
Abstract
Objective
Both obesity and insulin resistance are characterized by severe long-term changes in the expression of many genes of importance in the regulation of metabolism. Because these changes occur throughout life, as a result of external factors, the disorders of gene expression could be epigenetically regulated.
Materials/methods
We analyzed the relationship between obesity and insulin resistance in enrolled patients by means of evaluation of the expression rate of numerous genes involved in the regulation of adipocyte metabolism and energy homeostasis in subcutaneous and visceral adipose tissue depots. We also investigated global and site-specific DNA methylation as one of the main regulators of gene expression. Visceral and subcutaneous adipose tissue biopsies were collected from 45 patients during abdominal surgery in an age range of 40–60 years.
Results
We demonstrated hypermethylation of PPARG, INSR, SLC2A4, and ADIPOQ promoters in obese patients with insulin resistance. Moreover, the methylation rate showed a negative correlation with the expression of the investigated genes. More, we showed a correlation between the expression of PPARG and the expression of numerous genes important for proper insulin action. Given the impact of PPARγ on the regulation of the cell insulin sensitivity through modulation of insulin pathway genes expression, hypermethylation in the PPARG promoter region may constitute one of the epigenetic pathways in the development of insulin resistance in obesity.
Conclusions
Our research shows that epigenetic regulation through excessive methylation may constitute a link between obesity and subsequent insulin resistance.
Similar content being viewed by others

Introduction
Obesity is considered a strong risk factor in insulin resistance (IR) [1]. Excessive accumulation of adipose tissue, both visceral (VAT) and subcutaneous (SAT), is associated with metabolic, psychological, endocrine, and genetic factors [2]. Obesity causes changes in cell metabolism, which can lead to IR.
Metabolic disorders induced by obesity and IR are characterized by severe long-term changes in the expression of many genes important in metabolism regulation. Because these changes occur throughout life as a result of external factors (high-fat diet, sedentary lifestyle, stress), gene expression disorders have been regarded as being influenced by epigenetic modifications [3]. Epigenetics is defined as the heritable and reversible modification of gene expression without changes in the DNA sequence, maintained over a generation. One of the basic manifestations of epigenetics is tissue-specific gene regulation, which is mainly connected with the presence of methylation within DNA [4, 5].
The relationship between obesity and DNA methylation within both nuclear and mitochondrial DNA has been shown in numerous scientific reports [6,7,8,9]. An increased global methylation level was observed in the DNA of B cells from obese and type 2 diabetic patients, as compared to lean subjects [10]. Increased site-specific methylation in obese individuals was observed in the promoters of the genes regulating insulin sensitivity and the insulin-signaling pathway [7, 9, 11, 12].
In the present study, we analyzed the influence of obesity and obesity with concomitant IR on global and site-specific DNA methylation and the expression of genes involved in the insulin-signaling pathway, adipogenesis, lipid metabolism, inflammation, and the DNA methylation process in human adipose tissue. Both types of adipose tissue, VAT and SAT, were subjected to examination.
Materials and methods
The research protocols and all procedures were approved by the Ethical Review Board of Wroclaw Medical University, approval no. KB-124/2017.
Biological material
VAT and SAT biopsies were collected from 45 patients in an age range of 40–60 years during abdominal surgery, following written agreement. For each enrolled subject, the following parameters were assessed: fasting glucose, lipids panel, and body weight and height for calculation of BMI. In addition, a questionnaire regarding other metabolic diseases (type 2 diabetes, hypertension, sclerosis) and medications was completed.
Criteria for excluding patients from the study included other IR-related diseases (PCOS, Cushing’s syndrome), thyroid dysfunction, hepatitis, chronic inflammatory or infectious diseases, tumors, heavy drinking or a positive history thereof, and use of insulin or metformin.
Insulin level
Insulin levels were measured in plasma, using a Human Insulin ELISA Kit (Sigma-Aldrich). Absorbance was read using a Victor3 1420 Multilabel Counter.
