

Abstract
ABCG2 is a constitutively expressed ATP-binding cassette (ABC) transporter that protects many tissues against xenobiotic molecules. Its activity affects the pharmacokinetics of commonly used drugs and limits the delivery of therapeutics into tumour cells, thus contributing to multidrug resistance. Here we present the structure of human ABCG2 determined by cryo-electron microscopy, providing the first high-resolution insight into a human multidrug transporter. We visualize ABCG2 in complex with two antigen-binding fragments of the human-specific, inhibitory antibody 5D3 that recognizes extracellular loops of the transporter. We observe two cholesterol molecules bound in the multidrug-binding pocket that is located in a central, hydrophobic, inward-facing translocation pathway between the transmembrane domains. Combined with functional in vitro analyses, our results suggest a multidrug recognition and transport mechanism of ABCG2, rationalize disease-causing single nucleotide polymorphisms and the allosteric inhibition by the 5D3 antibody, and provide the structural basis of cholesterol recognition by other G-subfamily ABC transporters.
Similar content being viewed by others
Main
Humans have 48 distinct ABC transporters that belong to seven subfamilies (A–G) and differ in size, architecture, and domain arrangement1,2. ABCG2, also known as breast cancer resistance protein (BCRP), is a multidrug transporter that has physiological roles in many tissues including the mammary gland and the blood–brain, blood–testis, and maternal–fetal barriers3,4,5,6. ABCG2 has a minor role in uric acid transport, but its dysfunction is associated with several disease states linked to hyperuricaemia including gout, kidney disease, and hypertension7. Studies with ABCG2 knockout mice have revealed the effect of the protein on the oral availability of drugs in the gastrointestinal tract, whereas ABCG2 expressed in the canalicular membrane and in the apical membrane of kidney cells secretes diverse compounds into bile or urine. These two effects emphasize the impact of ABCG2 function on the pharmacokinetics of commonly used drugs. Consequently, ABCG2 is on the US Food and Drug Administration and the European Medicines Agency lists of transporters to be checked for drug–drug interactions8. Like its functional homologues ABCB1 and ABCC1, ABCG2 also has a notorious function in extruding anti-tumour drugs from a variety of cancer cells, which can result in multidrug resistance, a serious obstacle in cancer treatment9,10,11,12. While its remarkable substrate polyspecificity and widespread localization make it a contributor to multidrug resistance, it has so far failed to emerge as a viable therapeutic target and thus an understanding of its drug-binding pocket is of paramount importance6,7,13,14,15. ABCG2 was also considered a marker for haematopoietic stem cells, and an inhibitory conformational antibody (5D3) against the human protein was developed16.
Over recent decades, in vivo studies have provided detailed insight into the localization, physiological functions, trafficking, substrate specificity, and single nucleotide polymorphisms of ABCG2 (refs 15, 16, 17). However, our mechanistic understanding of ABCG2 is limited because no high-resolution structure is available, which is partly because of the challenges associated with its overexpression and functional purification. Whereas a crystal structure of the human sterol transporter ABCG5/G8 was recently reported18, direct structural insight into ABCG2 was limited to low-resolution electron microscopy imaging19 and an in silico model based on the structure of ABCG5/G8 (ref. 20). In this study, we used single-particle cryo-electron microscopy (cryo-EM) to determine the structure of wild-type, human ABCG2 in complex with two antigen-binding fragments of 5D3 (5D3-Fab).
In vitro function and structure determination
ABCG2 was expressed in human embryonic kidney cells (HEK293), purified in detergent solution, and reconstituted both into proteoliposomes and into lipidic nanodiscs for functional studies. The reconstituted protein catalysed transport of estrone-3-sulfate (E1S), a bona fide substrate of ABCG2, into proteoliposomes, with Michaelis constant (Km) values comparable to those previously reported using membrane vesicles derived from insect or human cells21,22 (Fig. 1a, b and Extended Data Fig. 1). E1S transport was inhibited by 5D3-Fab, but only if the antibody fragments were present in the lumen (inside) of the proteoliposomes. Thus our in vitro assays are consistent with earlier conclusions that 5D3 bound the extracellular surface of ABCG2 (ref. 16). Transport of E1S was completely abolished by the synthetic inhibitor Ko143 (Fig. 1b and Extended Data Fig. 1)23. Both 5D3-Fab and Ko143 reduced the ATPase activity of ABCG2 in liposomes while a mutation of the Walker-B residue Glu211 to a glutamine (E211Q) abolished activity (Fig. 1c).

