
Abstract
MicroRNAs (miRNAs) are small RNAs that suppress gene expression by their interaction with 3’untranslated region of specific target mRNAs. Although the dysregulation of miRNAs has been identified in human cancer, only a few of these miRNAs have been functionally documented in breast cancer. Thus, defining the important miRNA and functional target involved in chemoresistance is an urgent need for human breast cancer treatment. In this study, we, for the first time, identified a key role of miRNA 520h (miR-520h) in drug resistance. Through protecting cells from paclitaxel-induced apoptosis, expression of miR-520h promoted the drug resistance of human breast cancer cells. Bioinformatics prediction, compensatory mutation and functional validation further confirmed the essential role of miR-520h-suppressed Death-associated protein kinase 2 (DAPK2) expression, as restoring DAPK2 abolished miR-520h-promoted drug resistance, and knockdown of DAPK2 mitigated cell death caused by the depletion of miR-520h. Furthermore, we observed that higher level of miR-520h is associated with poor prognosis and lymph node metastasis in human breast cancer patients. These results show that miR-520h is not only an independent prognostic factor, but is also a potential functional target for future applications in cancer therapeutics.
Similar content being viewed by others

Introduction
Breast cancer is undoubtedly a major health threat to women worldwide. Breast cancer survival rates have significantly improved over the past several decades.1 However, a significant number of breast cancer patients still develop metastatic diseases and respond only transiently to conventional chemotherapy, becoming the major cause of cancer death. Traditional chemotherapeutic drugs are designed to induce apoptosis of cancer cells. However, drug resistance has been observed in cancer cells having dysregulated apoptosis- or antiapoptosis-related machinery, which is the leading cause of tumor recurrence leading to patient death. Despite the significant benefit of chemotherapy in breast cancer patients, only a small percentage respond even transiently to conventional treatments and targeted therapy, often resulting in metastatic disease and death.2, 3 Cancer cells escape from drug-induced cell death through the abnormal inhibition of proapoptotic signaling pathways or the activation of anti-apoptotic modulators.4, 5, 6 Because most conventional chemotherapeutic drugs induce apoptosis of cancer cells, the underlying mechanisms of the acquisition of resistance to chemotherapeutic agents remain a significant obstacle to successful breast cancer treatment.
MicroRNA (miRNA) is a novel class of gene regulators that are small but influential noncoding RNAs that posttranscriptionally regulate gene expression.7, 8 Accumulating evidence has indicated that miRNAs participate in various biological processes, such as embryo development, viral infections, cellular metabolism and human malignancies.9, 10 In addition, the functional roles of several miRNAs as master regulators of key genes implicated in chemoresistance have been identified in different types of cancer.11, 12, 13 Previous studies have proven that miR-520h acts as an oncomir in human lung cancer.14 In addition, the adenovirus type 5 E1A (E1A) protein executes its tumor suppressive activities through the downregulation of miR-520h, leading to the restored expression of the protein phosphatase 2A catalytic subunit (PP2A/C) and the subsequent inhibition of Twist-enhanced tumor growth and metastasis.15 To identify the role of miR-520h in the resistance to chemotherapeutic agents, we ectopically overexpressed or knocked down miR-520h in breast cancer cells and evaluated the paclitaxel sensitivity of these transfectants. In this study, we investigated the relationship of miR-520h with the paclitaxel sensitivity of human breast cancer cells and the underlying signaling pathway involved in the suppression of DAPK2.
Results
Expression of miR-520h enhances the drug resistance of breast cancer cells
To investigate whether miR-520h expression regulates the sensitivity of breast cancer cells to chemotherapeutic drugs, we established an miR-520h-expressing construct and successfully transfected it into MDA-MB-231 (MDA231/miR-520h) and MDA-MB-468 (MDA468/miR-520h) cells (Figure 1a). We used the soft agar assay to detect the effects of miR-520h on anchorage-independent cell growth. The clonogenic ability was found to be enhanced in MDA231/miR-520h cells, suggesting that miR-520h may promote the survival of cancer cells in an anchorage-independent condition (Figure 1b). Upon overexpression of miR-520h in MDA-MB-231 cells, these cells were observed to be more resistant to paclitaxel treatment (Supplementary Figure S1a). The IC50 values of vector control and miR-520h-overexpressed