DNA and RNA isolation
DNA was isolated using a commercial column spin method kit, the QIAamp DNA Mini Kit (QIAGEN) according to protocol. Total RNA was isolated using a combination of the trizol method and commercial spin column kits (Promega). The tissues samples (10–20 mg) were homogenized in 1 ml of Trizol, after extraction, RNA was precipitated with isopropanol and applied on the silica membrane column. Further extraction was carried out according to protocol.
Reverse transcription reaction and gene expression level
Reverse transcription was performed with the use of a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), using 200 ng of total RNA. Gene expression was done using Real-Time PCR based on a SYBR Green assay (Applied Biosystems). Primers were manually designed to flank two exons of mRNA (Supplementary Table 1). The specificity of primers was checked using Primer-BLAST; secondary structures were analyzed using OligoAnalyzer. Prior to real-time PCR, the efficiency of the primers was analyzed using the standard curve method; specificity was checked based on the denaturation curve. Only primers characterized by efficiency values higher than R2 > 0.95 were used for gene expression studies. A relative gene expression level, normalized to the housekeeping gene β-actin, was calculated using the delta-delta Ct (ΔΔCt) model.
Global DNA methylation analysis
The global methylation of DNA was measured using a commercial ELISA-based MethylFlash Methylated DNA Quantification Kit (Epigentek) in accordance with the manufacturer’s protocol. Absorbance was read using a Victor3 1420 Multilabel Counter.
Site-specific DNA methylation analysis and prediction of CpG islands
Prediction of CpG islands in the promoter region was accomplished using a USCS (University of California Santa Cruz) Genome Browser and MethPrimer software (UCSF). The prediction criteria: CG content >55% (region 500 bp in length), ObsCpG/ExpCpG >0.65.
Site-specific DNA methylation within the promoter region of the analyzed genes was carried out via methylated DNA precipitation (meDIP) using a MagMeDIP qPCR Kit (Diagenode) followed by a percentage of input measurements in real-time PCR according to the algorithm provided by the manufacturer. Primers were designed to amplify representative CG clusters located in the promoter regions of the analyzed genes (Supplementary Table 2). The results of the meDIP analysis were confirmed by the bisulfite sequencing technique. Bisulfite treatment of genomic DNA (500 ng) was performed using an EpiJET Bisulfite Conversion Kit (Thermo Fisher Scientific). Amplification of the CpG islands for the appropriate gene was done using a QIAGEN Multiplex PCR Kit (QIAGEN). The primers for PCR were selected based on data obtained during the prediction of the CpG islands using MethPrimer software (UCSF). Prior to the actual experiments, the temperature condition of PCR for each CpG island was determined. The amplification results were checked by means of gel electrophoresis. The amplified CpG islands were sequenced using the Sanger method. The results of sequencing were analyzed using QUMA (a quantification tool for methylation analysis) (Riken).
Assessment of IR and obesity
BMI was calculated as weight in kilograms divided by squared of height in meters [kg/m2]. IR rate was assessed using IR ratios calculated according to the following formulas:
(1) HOMA-IR [(glucose [mmol/l] × insulin [μU/ml])/22.5],
(2) QUICKI [1/(log glucose [mg/dl] + log insulin [μU/ml])].
Statistical analysis
Statistical analysis was performed using STATISTICA 13.1 and Microsoft Office Excel 2007. ANOVA and a post-hoc test (the NIR-Fisher test) were used to assess the difference between studied groups. The correlation between numerical values was made using a correlation coefficient. Statistical significance was set at p < 0.05.
Results
The enrolled patients were divided into three study groups (LH—lean healthy, OH—obese healthy, with normal values of IR ratios, OR—obese with IR) depending on the BMI, HOMA-IR and QUICKI parameters. Patients with BMI > 25 were classified in the obese group. IR was diagnosed based on HOMA-IR > 2.5 and QUICKI < 0.321. We performed all analysis for both VAT and SAT tissues. We treated LH group as a control group and compared the other groups to it.
Study cohort characterization
The groups were characterized according to obesity level, IR ratios, lipid metabolism, age, and sex. The results are presented in Table 1.