a, Schematic of ATP-driven transport of E1S. 5D3-Fab (depicted as yellow surface) inhibits E1S transport only when bound to the extracellular side of ABCG2 in the lumen of the proteoliposomes. Pi, inorganic phosphate. b, Normalized initial E1S transport rates. FT, freeze thawing allowing 5D3-Fab access to the liposome lumen; G2, ABCG2. c, Normalized E1S-stimulated ATPase activity of ABCG2. In b and c, two separate experiments were performed in triplicate (error bars, s.d.).
Because nanodiscs contain a patch of lipid bilayer, they represent a physiologically relevant environment for structural studies of membrane proteins24,25. Negative-stain EM revealed that two 5D3-Fab fragments bound to a single, nanodisc-reconstituted ABCG2 homodimer. The resulting ABCG2–5D3(Fab) complex had a molecular mass of ~250 kDa and maintained two-fold symmetry. Single-particle cryo-EM analysis resulted in a three-dimensional reconstruction at an overall resolution of 3.8 Å (Fourier shell correlation (FSC) = 0.143 criterion; Extended Data Fig. 2). The transmembrane domains (TMDs) and the variable domains of 5D3-Fab were better resolved, with local resolutions approaching ~3 Å (Extended Data Fig. 3). The quality of the EM density map allowed de novo building of an atomic model of the TMDs and of the variable domain part of 5D3-Fab (Extended Data Fig. 4). We independently determined the X-ray structure of 5D3-Fab at 1.5 Å resolution (Extended Data Fig. 5), which provided an initial model to be docked into the EM density map. In contrast, the nucleotide-binding domains (NBDs) of ABCG2 were resolved at lower resolution, most probably because of their inherent flexibility and a lack of particle side-views in the EM data. We therefore generated a homology model of the ABCG2 NBD based on the recent structure of ABCG5/G8 (ref. 18) and docked this into our EM density map, where modifications to secondary structure elements could be fitted.
Architecture of ABCG2
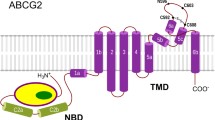
Like other G-subfamily ABC transporters, ABCG2 is a ‘half-transporter’ with one NBD and one TMD fused to a single polypeptide chain. The functional form of ABCG2 is a homodimer with a molecular mass of ~144 kDa. In the ABCG2–5D3(Fab) complex, the Fab fragments bind at an ~35° angle relative to the membrane plane (Fig. 2a). The fold of ABCG2 (Fig. 2b, c) is similar to that of ABCG5/G8, but distinct from that found in the B-subfamily of ABC transporters, exemplified by the bacterial multidrug transporter homologue Sav1866 (ref. 26) or the mouse ABCB1 (P-glycoprotein)27,28. The transmembrane helices and intracellular loops of ABCG2 are shorter than B-subfamily ABC transporters, resulting in a smaller distance between the NBDs and the membrane, thus resembling the more compact bacterial ABC importers29,30. The TMD interface is formed by TM2 and TM5a of opposing ABCG2 monomers. The two 406-GXXXG-410 motifs, previously postulated to be involved in dimerization31, are located on opposite sides of the transporter (Fig. 2b) and do not contact each other.

