
Abstract
Richter transformation (RT) is defined as development of aggressive lymphoma in patients (pts) with CLL. The incidence rates of RT among pts with CLL range from 2 to 10%. The aim of this analysis is to report the frequency, characteristics and outcomes of pts with RT enrolled in trials of the GCLLSG. A total of 2975 pts with advanced CLL were reviewed for incidence of RT. Clinical, laboratory, and genetic data were pooled. Time-to-event data, starting from time of CLL diagnosis, of first-line therapy or of RT diagnosis, were analyzed by Kaplan-Meier methodology. One hundred and three pts developed RT (3%): 95 pts diffuse large B-cell lymphoma (92%) and eight pts Hodgkin lymphoma (8%). Median observation time was 53 months (interquartile range 38.1–69.5). Median OS from initial CLL diagnosis for pts without RT was 167 months vs 71 months for pts with RT (HR 2.64, CI 2.09–3.33). Median OS after diagnosis of RT was 9 months. Forty-seven pts (46%) received CHOP-like regimens for RT treatment. Three pts subsequently underwent allogeneic and two pts autologous stem cell transplantation. Our findings show that within a large cohort of GCLLSG trial participants, 3% of the pts developed RT after receiving first-line chemo- or chemoimmunotherapy. This dataset confirms the ongoing poor prognosis and high mortality associated with RT.
Similar content being viewed by others


Introduction
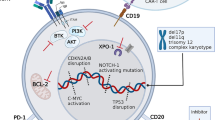
Richter transformation (RT) describes the development of a more aggressive lymphoma in patients with chronic lymphocytic leukemia (CLL) [1]. Transformation occurs due to the acquisition of multiple genetic defects that facilitate rapid proliferation [2]. RT is primarily defined as diffuse large B-cell lymphoma (DLBCL) variant, which accounts for ~90% of RT cases [3]. Hodgkin lymphoma (HL) variants are additionally recognized [4, 5]. Symptoms associated with RT are B-symptoms and progressive lymphadenopathy. A biopsy from suspect lymph nodes can ultimately confirm the diagnosis of RT based on histopathological features that include presence of enlarged CD20 + B cells with a diffuse growth pattern of large cells, similar to de novo DLBCL [1, 6, 7]. Distinction to de novo DLBCL, which has a better prognosis than RT, can be made via analysis of immunoglobulin genes. Based on the immunoglobulin gene sequence, 80% of DLBCL-type RT are detected as clonally related to the preceding CLL [2, 8]. Also, DLBCL-type RT harbors PD1 expression in ~80% of cases, which is uncommon in de novo DLBCL [9, 10].
The incidence rates of RT among CLL patients range from 2 to 10% [11, 12]. The median time from diagnosis of CLL to transformation ranges between 2 and 4 years [11, 13, 14]. Risk factors for development of RT include intrinsic biological features like TP53 aberrations, NOTCH1 mutation, subset eight stereotype, as well as therapy-related factors like exposure to purine analogues like fludarabine. However, up to one third of patients with RT are treatment-naïve patients with CLL [15, 16].
RT patients have a very poor prognosis with a median overall survival (OS) of 6–8 months. There is no established standard of care for RT and most patients are treated comparably to de novo DLBCL patients with chemoimmunotherapies like rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone (R-CHOP) or rituximab, dexamethasone, cytarabine, and cisplatin (R-DHAP) [15,16,17]. While this regimen achieves high response rates in de novo DLBCL and even cures up to 80% of patients, patients with RT are rarely cured by chemoimmunotherapy and response rates are considerably lower between 20–60% [16]. Given the poor prognosis, fit patients are considered for allogeneic transplantation once they respond to therapy. However, as CLL is a disease of the elderly with a median age of 72 years, most patients with RT are not fit enough to undergo allogeneic transplantation. Novel therapeutic strategies consider the use of targeted agents as well as immunotherapeutic approaches via checkpoint inhibition or chimeric antigen receptor T (CART) cell therapy [18].