Gene expression in IR and obesity
We measured the expression of genes important in adipocyte metabolism, i.e.: (1) insulin pathways: INSR, IRS1, IRS2, PIK3R1, AKT, SLC2A4; (2) lipid metabolism: LPL, ACACA, FASN, ACSS2, SCD1; (3) inflammation: IL10, IL1B; (4) adipokines: LEP, ADIPOQ, retinol-binding protein 4 (RBP4); (5) transcription factors: CEBPA, CEBPB, PPARG, IGF2, PPARGC1A, TNFA. We assessed the expression level of genes in all three study groups.
In the VAT samples of OR patients, we observed a statistically significant reduction in the expression rate of the following genes: INSR (p = 0.001), IRS1 (p = 0.012), IRS2 (p = 0.050), PIK3R1 (p = 0.009), SLC2A4 (p = 0.018), ACSS2 (p = 0.014), ACACA (p = 0.019), LPL (p = 0.030), FASN (p = 0.002), PPARG (p = 0.001), ADIPOQ (p = 0.031), RBP4 (p = 0.001). Contrastingly, we observed a significant increase of IL10 expression level in OR group (p = 0.017).
A similar profile of gene expression was noticed in VAT depots in OH group. Among others, reduction in expression rate was diagnosed mainly for INSR (p = 0.008), IRS1 (p = 0.025), PIK3R1 (p = 0.014), FASN (p = 0.069), PPARG (p = 0.057), ADIPOQ (p = 0.091) and RBP4 (p = 0.022).
In SAT samples in OR group, we observed statistically significant reductions in expression rate for the following genes: INSR (p = 0.001), SLC2A4 (p = 0.013), IRS1 (p = 0.062), IRS2 (p = 0.015), LPL (p = 0.039), FASN (p = 0.036), and PPARG (p = 0.017). Moreover, IL10 gene was overexpressed in the OR group (p = 0.034). The only genes that were downregulated in the SAT depots of the OH group were INSR (p = 0.008), SLC2A4 (p = 0.053), and IRS1 (p = 0.021).
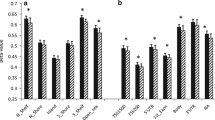
Next, we analyzed the correlation between the expression of all genes and BMI, HOMA-IR, and QUICKI values. The observed correlation between patients’ BMI and expression of numerous genes are presented in Fig. 1.

The results of correlation between BMI and the expression level of genes in visceral adipose tissue (VAT) and subcutaneous (SAT) adipose tissue, respectively.
A negative correlation was observed between QUICKI value and expression of IL10 (VAT: R = 0.410; p = 0.022, SAT: R = 0.435; p = 0.016). On the other hand, a positive correlation was seen between QUICKI and expression of PPARG (VAT: R = 0.312; p = 0.082, SAT: R = 0.353; p = 0.048), INSR (SAT: R = 0.306; p = 0.088), LPL (SAT: R = 0.308; p = 0.087), and ADIPOQ (SAT: R = 0.376; p = 0.034). In the case of HOMA-IR, we did not observe statistically significant correlations.
Global DNA methylation
We found differences in global DNA methylation between studied groups. We observed the highest level of DNA methylation in OR group in both fat depots (VAT: p = 0.000, SAT: p = 0.007). DNA methylation was also higher in the OH compared to the LH group, without, however, being significant (Fig. 2).

The results of DNMT1 gene expression are compared with the results of global DNA methylation in visceral (VAT) and subcutaneous (SAT) adipose tissue, respectively. Statistical significance in the comparative analysis was demonstrated for the OR group both in the analysis of DNMT1 gene expression (VAT: p = 0.039; SAT: p = 0.046) and global methylation (VAT: p = 0.001; SAT: p = 0.007).
Furthermore, in both VAT and SAT samples, we observed a positive correlation between the global DNA methylation level and BMI (VAT: R = 0.591; p = 0.000; SAT: R = 0.574; p = 0.000) as well as between the global DNA methylation level and HOMA-IR value (VAT: R = 0.380; p = 0.061; SAT: R = 0.431; p = 0.032). A negative correlation was observed between the global DNA methylation level and QUICKI value (VAT: R = −0.360; p = 0.077; SAT: R = −0.509; p = 0.009).