a, Ribbon diagram of ABCG2–5D3(Fab) complex. Protein subunits are labelled. G2′ refers to the second ABCG2 monomer. b, ABCG2 structure without 5D3-Fab, with one monomer coloured salmon. Motifs and residues associated with single nucleotide polymorphisms are coloured blue and labelled. The other ABCG2 monomer is coloured as a rainbow spectrum ranging from blue to red. c, Topology of the ABCG2 TMD coloured as in b. Cysteines forming inter- and intramolecular disulfide bonds (C592, C603, and C608) are indicated, as is the N-glycosylated residue N596.
While the architecture of ABCG2 is similar to that of ABCG5/G8 (Extended Data Fig. 6b), there are important differences that could not be predicted by the in silico model of ABCG2 (ref. 20) owing to model bias (root-mean-square deviation between TMDs of our structure and the in silico model is 2.2 Å) First, there is a backbone shift in TM2 and TM5a of both monomers. This causes ABCG2 to form an inward-facing cavity (see below), a feature that is not present in ABCG5/G8. Second, the structure of the external loop EL3 is distinct in the two structures.
Notably, the NBDs of ABCG2 remain in contact despite the absence of bound nucleotide. This was also observed in the structure of ABCG5/G8 but differs from the large conformational flexibility reported for B-subfamily ABC transporters32,33,34. The NBD of each ABCG2 monomer is linked to TM1a (known in ABCG5/G8 as connecting helix (CnH)) via a highly charged linker. This linker is not visible in the ABCG5/G8 X-ray structure or in the ABCG2 EM density, most probably because of its flexibility; therefore, it has not been modelled. The C2 motif of ABCG2, previously proposed to be important for ATP binding35, is located on an α-helix at the bottom of the NBDs, ~20 Å away from the Walker-A motif and ~45 Å from the signature motif, precluding its direct involvement in ATP binding to the canonical site (Fig. 2b).
Interaction of inhibitory 5D3 antibody with ABCG2
Each 5D3-Fab interacts with extracellular loops of both ABCG2 monomers (Fig. 3a). Most contacts are formed by EL3, the longest of the extracellular loops in ABCG2, connecting TM5c and TM6a. EL3 contains three cysteines (C592, C603, C608) and a single N-glycosylation site (N596; Fig. 3b). Our map showed clear density for up to two GlcNAc residues attached to N596, and there is equally clear density for two intramolecular disulfides (C592–C608 and C592′–C608′) as well as one intermolecular disulfide, C603–C603′, which is positioned on the two-fold symmetry axis of the ABCG2 homodimer. The proper formation of the intermolecular disulfide in our overexpression system was validated by SDS–polyacrylamide gel electrophoresis (SDS–PAGE), where fully dimeric ABCG2 was observed in non-reducing conditions both in the presence and absence of 5D3-Fab (Fig. 3c and Extended Data Fig. 7a). The intramolecular disulfides stabilize the structure of EL3, which is in line with previous reports that C592 and C608 are important for ABCG2 maturation and activity36,37. In contrast, the intermolecular disulfide did not seem to be critical for transport function in cellular assays, as mutation to various residues abolished the formation of the covalent dimer, but did not interfere with drug extrusion38.

a, Surface representation of ABCG2 and 5D3-Fab coloured as in Fig. 2a. Black lines indicate membrane boundaries. b, Structure of the external loops EL3 and EL3′ of the ABCG2 monomers and ribbon representation of the CDR regions of 5D3-Fab, as viewed from the extracellular side and coloured as in a. The inter- and intramolecular disulfides formed by C592, C603, and C608 are shown as sticks, as is N596 and attached GlcNAc moieties. c, Representative SDS–PAGE of purified ABCG2–5D3(Fab) complex. The same sample was applied after (lane 1) or before (lane 2) disulfide reduction using DTT (dithiothreitol), demonstrating disulfide-dependent dimerization of ABCG2 (black arrows) and 5D3-Fab (red arrows). M, marker proteins, with selected masses indicated on the left. d, ATPase activity of nanodisc-reconstituted ABCG2 at varying 5D3-Fab:ABCG2 ratios. For each point in the plot, error bars denote s.d. of triplicate measurements. e, Partial sequence alignment of human (Hs) and rat (Rn) ABCG2. The sequence corresponding to EL3 is boxed, red asterisks depict cysteines involved in inter- and intramolecular disulfides, green bars denote the ABCG2 EL3–5D3(Fab) interaction surfaces, and branches represent N-glycans.