MDA231 cells toward paclitaxel were 9.91 and 19.92 nM, respectively. Next we investigated whether the expression of miR-520 h is involved in the paclitaxel resistance of breast cancer cells. MDA231/miR-520h and MDA468/miR-520h cells were treated with various concentrations of paclitaxel and the number of surviving cells was determined using the MTS assay at 24 h after treatment. Overexpression of miR-520h in MDA-MB-468 cells was observed to significantly protect cells from paclitaxel-induced cell death, as MDA468/miR-520h cells more clearly exhibited increased cell viability compared with the vector-transfected cells (Figure 1c). The IC50 values of the MDA468/miR-520h cells toward paclitaxel were nearly three times higher than those of the MDA468/Vec cells (11.22 versus 4.03 nM). In addition, we used soft agar assay to detect the effects of miR-520h on anchorage-independent cell growth in MDA-MB-468 cells (Supplementary Figure S1b). The clonogenic ability was significantly enhanced in MDA-MB-468 cells overexpressing miR-520h, suggesting that miR-520h promotes survival of cancer cells in an anchorage-independent condition (Supplementary Figure S1b). We also investigated the effect of endogenous miR-520h on paclitaxel resistance using a specific antagomiR targeting miR-520h (anti-miR-520h). After depleting the endogenous miR-520h in MDA-MB-231 and Hs578T cells, these cells were noted to be more sensitive to paclitaxel treatment (Figure 1d). The IC50 values of scramble control and antagomirs-treated MDA231 cells toward paclitaxel were 10.84 and 4.17 nM, respectively, whereas the IC50 values of scramble control and antagomirs-treated Hs578T cells toward paclitaxel were 13.21 and 3.95 nM, respectively. Similar results were also found in the colony formation assay, as MDA231/miR-520h and MDA468/miR-520h cells formed more colonies than their control cells in the presence of paclitaxel (Figure 1e). Furthermore, colony formation was reduced in MDA231 and Hs578T cells treated with miR-520h antagomirs (Figure 1f). These findings indicate that the expression of miR-520h enhances resistance to paclitaxel treatment in human breast cancer cells. To confirm whether miR-520h enhancement also suppresses apoptosis, we performed flow cytometry to analyze cells in the sub-G1 phase of the cell cycle after treatment of paclitaxel. We observed that the paclitaxel-mediated dose-dependent increase in the sub-G1 population was attenuated in MDA231/miR-520h and MDA468/miR-520h cells (Figure 1g and Supplementary Figure S2). The knockdown of miR-520h was found to greatly promote paclitaxel-induced apoptosis in Hs578T cells (Figure 1h), suggesting that the protective effect of miR-520h to paclitaxel is mediated by its function in abrogating paclitaxel-induced apoptosis.

miR-520h expression enhances paclitaxel resistance in human breast cancer cells. (a) MiR-520h expression levels in transfected MDA231/Vec, MDA231/miR-520h, MDA468/Vec and MDA468/miR-520h cells were detected by using real-time reverse transcription-PCR. (b) The anchorage-independent colony-forming ability of the MDA231/Vec and MDA231/miR-520h cells was tested using a soft agar assay. (c) Viability of MDA468/Vec and MDA468/miR-520h cells after exposure to chemotherapy drugs. Cells were cultured in 96-well plates in the presence or absence of increasing concentrations of paclitaxel. Cell viability and IC50 values were then determined by using the MTS assay. (d) Viability of MDA231 and Hs578T cells in the presence of increasing concentrations of paclitaxel. Cells were transfected with scramble control oligos or miR-520h antagomirs and then cultured in 96-well plates. Cell viability and IC50 values were then determined by using the MTS assay. (e) Clonogenic ability of MDA231/Vec, MDA231/miR-520h, MDA468/Vec and MDA468/miR-520h cells. Cells were treated with various concentrations of paclitaxel for 24 h and then refed with fresh medium. After 2 weeks of culture, the colonies were stained with crystal violet and counted. Each experiment was done in triplicate. (f) Clonogenic ability of MDA231 and Hs578T cells treated with miR-520h antagomirs. Cells were transfected with scramble control oligos or miR-520h antagomirs and treated with various concentrations of paclitaxel for 24 h, and then refed with fresh medium. After 2 weeks of culture, the colonies were stained with crystal violet and counted. Each experiment was done in triplicate. (g, h) MiR-520h increased resistance to apoptosis induced by paclitaxel. MDA231/Vec and MDA231/miR-520h cells (g) or Hs578T cells with miR-520 antagomirs transfected (h) were treated with 5 nM of paclitaxel for 24 h and then assayed by flow cytometry analysis. Results were collected from three independent experiments, with triplicate repeats for each experiment. Data are shown as the mean±s.d. *P<0.05.