We also assessed the expression level of gene encoding DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b). In the case of DNMT3a and DNMT3b, we did not observe significant differences in gene expression between the study groups (data not shown). We showed that DNMT1 was overexpressed in OR patients in both fat depots (VAT: p = 0.039, SAT: p = 0.045; Fig. 2). Furthermore, we observed a positive correlation between the DNA methylation rate and the expression level of DNMT1 in both fat depots (VAT: R = 0.380; p = 0.029; SAT: R = 0.298; p = 0.087). We observed a similar relationship in the case of the expression level of DNMT1 and BMI (VAT: R = 0.346; p = 0.031; SAT: R = 0.372; p = 0.015). Moreover, the expression rate of DNMT1 correlated positively with IR assessed based on HOMA-IR value (VAT: R = 0.647; p = 0.000; SAT: R = 0.598; p = 0.000) and QUICKI (VAT: R = −0.441; p = 0.013; SAT: R = −0.487; p = 0.005).
Methylation of gene promoter region
Next, the site-specific methylation pattern within the promoter region of the selected genes was analyzed.
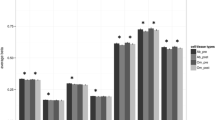
In VAT samples, we noted a significant increase in the methylation level of PPARG promoter in OR compared to the LH group (p = 0.009). What is more, the methylation rate of the PPARG promoter negatively correlated with the expression rate in this fat depot (R = −0.4702; p = 0.049). In both VAT and SAT samples, we observed an increased methylation rate of SLC2A4 (VAT: p = 0.041; SAT: p = 0.078) and ADIPOQ (VAT: p = 0.080; SAT: p = 0.013) promoter in the OR group. On the other hand, methylation of INSR increased in both obese groups in both VAT (OH: p = 0.002, OR—no statistical significance) and SAT (OH p = 0.016, OR—no statistical significance), which corresponded to expression rate of this gene. In SAT, we also observed a negative correlation between the INSR promoter methylation level and the expression of INSR (R = –0.3967; p = 0.068). The results are presented in Fig. 3A.

The results of the comparative analysis regarding the expression of INSR, SLC2A4, PPARG, and ADIPOQ and level of methylation of this gene promoter between three groups (LH, OH, and OR) in visceral (VAT) and subcutaneous (SAT) adipose tissue, respectively (A). The site-specific DNA methylation within the promoter region of PPARG gene in VAT and SAT—the results of the bisulfite sequencing technique (B).
We also wished to correlate the site-specific methylation pattern with clinical parameters of enrolled patients, such as BMI, HOMA-IR, QUICKI, and lipids panel.
In the case of VAT samples, we observed a negative correlation between the QUICKI value and level of promoter methylation of SLC2A4 (R = −0.4239; p = 0.055) and IL10 (R = −0.5010; p = 0.021). A positive correlation was seen between the HOMA-IR value and level of promoter methylation of SLC2A4 (R = 0.4841; p = 0.026) and IL10 (R = 0.5265; p = 0.014).
In SAT samples, we observed a positive correlation between BMI and the level of methylation of the promoters of the following genes: INSR (R = 0.5781; p = 0.006), ADIPOQ (R = 0.4784; p = 0.028), and IL6 (R = 0.8387; p = 0.000). The methylation rate of the promoter of INSR (R = 0.8341; p = 0.000) and IL6 (R = 0.8436; p = 0.001) correlated positively with triglyceride level in serum, similar to methylation of ADIPOQ promoter that correlated positively with the LDL cholesterol level in serum (R = 0.5697; p = 0.027).
To confirm the results of differentially methylated promoters, we performed a bisulfite sequencing study of representative samples from three investigated groups. In samples from both fat depots, VAT and SAT, we observed an increased site-specific methylation level within the promoter region of PPARG in OR compared to the LH group (Fig. 3B).
Gene expression and transcription factor
The nuclear peroxisome proliferator-activated receptor gamma (PPARγ) is a crucial transcription factor regulating adipocyte development and normal metabolism. Thus we were interested as to whether PPARγ could regulate the expression of genes necessary for normal adipocyte function. In both types of tissue, we observed a correlation between PPARG gene expression and several genes. The results are presented in Fig. 4.