So far, only a few collective analyses on the incidence of RT have been published; most reports on RT frequency are from separate clinical trials with limited follow-up and heterogenous treatments [12,13,14, 19]. The aim of this analysis was to assess frequency, characteristics an outcome of patients with RT based on a large number of patients with CLL treated within trials of the German CLL Study Group (GCLLSG).
Methods
Dataset
For this analysis, individual patient data of eight prospective multicenter phase II and phase III front line treatment trials with complete documentation and follow-up of the GCLLSG (CLL4, CLL5, CLL8, CLL10, CLL11, CLL2-BAG, CLL2-BIG, CLL2M; recruitment between 1999 and 2016; total N = 2975) [20,21,22,23,24,25,26,27] were reviewed for patients with a RT. Histopathological diagnosis of RT was done by local pathologists. Baseline characteristics and results of central diagnostics from serum markers (serum ß2 microglobulin [B2M], serum thymidine kinase [TK]) and genetic parameters (deletion 17p, deletion 11q, trisomy 12, deletion 13q, translocation 14q, IGHV status, including somatic hypermutation status, TP53 mutation, as previously described [28] as well as event-related data (time to RT, overall survival) were pooled. Median observation time for the whole study cohort was 53 months (interquartile range 38.1–69.5).
Statistical methods
Statistical analysis was performed based on all patients who received at least one dose of study medication. We compared categorical variables using the Fisher Exact test, and continuous variables using Mann-Whitney U test. Time to event parameters included time to RT and overall survival (OS) and were estimated using the Kaplan-Meier method. Time was measured starting from time of CLL diagnosis, time of first-line therapy and time of RT diagnosis. Hazard ratios (HR) and 95% confidence intervals (CI) were calculated using Cox proportional hazards regression modelling. Independent prognostic factors for PFS and OS were identified by multivariate analyses using Cox proportional hazards regression modelling with stepwise forward and backward selection procedures. All variables (age, sex, Binet stage, ECOG, comorbidities, B-symptoms, serum ß2-microglobulin and thymidine kinase, IGHV/TP53/Notch1/SF3B1 status, deletion 17p/11q/13q, trisomy 12, leukocyte and platelet count and LDH) with significant association in univariate Cox regression analyses were included into the multivariate analysis. Statistical tests were two-sided and statistical significance was defined as a p-value < 0.05 without adjustments for multiple testing. The analysis was performed with SPSS v23 (SPSS, Chicago, IL).
Results
Patient characteristics
Among a total of 2975 participants of eight GCLLSG trials, 103 (3.6%) developed RT during a median observation time of 53 months (interquartile range 38.1–69.5). Based on local histopathological examinations, 95 (92%) of those had transformed to aggressive NHL (diffuse large B-cell lymphoma) and eight (8%) had transformed to Hodgkin’s lymphoma. Characteristics of these 103 patients at baseline are listed in Table 1. Median age at first-line therapy was 62 years (range 36–81 years) in patients who later developed RT and 64 years (range 24–90 years) in patients who did not develop RT (nRT). Median age at diagnosis of RT was 65 years (range 41–86 years).
Frequency of some adverse risk factors at study entry were more common in RT patients than in nRT patients, including del(17p) (14.8 vs 6.3%, p = 0.006) and very high risk CLL-IPI (18.4 vs 7.8%, p = 0.004).
Stereotyped B-cell receptor was detected in 21.4% of patients with later development of RT. However, only one patient featured subset 8, which is particularly associated with RT (see supplementary Table 1) [29].
RT patients with HL (n = 8) in contrast to patients with NHL (n = 95) had a longer median time to RT (37.2 months vs 9.7 months), smaller proportion of deletion 17p (0 vs 12%) and higher proportion of mutated IGHV (57.1 vs 27%) (supplementary Table S2).
Treatment
Since this analysis focusses on patients with advanced stage and need of therapy, all patients have received at least one prior therapy. The majority of patients who were later diagnosed with RT had either received fludarabine-containing regimens as first-line treatment (76.7%) or rituximab (BR) (13.6%). The remaining patients received chlorambucil-containing regiments (9.7%) (Table 2). The median number of prior therapies was 1 (range 1–11).