miR-520h suppresses DAPK2 expression
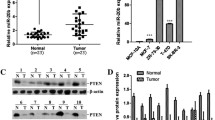
To further identify the functional target of miR-520h, we utilized the TargetScan algorithm (Release Version 5.1, http://www.targetscan.org/vert_50/) to isolate apoptosis-associated genes with a potential miR-520h-binding site in their 3’ untranslated region (3’UTR). One of the candidates, Death-associated protein kinase 2 (DAPK2), was of particular interest in light of its reported classification as a proapoptotic gene that enhances drug sensitivity of cancer cells. DAPK2, which belongs to the DAPK protein family, is involved in apoptosis and drug resistance.16, 17, 18 Specifically, DAPK2 contains an N-terminal protein kinase domain followed by a conserved calmodulin-binding domain similar to DAPK1, and thus functions as a positive regulator of cell death, in which its overexpression was shown to induce apoptosis of cancer cells.16, 17 We determined the steady-state levels of miR-520h and its downstream target DAPK2 in a breast cancer cell panel (Figures 2a and b). The levels of DAPK2 in BT483, T47D and MDA-MB-361 cells expressing lower miR-520h are markedly higher than that of Hs578T, MDA-MB-453, MDA-MB-231, MDA-MB-468 and HBL-100 cells expressing higher levels of miR-520h (Figures 2a and b). Although the three of five cell lines (MDA-MB-231, MDA-MB-468 and HBL-100) expressing lower DAPK2 protein (Figure 2a) seem to express lower miR-520h compared with Hs578T cells (Figure 2b), their inherent levels of miR-520h are still markedly higher than BT483, T47D and MDA-MB-361 cells. These results suggest that among these breast cancer cell lines we randomly assayed, there is an inversed association between DAPK2 and miR-520h expression. We further investigated the miR-520h-mediated DAPK2 regulation and observed that expression of the DAPK2 protein was significantly reduced in miR-520h-overexpressing MDA-MB-231 and MDA-MB-468 cells (Figure 2c). Moreover, blocking the endogenous miR-520h led to the upregulation of DAPK2 in MDA-MB-231 and Hs578T cells (Figure 2d). To further investigate whether the suppressive effects of miR-520h act through an miRNA-mediated regulation, we established a luciferase reporter possessing the 3’UTR of DAPK2 (Figure 2e), and observed a significant suppression of luciferase activity in cells overexpressing miR-520h. This indicates that miR-520h is able to downregulate gene expression via the DAPK2 3’UTR (Figure 2f). To investigate whether this suppression was due to a direct interaction between miR-520h and its binding site in the DAPK2 3’UTR, we constructed an miR-520h-binding site based on a mutated 3’UTR reporter in which the interaction between miR-520h and the 3’UTR was disrupted (Figure 2e). In addition, miR-520h with a compensatory mutation that restored base pairing to the DAPK2 3’UTR was also created to confirm their interaction (Figure 2e). As expected, in contrast to the wild-type DAPK2 3’UTR, the mutated DAPK2 3’UTR failed to be suppressed by miR-520h, but was sensitive to expression via Mut-miR-520h (Figure 2f). These results demonstrate that miR-520h directly hinders DAPK2 expression through a miRNA-mediated pathway.

DAPK2 is downregulated by miR-520h. (a) Expression of DAPK2 in breast cancer cells were detected by western blot. The intensities of individual bands were analyzed by Image J software and normalized with GAPDH to calculate the relative levels of DAPK2, as shown at the bottom. (b) Quantitative reverse transcription-PCR analysis was used to determine the miR-520h levels. Results were collected from three independent experiments with triplicate repeats. Data are shown as the mean±s.d. (c) Protein levels of DAPK2 in MDA231/Vec and MDA231/miR-520h were measured by western blot. (d) Effect of miR-520h depletion on the expression of DAPK2 protein. Protein levels of DAPK2 in miR-520h antagomirs-transfected MDA-MB-231 and Hs578T cell were measured by western blot using DAPK2-specific antibody. The intensities of individual bands were analyzed by Image J software and normalized with α-tubulin to calculate the relative levels of DAPK2, as shown at the bottom. (e) Schematic diagram showing the design of constructs containing wild-type and mutant miR-520h and DAPK2 3’UTR, respectively. The 3’UTR of DAPK2 was appended to the luciferase open reading frame of the pmirGLO vector. The miR-520h complementary seed region and the compensatory mutation sites are indicated in red. (f) DAPK2 wt-3’UTR or DAPK2 Mut-3’UTR reporter gene along with the indicated plasmids (Control, miR-520h and Mut-miR-520h) were co-transfected into MDA231 cells for 48 h and luciferase assays were performed. Luciferase activity was assessed by normalization of firefly luciferase activity to Renilla luciferase activity. Results were collected from three independent experiments, with triplicate repeats for each experiment. Data are shown as the mean±s.d. *P<0.05.
DAPK2 is involved in miR-520h-mediated paclitaxel resistance
Having shown the suppressive effect of miR-520h on DAPK2 expression, we further validated whether DAPK2 is functionally involved in miR-520h-enhanced paclitaxel resistance. We devised a DAPK2-expressing construct that lacked the DAPK2 3’UTR of the miR-520h target site and transfected it into MDA-MB-231 and MDA-MB-468 cells. The transfection of the DAPK2-expressing vector successfully restored the expression of the DAPK2 protein suppressed by miR-520h both in MDA-MB-231 and MDA-MB-468 cells (Figures 3a and b). Upon paclitaxel treatment, restoration of DAPK2 significantly antagonized miR-520h-enhanced cell survival in both MDA231/miR-520h and MDA468/miR-520h cells (Figures 3c and d). A lower IC50 was observed in MDA231/miR-520h/DAPK2 cells compared with MDA231/miR-520h/pcDNA6 cells (Figure 3c; 11.22 versus 18.92 nM), and a similar result was also observed when MDA468/miR-520h/DAPK2 and MDA468/miR-520h/pcDNA6 cells were compared (Figure 3d; 8.27 versus 21.35 nM). Flow cytometry analysis further indicated that the re-expression of DAPK2 in MDA231/miR-520h and MDA468/miR-520h cells abrogated resistance to paclitaxel-induced apoptosis (Figures 3e and f). The paclitaxel-induced sub-G1 population of MDA231/miR-520h/DAPK2 cells was increased