The results of correaltion between PPARG expresiion and the expression of other genes in visceral (VAT) and subcutaneous (SAT) adipose tissue, respectively.
Discussion
In the present study, we analyzed the influence of obesity on global and site-specific DNA methylation as well as the potential influence of these epigenetic modifications on the expression of genes involved in the insulin-signaling pathway, adipogenesis, lipid metabolism, inflammation, and DNA methylation process in human adipose tissue. The link between obesity and IR, which is reflected in disorders in the expression of genes relevant to the insulin-signaling pathway, is well described. Indeed, in the present study, we demonstrated a strong negative correlation between BMI and expression of genes important for the insulin pathway (INSR, IRS1, IRS2, PIK3R1, AKT, SLC2A4). The normal expression of all of these genes is extremely important for correct insulin signaling [13].
Numerous factors may be responsible for aberrant gene expression, ranging from intracellular to extracellular factors such as nutrition, age, or physical activity. Considering that obesity and IR have genetic and environmental backgrounds, we hypothesized that epigenetic modification might also be involved. In the present study, we showed the association between global DNA methylation level and obesity, which had also been confirmed previously [7, 9, 14, 15]. The observed increased level of methylation with increased BMI suggests a link between epigenetic modifications and obesity. What is more, we also demonstrated positive correlations between global DNA methylation and DNMT1 expression and between BMI and DNMT1 expression, which would explain the potential role of this enzyme in creating epigenetic modification in adipocyte DNA.
We observed a similar relationship between DNA methylation and IR (a positive correlation between DNA methylation and HOMA-IR value and a negative correlation between DNA methylation and QUICKI value). Moreover, comparative analysis between the groups showed that in both tissues the level of DNA methylation was significantly higher in the group of obese people with IR compared to lean, healthy people, which may suggest the role of epigenetic regulation in insulin sensitivity disorders in obese patients.
The detected relationships between increased global DNA methylation level in adipocytes and obesity and IR led us to take a closer look at site-specific methylation, especially within the promoter region of genes important for the insulin pathway or genes connected with the regulation of this pathway. We showed increased methylation levels within the promoter region of the INSR and SLC2A4 genes in the group of obese patients with IR compared to the lean group, in both VAT and SAT. The insulin receptor encoded by the INSR gene constitutes the first stage of the insulin pathway, but SLC2A4 encodes the glucose transporter 4, the major insulin-regulated glucose intercellular transporter, which is the last stage of this pathway. The expression level of these genes was also significantly reduced in the OR group, which may explain the silencing role of DNA methylation on gene expression. This is also confirmed by the demonstrated a significant negative correlation between methylation of the INSR promoter region and expression of this gene in SAT. Interestingly, in SAT we also observed a positive correlation between the methylation promoter level of INSR and BMI, suggesting that epigenetic modifications are connected with obesity, and in the next step lead to IR development via methylation of the promoter region of the genes, which are important for the insulin pathway, such as INSR.