N-glycosylation at N596 has been found to be essential for ABCG2 maturation, and mutations of this residue have resulted in protein destabilization and enhanced ubiquitin-mediated degradation39. N596 is not part of the ABCG2–5D3(Fab) interface, explaining why N-glycosylation of ABCG2 does not affect the binding of 5D3 (Fig. 3b)40. The physiological relevance of the interface observed in our structure is supported by the finding that 5D3-Fab is selective for human ABCG2 and does not bind to purified rat ABCG2 (Extended Data Fig. 7b). This can be rationalized by differences in the EL3 sequences of rat and human ABCG2, in particular those residues involved in the interface with 5D3 (N600, N601, and N604) (Fig. 3e). In addition, rat ABCG2 contains a second N-glycosylation site (N600), which probably interferes with 5D3 binding.
The binding of two 5D3-Fab fragments is consistent with the two-fold symmetry of the ABCG2 homodimer. In contrast, only one Fab fragment of the inhibitory antibody UIC2 bound to the multidrug transporter ABCB1, which is asymmetric32. We investigated whether two copies of 5D3-Fab are required for inhibition. Using nanodisc-reconstituted ABCG2, we found that only one 5D3-Fab per ABCG2 homodimer is required to observe maximal inhibition of the ATPase activity (Fig. 3d). This suggests that a single 5D3 molecule can inhibit transport activity of ABCG2 by clamping the ABCG2 monomers together, precluding the formation of an outward-facing conformation that is essential for active transport. In addition, the inhibition of ATPase activity suggests relatively strict conformational coupling of the TMD and NBD motions, given that the antibody-binding site and the ATPase sites are some ~80 Å apart and on opposite sides of the membrane. It was previously shown that, whereas AMP, ADP, and ATP did not appear to affect 5D3 binding in vitro, binding of 5D3 was reduced by the presence of AMP-PNP (adenosine 5′-(β-γ-imido)triphosphate) or orthovanadate40.
Translocation pathway and substrate-binding site
ABCG2 can bind or transport structurally diverse molecules ranging from hematoporphyrin, mitoxantrone, and methotrexate to inhibitors such as the tyrosine kinase inhibitor gefitinib or the fumitremorgin C-derived molecule Ko143 (ref. 23). The structure of ABCG2–5D3(Fab) revealed an inward-open conformation with a deep, slit-like cavity that is primarily lined by TM2 and TM5a from the two ABCG2 monomers. This cavity (cavity 1) opens to the cytoplasm and to the inner leaflet of the lipid bilayer, and it reaches more than halfway across the membrane to residues L554 and L554′ of opposing ABCG2 monomers. These leucine residues form a plug that separates cavity 1 from a secondary, smaller cavity (cavity 2) located below the EL3 external loops (Fig. 4a). Cavity 2 is inaccessible in the present conformation and is likely to have a lower affinity for substrate owing to its less pronounced hydrophobic surface, which may help promote substrate release to the extracellular side during the transport cycle.

a, Sagittal slice through a surface representation of the ABCG2 TMD with cavities 1 and 2 indicated and the leucine plug depicted by a dashed circle. b, Electrostatic surface potential of cavity 1 viewed from the cytoplasm with NBDs removed. Two bound cholesterol molecules are shown as green sticks. Arrows indicate entry sites into cavity 1 from the inner leaflet of the membrane. c, Ribbon diagram of the TMDs of ABCG2, with bound cholesterol molecules shown as green sticks and labelled. Helices are numbered as in Fig. 2c. d, EM density (white surface) around cholesterol molecule bound between TM2 and TM5a′. Residues within 4 Å of bound cholesterol are shown as sticks and labelled. Panels a, c, and d are coloured as in Fig. 2a. e, f, Sequence alignments of TM2 and TM5a regions of human ABCG subfamily members. Residues within 4 Å of the bound cholesterol are marked with red asterisks.