to 40.0±9.2%, compared with that of MDA231/miR-520h/pcDNA6 cells, which was only 17.4±1.6% (Figure 3e). In addition, MDA468/miR-520h/DAPK2 cells contained more sub-G1 cells than MDA468/miR-520h/pcDNA6 cells in the presence of paclitaxel (31.0±3.2% versus 14.3±3.8%; Figure 3f). To support these findings using ectopic overexpression models, we silenced the expression of both DAPK2 and miR-520h by using short hairpin RNA targeting DAPK2 and antagomirs blocking miR-520h, respectively. Consistent with our previous results (Figures 2a, b), antagonizing endogenous miR-520h significantly enhanced DAPK2 expression and this effect was further mitigated by shDAPK2 (Supplementary Figure S3a). Upon the treatment of paclitaxel, inhibition of DAPK2 significantly decreased miR-520h antagomirs-mediated cell death in MDA-MB-453 cells (Supplementary Figure S3b and c). Higher IC50 was observed in MDA453/miR-520h antagomirs/shDAPK2 cells compared to MDA453/miR-520h antagomirs/shGFP cells (Supplementary Figure S3b; 10.60 versus 5.80 nM). In addition, flow cytometry analysis further indicated that suppressing the miR-520h antagomirs-induced DAPK2 expression restored resistance to paclitaxel-induced apoptosis (Supplementary Figure S3c). The paclitaxel-induced sub-G1 population of MDA453/miR-520h antagomirs/shDAPK2 cells was reduced to 29±4.6%, compared with the level of MDA453/miR-520h antagomirs/shGFP cells is 48±2.66% (Supplementary Figure S3c). Thus, the suppression of endogenous DAPK2 is essential for miR-520h-mediated paclitaxel resistance of breast cancer cells. These data indicate that DAPK2 has a functional role in miR-520h-mediated paclitaxel resistance of breast cancer cells. To ascertain whether miR-520h-regulated DAPK2 modulates caspase-dependent apoptosis, we determine the activities of caspase-3 and 7 (Figure 3g). We found that caspase activity was significantly induced upon paclitaxel treatment (Figure 3g). The paclitaxel-mediated caspase induction was reduced upon miR-520h overexpression, and this effect was further mitigated by restoring DAPK2 expression in MDA231/miR-520h cells (Figure 3g). These results suggest that expression of miR-520h suppresses DAPK2 to desensitize cells from paclitaxel-induced caspase-dependent cell death. We also analyzed the expression of both pro-caspase and active caspase by anti-pro Caspase-3 antibody and anti-active Caspase-3 antibody (Figure 3h). A similar expression pattern was observed as caspase-3 was activated by paclitaxel treatment (Figure 3h). The paclitaxel-induced caspase activation was abolished upon miR-520h overexpression, and this effect was further mitigated by restoring DAPK2 expression in MDA231/miR-520h cells (Figure 3h). Again, these data suggest that miR-520h suppresses DAPK2 expression to protect cells from paclitaxel-induced caspase-dependent cell death. In addition, we treated cells with z-VAD-FMK, a pan-caspase inhibitor, and assayed the levels of paclitaxel-induced apoptosis among these cells. Our results showed that z-VAD-FMK treatment significantly repressed paclitaxel-induced apoptosis among all of the test cells including those transfected with miR-520h antagomirs (Figure 3i), further supporting our findings of miR-520h antagomirs-enhanced caspase-dependent apoptosis triggered by paclitaxel.


DAPK2 is a functional effector in miR-520h-mediated drug resistance. (a, b) Restoration of DAPK2 by transfection of DAPK2 plasmid into MDA231/Vec, MDA231/miR-520h (a) and MDA468/Vec, MDA468/miR-520h (b) cells. DAPK2 protein expression was determined by Western blot. (c– f) Effect of DAPK2 overexpression on paclitaxel-induced apoptosis. After transfection as indicated in a, b, these cells were treated with paclitaxel for 24 h and then subjected to MTS assay (c, d) or treated with 5 nM of paclitaxel for flow cytometry analysis (e, f). Results were collected from three independent experiments, with triplicate repeats for each experiment. Data are shown as the mean±s.d. *P<0.05. (g) miR-520 h suppresses the caspase-dependent cell death induced by paclitaxel. Cells were treated with 5 nM of paclitaxel for 24 h and then subjected to caspase activity assay. Results were collected from three independent experiments, with triplicate repeats for each experiment. Data are shown as the mean±s.d. *P<0.05. (h) miR-520 h suppresses paclitaxel-induced caspase activation. Cells were treated with 5 nM of paclitaxel for 24 h. Cell lysates were collected and then subjected to western blot using anti-pro Caspase-3 antibody and anti-active Caspase-3 antibody to detect the pro-form and active form of caspase-3, respectively. (i) Effect of caspase inhibition on miR-520 h antagomirs-induced apoptosis upon paclitaxel treatment. Cells were treated with paclitaxel for 24 h and then subjected to apoptosis assay by detection the sub-G1 population using flow cytometry. Results were collected from three independent experiments with triplicate repeats. Data are shown as the mean±s.d. *P<0.05.
Elevated level of miR-520h is associated with poor prognosis and cancer metastasis in human breast cancer patients
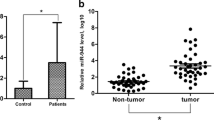
Having identified the functional roles of miR-520h in breast cancer cells, we next asked whether the expression of miR-520h is correlated with the clinical outcome in patients with breast cancer. A log-rank analysis indicated that the patient survival is significantly worse in patients with high miR-520h expression (n=95) in their tumor tissues, whereas those with low miR-520h (n=51) levels had a better survival probability (Figure 4). To further ascertain whether the expression of miR-520h is also associated with the clinicopathological characteristics of breast cancer, we examined their TNM status and found that patients whose tumors expressed higher levels of miR-520h exhibited advanced tumor status and increased lymph node metastases (Table 1). In addition to clarifying the oncogenic roles of miR-520h in breast cancer, the clinical evidence further confirmed the impact of miR-520h on human breast tumors. These results indicate that level of miR-520h is positively corrected with tumor and lymph node status, as well as with poor survival outcome in patients with breast cancer.