PPARγ is considered a transcriptional regulator of adipogenesis and lipid and glucose metabolism. The PPARG gene is expressed especially in both white and brown adipose tissue [16, 17]. What is more, its synthetic ligands, such as glitazones, are used in the treatment of diabetes, as they improve insulin and glucose parameters and increase insulin sensitivity [18, 19]. Dysregulation of PPARγ can lead to the development of obesity, IR, and type 2 diabetes [17]. Studies have shown that changes in PPARG expression, e.g., gene knockout, cause IR and dysregulation of adipogenesis in mice [20, 21]. In our research, we also observed a relationship, in both VAT and SAT, between PPARγ and obesity and IR, which is shown by a negative correlation between PPARG expression and BMI as well as by a positive correlation between PPARG expression and QUICKI value. Moreover, our comparative analysis also confirms the association of both diseases with PPARγ. The expression of PPARG was significantly reduced in the OR group. Interestingly, we also demonstrated that changes in PPARG expression are based on epigenetic regulation, which confirms the very high level of methylation within the promoter region of this gene. The level of methylation was at least several times (in VAT about 5.5 times, in SAT 2.5 times) higher in the OR group compared to the LH group. What is more, with an increase of methylation within the promoter region of PPARG, we observed decreased expression of this gene (a negative correlation between the level of methylation and expression of PPARG), which confirms that PPARG is subject to epigenetic regulation. PPARγ as a transcription factor can influence regulation of the expression of other genes important for the insulin pathway or adipogenesis. In order to better elucidate the regulation of its role, we performed correlation analysis. We observed a strong positive correlation between the expression of PPARγ and numerous genes. It has been shown that PPARγ takes part in the regulation of INSR, IRS1, IRS2, PIK3R1, and SLC2A4, which make up a significant part of the genes involved in the transmission of the insulin pathway signal, and also in the regulation of genes involved in lipid metabolism, namely LPL, ACACA, ACSS2, SCD1, and FASN, and a gene coding, a transcriptional factor important for adipogenesis, CEBPA. These results show the important role of PPARγ in regulating metabolic pathways and in potential disorders. Changes in PPARG expression caused by epigenetic modifications can significantly interfere with the expression of other genes relevant for the insulin pathway, lipogenesis, or adipogenesis.
In addition, we examined the effect of epigenetic regulation on adiponectin, because PPARγ directly regulates expression of the ADIPOQ gene, as we have shown in this study (a strong positive correlation between the expression of PPARG and of ADIPOQ) as was proved earlier [22]. Adiponectin, one of the adipokines produced by adipocytes of white adipose tissue, is involved in insulin sensitivity, glucose uptake, and lipid metabolism. Some studies have shown that adiponectin improves insulin sensitivity by reducing the amount of intercellular fat and enhancing the insulin receptor substrate [23,24,25]. Other studies have shown that adiponectin gene expression is downregulated in IR and obesity [26,27,28,29]. We also demonstrated an association between BMI and ADIPOQ expression (negative correlation) and between QUICKI value and ADIPOQ expression (positive correlation). What is more, we showed that the expression level of the adiponectin gene is strongly downregulated in the OR and OH group compared to LH group, however only in VAT depots. Interestingly, we also demonstrated that changes in ADIPOQ expression are based on epigenetic regulation, which confirms the very high level of methylation within the promoter region of ADIPOQ. The level of methylation was at least several times (in VAT about 4.5 times, in SAT 4 times) higher in the OR group compared to the LH group. A published study also demonstrated a decrease in ADIPOQ expression in mice with hypermethylation of the promoter region of the ADIPOQ gene [30].
We also took a closer look at another potential regulatory factor of insulin sensitivity in obesity. RBP4 has been identified as an adipokine with potential involvement in the development of impaired glucose metabolism. It has been shown that the serum level of RBP4 positively correlates with obesity and IR, and induces IR through preventing insulin-initiated phosphorylation of insulin receptor substrate 1 [31,32,33]. Interestingly, in the present study, we demonstrated completely different relationships, namely, a negative correlation in VAT samples between BMI and RBP4 expression. More significantly, the downregulation of the expression of this gene in OR compared to LH group indicates that RBP4 may play an important role in regulating insulin sensitivity in obese patients with IR. Moreover, we showed an association between the expression of PPARG and RBP4. The strong positive correlation between these genes could suggest that RBP4, similar to adiponectin, may be under the control of PPARγ, which is considered a positive regulator of insulin sensitivity. It is, therefore, possible that the nature of the effect of RBP4 on the development of IR should be investigated further.
Noteworthy are also the slight differences we observed in the expression of genes relevant to the insulin pathway between the group of obese patients with normal glucose tolerance and the obese group with IR. These small differences may indicate that obesity itself generates disorders in the insulin pathway at the molecular level that are visible in gene expression but are not yet visible in metabolic parameters such as QUICKI and HOMA-IR. We suggest that a very important role in this process may be played by epigenetic regulation, as we observed a higher level of site-specific methylation in the INSR gene in OH compared to OR. Thus obese patients may experience disorders in the insulin pathway long before their clinical manifestation and complete development of IR.