Data on RT treatment were available for 62 patients (60.2%). For patients with DLBCL RT, 47 patients (45.6%) received CHOP-like regimens (cyclophosphamide, doxorubicin, vincristine, prednisolone) as initial treatment for RT. Of the eight patients with HL RT two patients (1.9%) had received BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisolone) and one patient (1.0%) had received ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine). The remaining patients (11.7%) received other combinations of chemotherapeutic agents (Table 3). Three patients (2.9%) underwent allogeneic hematopoietic stem cell transplantation and 2 (1.9%) underwent autologous stem cell transplantation after RT diagnosis.
Time to transformation
Median time from CLL diagnosis to start of first-line treatment was 12.4 months for patients who later developed RT, whereas time to first-line treatment was 90.2 months in patients who did not develop RT. Median time from CLL first-line treatment to RT diagnosis was not reached. After 3 years, 2.1% of patients with advanced stage CLL had developed RT and after 12 years, 8.2% (Fig. 1).

RT Richter Transformation, Pts Patients.
Multivariate analysis, including B-symptoms, deletion 17p, deletion 13q, and serum thymidine kinase, was performed to identify independent prognostic factors for time to RT from first-line treatment. According to a final multivariable model, deletion 17p (HR 3.457, [1.561–7.655], p = 0.002), thymidine kinase > 10 U/L (HR 3.991, [1.823–8.739], p = 0.001) and presence of B-symptoms (HR 1.942, [1.200–3.145], p = 0.007) were independent prognostic factors.
Overall survival
Median overall survival (OS) after diagnosis of CLL was 166.8 months in patients without RT and 71 months in patients with RT (HR 2.639, [2.094–3.325]) (Fig. 2a). Median OS after first-line treatment of CLL was 99.9 months in patients without RT and 53.7 months in patients with RT (HR 2.710, [2.151–3.412]) (Fig. 2b). Median OS after diagnosis of RT was 9.4 months (Fig. 2c). Patients with transformation to aggressive NHL had a significantly shorter OS after transformation of 8.7 months, whereas patients with transformation to Hodgkin lymphoma had a median OS after transformation of 82.6 months (HR 0.182, [0.044–0.748]) (Fig. 2d). Patients who underwent allogeneic stem cell transplantation (n = 3) had a median OS of 17.9 months.

Overall survival with and without RT after diagnosis of CLL (a), after first-line treatment of CLL (b), after diagnosis of RT (c), and according to type of RT after diagnosis of RT (d). RT Richter Transformation, nRT No Richter Transformation, HL Hodgkin’s lymphoma, NHL Non-Hodgkin’s Lymphoma, OS overall survival.
The standardized mortality ratio (SMR), calculated to show the risk of death in comparison to a healthy, aged-matched German population, indicated an increased risk of death by 8.66 (CI 8.86–10.80) in patients with RT, while CLL patients without RT had an increased risk of death by 2.62 (CI 2.44–2.80).
Discussion
Over the past years, substantial progress has been achieved in the management and treatment of patients with advanced CLL. In contrast, one of the biggest remaining challenges in the management of CLL are transformations into an aggressive phenotype. The aim of this analysis was to systematically evaluate the incidence and outcome of patients with RT within a large cohort of participants in front line treatment trials on CLL.
In this analysis, which covered 2975 study participants from 1999 to 2016, 3.5% of patients developed RT at a median observation time of 53 months. This is comparable to other reports where an incidence between 2 and 9% has been reported [11, 12, 14, 15, 30, 31, 32, 33]. Patients had a median age of 65 years at the time of transformation, which is also similar to previous reports [11, 12, 17, 30], although one retrospective analysis suggested transformations to be more prevalent at a younger age of ≤55 years [34].