miR-520h expression level predicts poor clinical outcome in patients with breast cancer. The log-rank test (two-sided) was used to compare the differences between groups. The Kaplan–Meier curves show analyses of overall survival in 146 patients with breast cancer. High and low miR-520h expression is defined as an expression being higher and lower than the mean of miR-520h from total patients by quantitative reverse transcription-PCR, respectively. Results were collected from three independent experiments, with triplicate repeats for each experiment. **P<0.01.
Discussion
Failure of cancer treatment is not usually due to a lack of initial response but to tumor recurrence after the primary therapy. Even if the survival rate of breast cancer patients has been significantly improved, the metastatic breast cancer remains a problem for current treatment. In this study, we not only observed the elevated level of miR-520h in advanced breast cancer tissues, which may serve as a potential biomarker, but also identified miR-520h as a functional miRNA in protecting cancer cells from drug-induced cell death. This new information regarding miRNA function and its importance to the underlying mechanisms in human breast cancer suggests that it is a novel target for management of tumor recurrence and metastasis.
Although hundreds of miRNAs have been identified in humans, our knowledge of their biological functions and specific targets remains limited. Recently, several miRNAs have been found to regulate drug resistance of cancer cells,11 with miR-15a and miR-16-1 being identified as tumor suppressors in chronic lymphocytic leukemia.19 Deficiency of these suppressors enhances BCL2 expression and prevents the cleavage of pro-caspase 9 and poly-ADP-ribose polymerase, which is required for the activation of the intrinsic apoptosis pathway. Other studies also found that miR-15b and miR-16 negatively regulate Bcl-2 protein expression and subsequently sensitize gastric cancer cells to anticancer agents.20 In addition, miR-21 has been proven to enhance the resistance of breast cancer cells to topotecan treatment.21 Herein, we provide the first evidence for an antiapoptosis activity of miR-520h in human breast cancer. Whereas most of the miRNAs known to be involved in mediating drug resistance tend to act through controlling the expression of drug pumps, such as miR-451-mediated Mdr1 inhibition and miR-519c-mediated suppression of ABCG2 expression in cancer cells,22, 23 our results demonstrate a novel mechanism of miRNA-mediated drug resistance. Specifically, miR-520h directly targeted DAPK2 3’UTR and suppressed the expression of DAPK2 protein, which led to breast cancer cells being insensitive to drug-induced apoptosis. Although DAPK2 is a well-documented proapoptotic protein in human malignancies, the regulation of DAPK2 by miRNA has never been identified in breast cancer. This study marks the first time that a miRNA, miR-520h, has been identified as having the capacity to repress DAPK2 expression by epigenetic regulation. This mechanism thus represents a new layer of DAPK2 regulation and drug resistance in breast cancer.
Recent studies have revealed the phosphatidylinositol 3-kinase/Akt pathway as a key player in the pathogenesis of triple-negative breast cancer.24, 25 In our previous studies, the expression and oncogenic functions of miR-520h have been shown to be involved in the phosphatidylinositol 3-kinase/Akt pathway.14, 15 This molecule posttranscriptionally suppresses PP2A/C to activate the Akt and NF-κB pathways, leading to an increased expression of forkhead box C2, which further facilitates metastatic ability.14 Moreover, miR-520h has an essential role in E1A-mediated tumor suppression, as shown by the downregulation of the expression of Twist, an oncogenic epithelial-to-mesenchymal transition gatekeeper, by E1A-suppressed miR-520h in breast cancer cells.15 Epithelial-to-mesenchymal transition is positively correlates with multidrug resistance and poor clinical outcome of cancers. In this study, in addition to demonstrating the novel mechanism of miR-520h-mediated DAPK2 expression, we observed that the expression of miR-520h is an indicator of poor prognosis in breast cancer patients and is associated with more aggressive breast cancer. Because loss of drug sensitivity is the major causes of tumor recurrence and distant metastasis, these results provide an important clue that miR-520h has a dual role in drug resistance and metastasis as well as an effect on the prognosis of breast cancer patients.
Materials and methods
Reagents and antibodies
Paclitaxel, polyethyleneimine, polybrene, RNase A, propidium iodide were purchased from Sigma-Aldrich (St Louis, MO, USA). Protease inhibitor cocktail and FuGENE 6 were purchased from Roche (Basel, CH, USA). Noble agar was purchased from DIFCO Laboratories (Detroit, MI, USA). All cell culture-related reagents were purchased from Invitrogen (Carlsbad, CA, USA). Western blotting was performed with the use of the following antibodies: DAPK2 (GTX113947, GeneTex, Irvine, CA, USA), Pro Caspase-3 (Clone E61, ab32150, Abcam, Cambridge, UK) and active Caspase-3 (Clone E83-77, ab32042, Abcam), α-tubulin (T-5168, Sigma-Aldrich) and GAPDH (GTX627408, GeneTex). All secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA, USA).