The value and strength of presented expression results would be increased by the western blot analysis of the protein; however, the limited amount of biological material made it impossible to perform the research. In the future, we plan to take care of the increased amount of collected biological material to extend the research to protein analysis while enhancing the power of the results.
Summarizing, our research shows that epigenetic regulation through excessive methylation may constitute a link between obesity and subsequent IR. Moreover, our research confirms that, in obese patients with co-existing IR, the expression of genes relevant to the insulin pathway is significantly reduced compared to lean healthy patients, and the expression was shown to be epigenetically regulated. Interestingly, the differences in gene expression between the group of obese patients (with normal insulin sensitivity and with IR) were not very large except in the case of the SLC2A4 gene. This observation shows how obesity adversely affects the insulin pathway, creating the necessary conditions for the development of cells’ resistance to insulin through disorder in gene expression, even where IR has not yet been diagnosed.
References
Aung K, Lorenzo C, Hinojosa MA, Haffner SM. Risk of developing diabetes and cardiovascular disease in metabolically unhealthy normal-weight and metabolically healthy obese individuals. J Clin Endocrinol Metab. 2014;99:462–8.
Nguyen DM, El-Serag HB. The epidemiology of obesity. Gastroenterol Clin N Am. 2010;39:1.
Smith CJ, Ryckman KK. Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diabetes Metab Syndr Obes. 2015;8:295–302.
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781.
Maor GL, Yearim A, Ast G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015;31:274–80.
Zheng LD, Linarelli LE, Liu L, Wall SS, Greenawald MH, Seidel RW, et al. Insulin resistance is associated with epigenetic and genetic regulation of mitochondrial DNA in obese humans. Clin Epigenetics. 2015;7:60.
Arner P, Sinha I, Thorell A, Rydén M, Dahlman-Wright K, Dahlman I. The epigenetic signature of subcutaneous fat cells is linked to altered expression of genes implicated in lipid metabolism in obese women. Clin Epigenetics. 2015;7:93.
Pietiläinen KH, Ismail K, Järvinen E, Heinonen S, Tummers M, Bollepalli S, et al. DNA methylation and gene expression patterns in adipose tissue differ significantly within young adult monozygotic BMI-discordant twin pairs. Int J Obes. 2016;40:654–61.
Barberio MD, Nadler EP, Sevilla S, Lu R, Harmon B, Hubal MJ. Comparison of visceral adipose tissue DNA methylation and gene expression profiles in female adolescents with obesity. Diabetol Metab Syndr. 2019;11:98.
Simar D, Versteyhe S, Donkin I, Liu J, Hesson L, Nylander V, et al. DNA methylation is altered in B and NK lymphocytes in obese and type 2 diabetic human. Metabolism. 2014;63:1188–97.
Drogan D, Boeing H, Janke J, Schmitt B, Zhou Y, Walter J, et al. Regional distribution of body fat in relation to DNA methylation within the LPL, ADIPOQ and PPARγ promoters in subcutaneous adipose tissue. Nutr Diabetes. 2015;5:168.
Zhang Q, Xiao X, Zheng J, Li M, Yu M, Ping F, et al. A maternal high-fat diet induces DNA methylation changes that contribute to glucose intolerance in offspring. Front Endocrinol. 2019;10:871.
Choi K, Kim Y-B. Molecular mechanism of insulin resistance in obesity and type 2 diabetes. Korean J Intern Med. 2010;25:119–29.
Cash HL, McGarvey ST, Houseman EA, Marsit CJ, Hawley NL, Lambert-Messerlian GM, et al. Cardiovascular disease risk factors and DNA methylation at the LINE-1 repeat region in peripheral blood from Samoan Islanders. Epigenetics. 2011;6:1257–64.
Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW. DNA methylation as a biomarker for cardiovascular disease risk. PLoS ONE. 2010;5:3.
Medina-Gomez G, Gray S, Vidal-Puig A. Adipogenesis and lipotoxicity: role of peroxisome proliferator-activated receptor γ (PPARγ) and PPARγcoactivator-1 (PGC1). Public Health Nutr. 2007;10:1132–7.