Given the size, hydrophobic nature, and accessibility of cavity 1 to the cytoplasm and the membrane, we assign it the role of a multidrug-binding pocket. Its geometry makes it suitable to accommodate flat, polycyclic and hydrophobic substrates (Fig. 4b). We observed two large density features that were symmetrically located in cavity 1. Their size and shape were inconsistent with bound phospholipids and suggested a polycyclic molecule (Fig. 4c, d). Only three such molecules were present during ABCG2 reconstitution into nanodiscs: cholesterol, cholesteryl hemisuccinate, and cholate. Of these, hemisuccinate could be excluded as a candidate because its substituents would cause a steric clash with the L554 plug at the top of cavity 1. While cholate, used for the preparation of nanodiscs, would fit the density, functional data suggest that ABCG2 is not responsive to cholate in the presence or absence of E1S (Extended Data Fig. 1f). We therefore interpreted the density features as two cholesterol molecules that are in contact with six hydrophobic residues from each ABCG2 monomer (F432 and F439 from TM2 and L539, I543, V546, and M549 from TM5a). The shape of the density guided us to fit the cholesterols with their hydroxyl groups pointing into the cavity, as the opposite orientation would generate a steric clash with the L554 plug (Extended Data Fig. 4e). We conclude that cholesterol, which is not a bona fide substrate, can nevertheless bind to ABCG2 and reveals where the chemically similar, physiological substrate E1S would bind. Two E1S molecules could indeed be fitted in the substrate-binding pocket where the cholesterol molecules are bound, with their sulfate groups pointing towards the cytoplasm. The conclusion that cavity 1 represents an ABCG2 substrate-binding site is supported by the observation that the basal ATP hydrolysis rate of cholesterol-bound, nanodisc-reconstituted ABCG2 is indistinguishable from the E1S-stimulated ATPase activity in liposomes (Extended Data Fig. 1f).
While ABCG2 does not catalyse significant cholesterol transport in vivo, its function was previously shown to be regulated by cholesterol41. In contrast, the other members of the ABCG subfamily (ABCG1 (ref. 42), ABCG4 (ref. 43), and ABCG5/G8) are reported sterol transporters. The residues in contact with bound cholesterol in ABCG2 are either fully conserved or substituted by other hydrophobic residues in the other G-subfamily ABC transporters, with the exception of V546, which corresponds to N568 in ABCG8 (Fig. 4e, f). Intriguingly, two of these six residues have been investigated in human ABCG5/G8. A mutation of Y432 in ABCG5 (corresponding to F439 in ABCG2) to an alanine disrupted in vivo cholesterol transport18. Our structure reveals that F439 forms a stacking interaction with the B-ring of bound cholesterol (Fig. 4d). In addition, a mutation of A540 in ABCG5 (corresponding to V546 in ABCG2) to a phenylalanine also interfered with biliary cholesterol transport, which was interpreted as a possible steric clash with bound cholesterol by the larger, aromatic residue18. These results are fully in line with our structural data and validate our identification of a conserved sterol-binding cavity that is probably shared by other G-subfamily ABC transporters, even though the orientation of cholesterol may be different when recruited from the lipid bilayer (hydroxyl group pointing to the cytoplasm).
Cavity 2 observed in ABCG2 is not present in the ABCG5/G8 structure owing to differences in the structure of EL3. The sequence similarity in EL3 is low among ABCG subfamily members (Extended Data Fig. 6c). Whereas ABCG1, ABCG4, and ABCG5 each have two cysteines in similar positions to ABCG2 (C587, C600), ABCG8 has no cysteines in EL3. It is conceivable that the formation of intramolecular disulfides in EL3 of ABCG2 contributes to its function as a multidrug transporter, because EL3 is a key element in forming cavity 2.
