SH, Lee CT, et al. Synthesis and biological evaluation of manassantin analogues for hypoxia-inducible factor 1alpha inhibition. J Med Chem. 2015;58:7659–71.
Lee K, Lee JH, Boovanahalli SK, Jin Y, Lee M, Jin X. et al. (Aryloxyacetylamino)benzoic acid analogues: a new class of hypoxia-inducible factor-1 inhibitors. J Med Chem. 2007;50:1675–84.
Naik R, Won M, Kim BK, Xia Y, Choi HK, Jin G, et al. Synthesis and structure-activity relationship of (E)-phenoxyacrylic amide derivatives as hypoxia-inducible factor (HIF) 1alpha inhibitors. J Med Chem. 2012;55:10564–71.
Kothari S, Cizeau J, McMillan-Ward E, Israels SJ, Bailes M, Ens K, et al. BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene. 2003;22:4734–44.
Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett. 2006;237:10–21.
Liu YB, Mei Y, Tian ZW, Long J, Luo CH, Zhou HH. Downregulation of RIF1 enhances sensitivity to platinum-based chemotherapy in epithelial ovarian cancer (EOC) by regulating nucleotide excision repair (NER) pathway. Cell Physiol Biochem. 2018;46:1971–84.
Qiang L, Zhao B, Shah P, Sample A, Yang S, He YY. Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy. 2016;12:357–68.
Teng X, Fan XF, Li Q, Liu S, Wu DY, Wang SY, et al. XPC inhibition rescues cisplatin resistance via the Akt/mTOR signaling pathway in A549/DDP lung adenocarcinoma cells. Oncol Rep. 2019;41:1875–82.
Acknowledgements
We are grateful to Dr. Feihong Chen (Southeast University at Nanjing, China) for kindly providing CDDP-resistant A549/CDDP cells and Dr. Zhaoqiu Wu (China Pharmaceutical university, Nanjing, China) for kindly providing pGEX-6P-1 plasmids.
Funding
This work was supported by the China Postdoctoral Science Foundation Grant (2019M652035); the General Program of National Natural Science Foundation of China (81772911, 81974425, and 81903648); the Natural Science Foundation of Jiangsu Province (BK20170744); the Six Talent Peaks Project in Jiangsu Province (SWYY-095).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, Y., Wang, X., Li, W. et al. Oroxylin A reverses hypoxia-induced cisplatin resistance through inhibiting HIF-1α mediated XPC transcription. Oncogene 39, 6893–6905 (2020). https://doi.org/10.1038/s41388-020-01474-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-01474-x
- Springer Nature Limited