Lehrke M, Lazar MA. The many faces of PPARγ. Cell. 2005;123:993–9.
Elstner E, Müller C, Koshizuka K, Williamson EA, Park D, Asou H, et al. Ligands for peroxisome proliferator-activated receptorγ and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. PNAS. 1998;95:8806–11.
Deeg MA, Tan MH. Pioglitazone versus Rosiglitazone: effects on lipids, lipoproteins, and apolipoproteins in head-to-head randomized clinical studies. PPAR Res. 2008;2008:520465.
He W, Barak Y, Hevener A, Olson P, Liao D, Le J, et al. Adipose-specific peroxisome proliferator-activated receptor γ knockout causes insulin resistance in fat and liver but not in muscle. PNAS. 2003;100:15712–7.
Koutnikova H, Cock T-A, Watanabe M, Houten SM, Champy M-F, Dierich A, et al. Compensation by the muscle limits the metabolic consequences of lipodystrophy in PPARγ hypomorphic mice. PNAS. 2003;100:14457–62.
Gustafson B, Jack MM, Cushman SW, Smith U. Adiponectin gene activation by thiazolidinediones requires PPARγ2, but not C/EBPα—evidence for differential regulation of the aP2 and adiponectin genes. Biochem Biophys Res Commun. 2003;308:933–9.
Fischer S, Navarrete Santos A, Thieme R, Ramin N, Fischer B. Adiponectin stimulates glucose uptake in rabbit blastocysts. Biol Reprod. 2010;83:859–65.
Berendoncks AMV, Stensvold D, Garnier A, Fortin D, Sente T, Vrints CJ, et al. Disturbed adiponectin—AMPK system in skeletal muscle of patients with metabolic syndrome. Eur J Prev Cardiolog. 2015;22:203–5.
Schindler M, Pendzialek M, Grybel KJ, Seeling T, Gürke J, Fischer B, et al. Adiponectin stimulates lipid metabolism via AMPK in rabbit blastocysts. Hum Reprod. 2017;32:1382.
Fasshauer M, Klein J, Neumann S, Eszlinger M, Paschke R. Hormonal regulation of adiponectin gene expression in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2002;290:1084–9.
Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes. 2003;52:1779–85.
Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86:1930–5.
Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. 1996;271:10697–703.
Kim AY, Park YJ, Pan X, Shin KC, Kwak S-H, Bassas AF. et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat Commun. 2015;6:7585.
Cho YM, Youn B-S, Lee H, Lee N, Min S-S, Kwak SH, et al. Plasma retinol-binding protein-4 concentrations are elevated in human subjects with impaired glucose tolerance and type 2 diabetes. Diabetes Care. 2006;29:2457–61.
Mohapatra J, Sharma M, Acharya A, Pandya G, Chatterjee A, Balaraman R, et al. Retinol-binding protein 4: a possible role in cardiovascular complications. Br J Pharmacol. 2011;164:1939.
Öst A, Danielsson A, Lidén M, Eriksson U, Nystrom FH, Strålfors P. Retinol-binding protein-4 attenuates insulin-induced phosphorylation of IRS1 and ERK1/2 in primary human adipocytes. FASEB J. 2007;21:3696–704.
Acknowledgements
The project was funded by The National Science Centre, Poland. The number of the research project: 2016/21/D/NZ5/00155.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Cierzniak, A., Pawelka, D., Kaliszewski, K. et al. DNA methylation in adipocytes from visceral and subcutaneous adipose tissue influences insulin-signaling gene expression in obese individuals. Int J Obes 45, 650–658 (2021). https://doi.org/10.1038/s41366-020-00729-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-020-00729-7
- Springer Nature Limited
This article is cited by
-
Trends in insulin resistance: insights into mechanisms and therapeutic strategy
Signal Transduction and Targeted Therapy (2022)
-
Prevalence of obesity and diabetes in older people with sarcopenia defined according to EWGSOP2 and FNHI criteria
Aging Clinical and Experimental Research (2022)