Cell culture and transfection
The human breast cancer cell lines (MDA-MB-231, MDA-MB-468, Hs578T, BT483, T47D, MCF-7, MDA-MB-361, MDA-MB-453 and HBL-100) were purchased from the American Type Culture Collection (ATCC) and grown in Dulbecco's Modified Eagle's Medium/F12 supplemented with 10% fetal bovine serum. All cell lines were grown at 37 °C in a humidified 5% CO2 atmosphere. These cells were free of mycoplasma contamination and were confirmed by STR profiling at Center for Genomic Medicine, NCKU (Tainan, Taiwan). Transfections were carried out using FuGENE 6 (Roche) according to the manufacturer’s instructions. Established stable cell lines were generated by transfection with FuGENE 6 followed by selection with blasticidin (Invitrogen).
Specimens
The tissues used were from the National Taiwan University Hospital with Institutional Review Board approval. Written informed consent was obtained from all patients. None of the patients had received preoperative neoadjuvant chemotherapy or radiation therapy. The surgical specimens had been fixed in formalin and embedded in paraffin before they were archived. The tumor status, node status and distant metastasis were defined according to the International Union Against Cancer for breast cancer.26 Follow-up of patients was carried out up to 10 years. Patients who died of postoperative complications within 30 days after surgery were excluded from the survival analysis. We analyzed data for patients whose TNM and survival information was available (n=146) and divided the patients by miR-520h levels: 95 above the mean and 51 below the mean for log-rank analysis. We calculated the power of our main analysis and performed at least 80% power to analyze survival studies as described.27
RNA extraction and miRNA analysis
Total RNA was isolated from cell lines using Trizol reagent (Invitrogen) and from paraffinized tissues using the High Pure FFPE RNA Micro Kit (Roche) according to the manufacturer's instructions. The expression of human miRNA was assayed by using real-time PCR14, 28 and was normalized to internal control RNU6B. To determine the expression of miR-520h, we used the TaqMan MicroRNA Assay kit (Applied Biosystems, Carlsbad, CA, USA) following the manufacturer's protocol. RNA was reverse transcribed using 7 μl of RT mixture containing dNTPs, RT and RNase inhibitor and 3 μl of respective primer. The mixture was incubated at 16 °C for 30 min, 42 °C for 30 min, followed by 85 °C for 5 min. Real-time PCR reactions were then carried out in a total volume of 20 μl reaction mixture containing 1.33 μl of RT product, 10 μl of 2 × Taqman universal PCR master mix, 7.67 μl of water and 1 μl of TaqMan assay probe. All reactions, including controls, were performed in triplicate.
Construct of expression vectors, plasmids and anti-miR
The hsa-mir-520h stem-loop sequence plus its up- and downstream-flanking regions was amplified by PCR using genomic DNA of HeLa cells and cloned into the XhoI and NotI sites of pLemiR-empty vector (Open Biosystem, Huntsille, AL, USA). The human full-length DAPK2 (GenBank: NM_014326) was amplified by PCR using the cDNA of HeLa cells and cloned into the BamHI and NotI sites of pcDNA6/myc-His (Invitrogen). All primer sequences of miRNA and cDNA constructs are shown in Supplementary Table S1. The lowercase was representative of the additional sequence, and the restriction enzyme site was underlined. All constructs were confirmed by DNA sequencing. The pMD2.G and pCMV-ΔR8.91 plasmids were purchased from the National RNAi Core Facility at Academia Sinica, Taipei, Taiwan. The pmirGLO Dual-Luciferase vector was obtained from Promega (Madison, WI, USA). miR-520h antagomomirs, miRNA inhibitor (Ambion, Foster City, CA, USA) is single-stranded chemically modified oligonucleotides designed to inhibit endogenous miR-520h. Scramble control is a random sequence that has been extensively tested in human cell lines and tissues and validated to produce no identifiable effects on known miRNA function.
Lentiviruses infection
Recombinant lentiviruses were produced by co-transfecting a mixture of the indicated expression plasmid, the envelope plasmid (pMD2.G) and the packaging plasmid (pCMV-ΔR8.91) into HEK293T cells using polyethyleneimine (Sigma-Aldrich) according to the manufacturer's instructions. The viruses were harvested from the culture medium on day 2 after transfection and filtered using a 0.45-μm filter. Cultured cells were incubated with lentivirus containing 8 μg/ml polybrene for 24 h, replaced with fresh medium and incubated for another 48 h. For stable cell lines, cells were selected by puromycin.
Soft agar assays
Cells were seeded in six-well culture dishes in suspensions of 0.35% Noble Agar (DIFCO) in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum on top of a bed of 0.7% Noble Agar in the same complete medium. After 3 weeks, tumor cell colonies were stained with 0.05% crystal violet and colonies larger than 0.05 mm were counted under a dissecting microscope.
Cell viability by MTS test
Cells (5 × 103) per well were seeded in 96-well plates in medium containing 1% fetal bovine serum. Cells were allowed to adhere for 24 h and were subsequently incubated with the indicated drug concentrations. Each condition was tested in triplicate wells. Viability was assessed by Celltiter96 Aqueous1 solution reagent (Promega) according to the manufacturer's instructions. The amount of MTS formazan product was measured at a wavelength of 490 nm by a PowerWave X Microplate ELISA Reader (Bio-TeK Instruments, Winoski, VT, USA).