Among RT patients adverse prognostic factors as TP53 aberrations, deletion 11q or unmutated IGHV status were common at the time, when they received first-line treatment of CLL. However, patients were not systematically re-examined at the time of transformation, hence, the rate of chromosomal aberrations and TP53 mutations at time of transformation cannot be calculated reliably. Some studies have indicated that the rate of deletion 17p and TP53 mutations increases by the line of therapy and can go up to 30 or 40% of patients [35, 36]. Moreover, stereotyped BCR has been associated with increased risk of RT and frequencies of up to 43% have been observed in RT patients [29, 37]; in our cohort 21.3% of patients with RT had a stereotyped BCR, but only one patient carried subset 8, which has been particularly associated with an increased risk of RT.
In contrast to this pooled analysis, a review of RT patients at the MD Anderson Cancer Center showed a broad range of clinical features associated with RT, e.g. elevated LDH or progressive lymphadenopathy, but no association of chromosomal groups with development of RT was observed [12].
Based on a multivariate analysis of our cohort, independent prognostic factors for RT development were deletion 17p, elevated thymidine kinase > 10 U/L and presence of B-symptoms at the time of first-line treatment of CLL. While deletion 17p has been known to be a risk factor for genetic instability and therefore RT, elevated TK and B-symptoms have not been known as distinct risk factors yet [1, 19].
The majority of RT patients for which treatment data were available received CHOP-like regimens, which is in line with current national and international recommendations. Interestingly, allogeneic stem cell transplantation was only performed in three patients and survival was considerably longer than in those who did not receive transplantation (although number of patients with very limited). As the median age of RT patients was 65 years in this analysis, age was probably not the leading cause for transplant ineligibility. Response rates to RT therapy were not available for this meta-analysis. However, estimated ORR to CHOP-like regimens in RT patients reportedly was between 30–50% [38, 39] and of 67% for one trial [40]. Thus, it is possible that due to a lack of sufficient remissions, patients could not be transferred to allogeneic stem cell transplantation. This explains the particularly poor overall survival after RT diagnosis with a median of 9.4 months. Comparable survival rates between 6 and 12 months have been observed in other retrospective analyses [11, 12, 41]. Interestingly, based on histological distinction, patients with transformation to HL had a median OS of 82.6 months in contrast to those with NHL of only 8.7 months. This underlines that the definition of the histological entity is crucial as treatment with ABVD or BEACOPP in these RT patients seems to achieve good results.
Due to the retrospective nature of this pooled analysis, some limitations have to be considered. While all transformations in this analysis were biopsy-confirmed, no central histopathological examinations were performed. As diagnostic work-up of RT can be challenging, there is a chance of false-positive but also false-negative cases [7]. As data on treatment and response to treatment were not available for all patients at the time of RT, information on the therapeutic efficacy of regimens for RT, such as R-CHOP, are limited. Moreover, all patients in this RT cohort had received chemo- or chemoimmunotherapy as front-line regimen; thus, the results might differ from patients who received novel agents for first-line therapy. So far, clinical trials with front-line ibrutinib or venetoclax have reported a frequency of between 0.5 and 1.5% [42,43,44], although follow-up is very limited compared to chemoimmunotherapy trials. Data on distinct characteristics and outcome of these transformations have not been published yet.
Finally, data on clonal relationship of DLBCL were not available. However, as clonal unrelated de novo DLBCL has a median OS of ~5 years, the very short OS described here indicates that the majority were probably clonally related DLBCL cases, as those cases have been described in 80% of the cohort [2].
In conclusion, our data confirm that RT is a rare event but remains a clinical challenge with a disappointing outcome. Chemoimmunotherapy with R-CHOP is widely used as a first-line treatment for RT, however, despite good outcomes in de novo DLBCL, median OS was very poor with 8.7 months in DLBCL-RT patients. Clinical trials evaluating immunotherapeutic approaches with checkpoint inhibitors or CART cells together with targeted agents yield promising efficacy and warrant further investigations [45,46,47,48].
References
Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131:2761–72.