Cell apoptosis by flow cytometry analysis
Paclitaxel-induced cell death was analyzed as previously described.28 In brief, cells were treated with indicated paclitaxel concentrations and incubated for 24 h. Aliquots of 1 × 106 cells were collected and washed once with ice-cold phosphate-buffered saline (PBS) and then fixed with ice-cold 70% ethanol overnight. Fixed cells were then washed twice in PBS and stained with 1 μg/ml propidium iodide in PBS containing 10 μg/ml RNase A for 1 h and then analyzed using a flow cytometry with the FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
Colony formation assay
A total of 5 × 102 cells were seeded in six-well tissue culture dishes. After 2 weeks of culture, colonies were fixed with 4% paraformaldehyde and stained by 0.05% crystal violet. The total number of colonies in each well was counted.
Western blot
Cells were washed twice with PBS, lysed in NETN lysis buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% Nonidet P-40 and 1 mM EDTA) containing protease inhibitor cocktail (Roche) for 5 min with sonication and then centrifuged at 14 000 g for 30 min. An equal quantity of protein from cell lysates were resuspended in gel sample buffer, resolved by SDS–polyacrylamide gel electrophoresis, and transferred to PVDF membranes (Millipore, Billerica, MA, USA). After the blots were blocked in a solution of 10% skimmed milk, 0.1% Tween 20 and PBS, membrane-bound proteins were probed with primary antibodies overnight at 4 °C. The membranes were washed and then incubated with horseradish peroxidase-conjugated secondary antibodies for 45 min. Antibody-bound protein bands were detected using enhanced chemiluminescence reagents (Millipore) and photographed with Kodak X-Omat Blue autoradiography film (Perkin Elmer Life Sciences, Boston, MA, USA).
Construct of luciferase reporters and reporter assay
The 1390-bp 3’UTR of the DAPK2 gene (DAPK2 wt-3’UTR) containing miR-520h targeting site was amplified from genomic DNA of HeLa cells and cloned into the NheI and XhoI sites of pmirGLO Dual-Luciferase vector (Promega). The DAPK2 wt-3’UTR containing the mutant miR-520h target seed region sequences (TTT to CCC, DAPK2 Mut-3’UTR) and miR-520h containing the mutant DAPK2 3’UTR sequence (AAA to GGG, Mut-miR-520h) were mutated using a QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). All primers sequences of constructs are shown in Supplementary Table S1. The lowercase was representative to the additional sequence. Restriction enzyme site and mutation site sequences were underlined. All constructs were confirmed by DNA sequencing. For the DAPK2 3’UTR reporter assay, indicated DAPK2 3’UTR reporter gene constructs (0.1 μg) and miR-520 expression vectors (1 μg) were co-transfected into MDA-MB-231 cells (50% confluent in 24-well plates) using FuGENE 6 (Roche) according to the manufacturer's instructions. All cell extracts were prepared 48 h after transfection, and luciferase activities were determined by the Dual-Luciferase Reporter Assay System (Promega) following the protocols provided by manufacturer. Luciferase activity was assessed by normalization of firefly luciferase activity to Renilla luciferase activity.
Statistical analysis
All data of in vitro experiments were approximately normally distributed and presented as the mean±s.d. The variance of each experimental group was similar and performed two-tailed Student’s t-test for comparison of two groups. Statistical analyses of clinicopathological data were performed using Pearson’s X2-test. Survival curves were analyzed using the Kaplan–Meier method and log-rank test. P-values <0.05 were considered to be statistically significant.
References
Giordano SH, Buzdar AU, Smith TL, Kau SW, Yang Y, Hortobagyi GN . Is breast cancer survival improving? Cancer 2004; 100: 44–52.
Greenberg PA, Hortobagyi GN, Smith TL, Ziegler LD, Frye DK, Buzdar AU . Long-term follow-up of patients with complete remission following combination chemotherapy for metastatic breast cancer. J Clin Oncol 1996; 14: 2197–2205.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783–792.
Mehlen P, Puisieux A . Metastasis: a question of life or death. Nat Rev Cancer 2006; 6: 449–458.
Glinsky GV, Glinsky VV, Ivanova AB, Hueser CJ . Apoptosis and metastasis: increased apoptosis resistance of metastatic cancer cells is associated with the profound deficiency of apoptosis execution mechanisms. Cancer Lett 1997; 115: 185–193.
Del Bufalo D, Biroccio A, Leonetti C, Zupi G . Bcl-2 overexpression enhances the metastatic potential of a human breast cancer line. FASEB J 1997; 11: 947–953.
He L, Hannon GJ . MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004; 5: 522–531.
Krol J, Loedige I, Filipowicz W . The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010; 11: 597–610.
Bartel DP . MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–297.
Garzon R, Calin GA, Croce CM . MicroRNAs in Cancer. Annu Rev Med 2009; 60: 167–179.
Chen PS, Su JL, Hung MC . Dysregulation of microRNAs in cancer. J Biomed Sci 2012; 19: 90.
Kutanzi KR, Yurchenko OV, Beland FA, Checkhun VF, Pogribny IP . MicroRNA-mediated drug resistance in breast cancer. Clin Epigenet 2011; 2: 171–185.
Zheng T, Wang J, Chen X, Liu L . Role of microRNA in anticancer drug resistance. Int J Cancer 2010; 126: 2–10.
Yu YH, Chen HA, Chen PS, Cheng YJ, Hsu WH, Chang YW et al. MiR-520h-mediated FOXC2 regulation is critical for inhibition of lung cancer progression by resveratrol. Oncogene 2013; 32: 431–443.
Su JL, Chen PB, Chen YH, Chen SC, Chang YW, Jan YH et al. Downregulation of microRNA miR-520h by E1A contributes to anticancer activity. Cancer Res 2010; 70: 5096–5108.
Tanaka T, Bai T, Toujima S, Utsunomiya T, Utsunomiya H, Yukawa K et al. Impaired death-associated protein kinase-mediated survival signals in 5-fluorouracil-resistant human endometrial adenocarcinoma cells. Oncol Rep 2012; 28: 330–336.
Zhang X, Yashiro M, Qiu H, Nishii T, Matsuzaki T, Hirakawa K . Establishment and characterization of multidrug-resistant gastric cancer cell lines. Anticancer Res 2010; 30: 915–921.
Ogawa T, Liggett TE, Melnikov AA, Monitto CL, Kusuke D, Shiga K et al. Methylation of death-associated protein kinase is associated with cetuximab and erlotinib resistance. Cell Cycle 2012; 11: 1656–1663.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 2005; 102: 13944–13949.
Xia L, Zhang D, Du R, Pan Y, Zhao L, Sun S et al. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer 2008; 123: 372–379.
Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY . miR-21-mediated tumor growth. Oncogene 2007; 26: 2799–2803.
To KK, Robey RW, Knutsen T, Zhan Z, Ried T, Bates SE . Escape from hsa-miR-519c enables drug-resistant cells to maintain high expression of ABCG2. Mol Cancer Ther 2009; 8: 2959–2968.
Kovalchuk O, Filkowski J, Meservy J, Ilnytskyy Y, Tryndyak VP, Chekhun VF et al. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther 2008; 7: 2152–2159.
Gordon V, Banerji S . Molecular pathways: PI3K pathway targets in triple-negative breast cancers. Clin Cancer Res 2013; 19: 3738–3744.
Baselga J . Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist 2011; 16: 12–19.
Sobin LH, Fleming ID . TNM Classification of Malignant Tumors, fifth edition (1997). Union Internationale Contre le Cancer and the American Joint Committee on Cancer. Cancer 1997; 80: 1803–1804.
Barthel FM, Babiker A, Royston P, Parmar MK . Evaluation of sample size and power for multi-arm survival trials allowing for non-uniform accrual, non-proportional hazards, loss to follow-up and cross-over. Stat Med 2006; 25: 2521–2542.
Chen PS, Su JL, Cha ST, Tarn WY, Wang MY, Hsu HC et al. miR-107 promotes tumor progression by targeting the let-7 microRNA in mice and humans. J Clin Invest 2011; 121: 3442–3455.
Acknowledgements
This work was supported by the National Science Council grant from Taiwan (NSC 101-2320-B-400-016-MY3, NSC 102-2314-B-038-028-MY3, NSC 103-2314-B-038-059, NSC 101-2320-B-006-045-MY2, MOST 103-2628-B-006-003-MY3); National Health Research Institutes grant from Taiwan (CA-102-PP-41, CA-104-SP-01, CA-104-PP-12, MOHW104-TDU-B-212-124-008); Taipei Medical University-Shuang Ho Hospital, Ministry of Health and Welfare grant from Taiwan (103TMU-SHH-26). We thank National RNAi Core Facility (Academia Sinica, Taiwan) for providing specific shRNAs. We thank Ms Fang-Yu Tsai and Dr I-Shou Chang of Taiwan Bioinformatics Institute Core Facility for assistances on using Oncomine (National Core Facility Program for Biotechnology, NSC-100-2319-B-400-001). This research was, in part, supported by the Ministry of Education, Taiwan, R.O.C. The Aim for the Top University Project to the National Cheng Kung University (NCKU).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Su, CM., Wang, MY., Hong, CC. et al. miR-520h is crucial for DAPK2 regulation and breast cancer progression. Oncogene 35, 1134–1142 (2016). https://doi.org/10.1038/onc.2015.168
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.168
- Springer Nature Limited
This article is cited by
-
Expression study of microRNA cluster on chromosome 19 (C19MC) in tumor tissue and serum of breast cancer patient
Molecular Biology Reports (2023)
-
Breast Cancer Response to Therapy: Can microRNAs Lead the Way?
Journal of Mammary Gland Biology and Neoplasia (2021)
-
MAGI1 mediates tumor metastasis through c-Myb/miR-520h/MAGI1 signaling pathway in renal cell carcinoma
Apoptosis (2019)
-
The activation of microRNA-520h–associated TGF-β1/c-Myb/Smad7 axis promotes epithelial ovarian cancer progression
Cell Death & Disease (2018)
-
Selective activation of miRNAs of the primate-specific chromosome 19 miRNA cluster (C19MC) in cancer and stem cells and possible contribution to regulation of apoptosis
Journal of Biomedical Science (2017)