Rossi D, Spina V, Deambrogi C, Rasi S, Laurenti L, Stamatopoulos K, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117:3391–401.
Swerdlow SH. WHO classification of tumours of haematopoietic and lymphoid tissues. WHO classification tumours 2008. 2008;22008:439.
Bockorny B, Codreanu I, Dasanu CA. Hodgkin lymphoma as Richter transformation in chronic lymphocytic leukaemia: a retrospective analysis of world literature. Br J Haematol. 2012;156:50–66.
Brecher M, Banks PM. Hodgkin’s disease variant of Richter’s syndrome. Report of eight cases. Am J Clin Pathol. 1990;93:333–9.
Mato AR, Wierda WG, Davids MS, Cheson BD, Coutre SE, Choi M, et al. Utility of PET-CT in patients with chronic lymphocytic leukemia following B-cell receptor pathway inhibitor therapy. Haematologica. 2019;104:2258–64.
Federmann B, Mueller MR, Steinhilber J, Horger MS, Fend F. Diagnosis of Richter transformation in chronic lymphocytic leukemia: histology tips the scales. Ann Hematol. 2018;97:1859–68.
Mao Z, Quintanilla-Martinez L, Raffeld M, Richter M, Krugmann J, Burek C, et al. IgVH mutational status and clonality analysis of Richter’s transformation: diffuse large B-cell lymphoma and Hodgkin lymphoma in association with B-cell chronic lymphocytic leukemia (B-CLL) represent 2 different pathways of disease evolution. Am J Surg Pathol. 2007;31:1605–14.
Behdad A, Griffin B, Chen YH, Ma S, Kelemen K, Lu X, et al. PD-1 is highly expressed by neoplastic B-cells in Richter transformation. Br J Haematol. 2018;185:370–3.
He R, Ding W, Viswanatha DS, Chen D, Shi M, Van Dyke D, et al. PD-1 expression in chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and large B-cell richter transformation (DLBCL-RT): a characteristic feature of DLBCL-RT and potential surrogate marker for clonal relatedness. Am J Surg Pathol. 2018;42:843–54.
Parikh SA, Rabe KG, Call TG, Zent CS, Habermann TM, Ding W, et al. Diffuse large B-cell lymphoma (Richter syndrome) in patients with chronic lymphocytic leukaemia (CLL): a cohort study of newly diagnosed patients. Br J Haematol. 2013;162:774–82.
Tsimberidou AM, O’Brien S, Khouri I, Giles FJ, Kantarjian HM, Champlin R, et al. Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol Off J Am Soc Clin Oncol. 2006;24(May):2343–51.
Harousseau JL, Flandrin G, Tricot G, Brouet JC, Seligmann M, Bernard J. Malignant lymphoma supervening in chronic lymphocytic leukemia and related disorders. Richter’s syndrome: a study of 25 cases. Cancer. 1981;48:1302–8.
Robertson LE, Pugh W, O’Brien S, Kantarjian H, Hirsch-Ginsberg C, Cork A, et al. Richter’s syndrome: a report on 39 patients. J Clin Oncol Off J Am Soc Clin Oncol. 1993;11:1985–9.
Tsimberidou AM, Keating MJ, Wierda WG. Richter’s transformation in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2007;2(Oct):265–71.
Jain N, Keating MJ. Richter transformation of CLL. Expert Rev Hematol. 2016;9:793–801.
Parikh SA, Kay NE, Shanafelt TD. How we treat Richter syndrome. Blood. 2014;123:1647–57.
Ding W. Richter transformation in the era of novel agents. Hematol Am Soc Hematol Educ Program. 2018;2018:256–63.
Wang Y, Tschautscher MA, Rabe KG, Call TG, Leis JF, Kenderian SS, et al. Clinical characteristics and outcomes of Richter transformation: Experience of 204 patients from a single center. Haematologica 2019;105:765–73.
Cramer P, von Tresckow J, Bahlo J, Robrecht S, Langerbeins P, Al-Sawaf O, et al. Bendamustine followed by obinutuzumab and venetoclax in chronic lymphocytic leukaemia (CLL2-BAG): primary endpoint analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19:1215–28.
Eichhorst B, Fink AM, Bahlo J, Busch R, Kovacs G, Maurer C, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17:928–42.
Eichhorst BF, Busch R, Hopfinger G, Pasold R, Hensel M, Steinbrecher C, et al. Fludarabine plus cyclophosphamide versus fludarabine alone in first-line therapy of younger patients with chronic lymphocytic leukemia. Blood. 2006;107:885–91.
Eichhorst BF, Busch R, Stilgenbauer S, Stauch M, Bergmann MA, Ritgen M, et al. First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood. 2009;114:3382–91.
Fischer K, Cramer P, Busch R, Bottcher S, Bahlo J, Schubert J, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30:3209–16.
Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370:1101–10.
Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376:1164–74.
von Tresckow J, Cramer P, Bahlo J, Robrecht S, Langerbeins P, Fink AM, et al. CLL2-BIG: sequential treatment with bendamustine, ibrutinib and obinutuzumab (GA101) in chronic lymphocytic leukemia. Leukemia. 2019;33:1161–72.
Stilgenbauer S, Schnaiter A, Paschka P, Zenz T, Rossi M, Dohner K, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123:3247–54.
Rossi D, Spina V, Cerri M, Rasi S, Deambrogi C, De Paoli L, et al. Stereotyped B-cell receptor is an independent risk factor of chronic lymphocytic leukemia transformation to Richter syndrome. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15:4415–22.
Rossi D, Cerri M, Capello D, Deambrogi C, Rossi FM, Zucchetto A, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2008;142:202–15.
Yee KW, O’Brien SM, Giles FJ. Richter’s syndrome: biology and therapy. Cancer J. 2005;11:161–74.
Cheson BD, Vena DA, Barrett J, Freidlin B. Second malignancies as a consequence of nucleoside analog therapy for chronic lymphoid leukemias. J Clin Oncol Off J Am Soc Clin Oncol. 1999;17:2454–60.
Robak T, Blonski JZ, Gora-Tybor J, Kasznicki M, Konopka L, Ceglarek B, et al. Second malignancies and Richter’s syndrome in patients with chronic lymphocytic leukaemia treated with cladribine. Eur J Cancer. 2004;40:383–9.
Mauro FR, Foa R, Giannarelli D, Cordone I, Crescenzi S, Pescarmona E, et al. Clinical characteristics and outcome of young chronic lymphocytic leukemia patients: a single institution study of 204 cases. Blood. 1999;94:448–54.
Stilgenbauer S, Zenz T, Winkler D, Buhler A, Schlenk RF, Groner S, et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27:3994–4001.
Tam CS, Shanafelt TD, Wierda WG, Abruzzo LV, Van Dyke DL, O’Brien S, et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood. 2009;114:957–64.
Rossi D, Gaidano G. Biological and clinical significance of stereotyped B-cell receptors in chronic lymphocytic leukemia. Haematologica. 2010;95:1992–5.
Dabaja BS, O’Brien SM, Kantarjian HM, Cortes JE, Thomas DA, Albitar M, et al. Fractionated cyclophosphamide, vincristine, liposomal daunorubicin (daunoXome), and dexamethasone (hyperCVXD) regimen in Richter’s syndrome. Leuk lymphoma. 2001;42:329–37.
Tsimberidou AM, Wierda WG, Wen S, Plunkett W, O’Brien S, Kipps TJ, et al. Phase I-II clinical trial of oxaliplatin, fludarabine, cytarabine, and rituximab therapy in aggressive relapsed/refractory chronic lymphocytic leukemia or Richter syndrome. Clin Lymphoma Myeloma Leuk. 2013;13:568–74.
Langerbeins P, Busch R, Anheier N, Durig J, Bergmann M, Goebeler ME, et al. Poor efficacy and tolerability of R-CHOP in relapsed/refractory chronic lymphocytic leukemia and Richter transformation. Am J Hematol. 2014;89:E239–43.
Abrisqueta P, Delgado J, Alcoceba M, González M, Oliveira AC, Loscertales J, et al. Clinical Outcome and Prognostic Factors of Patients with Richter’s Syndrome: Retrospective Multicenter Study of the Spanish Chronic Lymphocytic Leukemia (CLL) Study Group (GELLC). Blood. 2017;130(Suppl 1):2995.
Burger JA, Barr PM, Robak T, Owen C, Ghia P, Tedeschi A, et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia. 2019;34:787–98.
Fischer K, Al-Sawaf O, Bahlo J, Fink AM, Tandon M, Dixon M, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380:2225–36.
Woyach JA, Ruppert AS, Heerema NA, Zhao W, Booth AM, Ding W, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379:2517–28.
Ding W, Le-Rademacher J, Call TG, Parikh SA, Leis JF, Shanafelt TD, et al. PD-1 blockade with pembrolizumab in relapsed CLL including Richter’s transformation: an updated report from a phase 2 trial (MC1485). Blood. 2016;128:4392.
Gauthier J, Hirayama AV, Hay KA, Li D, Lymp J, Sheih A, et al. Comparison of efficacy and toxicity of CD19-specific chimeric antigen receptor T-cells alone or in combination with ibrutinib for relapsed and/or refractory CLL. Blood. 2018;132(Suppl 1):299.
Gill SI, Vides V, Frey NV, Metzger S, O’Brien M, Hexner E, et al. Prospective clinical trial of anti-CD19 CAR T cells in combination with ibrutinib for the treatment of chronic lymphocytic leukemia shows a high response rate. Blood. 2018;132(Suppl 1):298.
Jain N, Ferrajoli A, Basu S, Thompson PA, Burger JA, Kadia TM, et al. A phase II trial of nivolumab combined with ibrutinib for patients with richter transformation. Blood. 2018;132(Suppl 1):296.
Acknowledgements
We thank the patients and their families for the trust and confidence they have placed in us as well as all participating sites for their active contributions to our studies. OA, SR, JB, BE designed the study, analysed the data and wrote the manuscript. AMF, PC, JvT, EL, MK, MD, MR, JD, ET, SS, CMW, VG and MH revised and discussed the data and reviewed the manuscript. ET and StSt were supported by the DFG (SFB1074, subprojects B1 and B2).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
OA reports personal fees and non-financial support from AbbVie, Roche, Gilead, Janssen. JB reports personal fees and non-financial support from Roche. PC reports personal fees and non-financial support from Roche, Janssen, Astellas, Gilead, Mundipharma, Novartis. JD reports personal fees and non-financial support from Celgene, Janssen and Abbvie. BE reports grants and personal fees from Roche, AbbVie, Gilead, Janssen. AMF reports personal fees from Janssen Pharmaceutical. KF reports non-financial support and personal fees from Roche and Abbvie. VG reports personal fees and non-financial support from Roche, Gilead, Janssen. MH reports personal fees and non-financial support from AbbVie, Roche, Gilead, Janssen, personal fees from Celgene, Boehringer Ingelheim. MR reports grants and personal fees from Roche/AbbVie. StSt reports grants, personal fees and non-financial support from AbbVie, Roche; grants, personal fees and non-financial support from Amgen, AstraZeneca, Celgene, Gilead, GSK, Janssen, Novartis, Pharmacyclics, Sunesis. ET reports personal fees from Roche, AbbVie. CMW reports grants and/or personal fees from Hoffmann-La Roche, Mundipharma, Servier; Janssen-Cilag, Novartis, Gilead, Morphosys and Abbvie. All others declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Al-Sawaf, O., Robrecht, S., Bahlo, J. et al. Richter transformation in chronic lymphocytic leukemia (CLL)—a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials. Leukemia 35, 169–176 (2021). https://doi.org/10.1038/s41375-020-0797-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-020-0797-x
- Springer Nature Limited