Single nucleotide polymorphisms of ABCG2
The mutation Q141K is an ABCG2 single nucleotide polymorphism that is linked to hyperuricaemia and gout44. It results in decreased ABCG2 expression, misfolding, and degradation by the endoplasmic-reticulum-associated protein degradation (ERAD) system. Nevertheless, residual ABCG2 (Q141K) in the plasma membrane shows transport activity45. Despite the lower resolution in the NBDs of our ABCG2 structure, we can place Q141 on the α-helix adjacent to TM1a (Fig. 2b). TM1a is an amphipathic α-helix, and its positively charged side faces Q141. We expect that the mutation of Q141 to a positively charged lysine would result in electrostatic repulsion that could distort TM1a, affecting ABCG2 folding. Another single nucleotide polymorphism-associated residue, R482, has been extensively studied and reported to be important for ABCG2 function. Its mutation to various residues affects substrate specificity46, although it was proposed that this is not through direct substrate interaction47. In our structure, R482 is located on TM3 (Fig. 2b) where it forms a stabilizing hydrogen bond with S521 on TM4, but is approximately 9 Å away from the substrate-binding pocket. The reported impact on substrate specificity is therefore probably caused by allosteric effects.
Proposed transport mechanism
Our results allow us to formulate a transport and inhibition mechanism that is consistent with the available biochemical and functional data. The mechanism invokes ATP-driven alternating access48, whereby the closing of the NBD interface in response to ATP binding converts the substrate-bound, inward-open conformation of ABCG2 to an outward-facing state that can release the substrate to the outside (Fig. 5). In the absence of bound ATP, the hydrophobic substrate-binding site (cavity 1) is exposed to the cytoplasm and the inner leaflet of the plasma membrane, from where substrate is recruited. In this state, cavity 2 is empty but occluded. Upon binding of substrate and ATP, conformational changes convert the transporter to an outward-facing state, where the substrate is moved into cavity 2, from where it is expelled into the extracellular medium by means of a hydrophobic mismatch. The transporter cannot convert to an inward-facing state if the substrate is present in cavity 2, owing to steric clashes. Thus, ATP hydrolysis and the conversion to an inward-facing state are directly linked to substrate release, providing the molecular basis of active transport. The inward-facing state can be inhibited by 5D3-Fab. Although two 5D3-Fab fragments can bind to the extracellular side, only one is required to clamp the ABCG2 monomers together, restricting their relative motion and allosterically impairing both substrate transport and ATP hydrolysis by shifting the equilibrium to the inward-facing conformation. This demonstrates strong coupling between TMDs and NBDs in ABCG2, which in turn can provide a molecular explanation for substrate-induced ATPase stimulation: substrate binding to cavity 1 probably stimulates the ATPase activity of the transporter by stabilizing the TMD interface, which results in closer proximity of the NBDs. The structure presented here reveals that inhibitory 5D3-Fab and substrate binding are not mutually exclusive, but transport from this state is impossible if the TMDs are externally clamped together by the antibody fragments.

ABCG2 monomers are coloured red and blue, with cysteines forming disulfide bonds (yellow dashed lines) labelled ‘s’. ATP-driven transport of substrate (orange hexagon labelled ‘S’) occurs between states 1 and 2 (states indicated by circled numbers). Substrate enters cavity 1 (labelled ‘1’) from the cytoplasm or from the inner leaflet of the membrane, but cannot access cavity 2 (labelled ‘2’) owing to the leucine plug (shown between ABCG2 monomers in states 1 and 3). Closing of the NBD interface converts state 1 to an outward-facing state 2 that can release substrate to the outside. The release of inorganic phosphate (Pi) is accompanied by the conversion of ABCG2 from state 2 to state 1. In state 3 (which corresponds to the structure presented here), two 5D3-Fab fragments (shaded green and yellow and labelled Fab) bind to the extracellular side of ABCG2. This leads to inhibition of transport and ATPase activity (indicated by red arrows to the left). While inhibition of transport occurs by clamping the ABCG2 monomers together, inhibiting ATP hydrolysis occurs allosterically (depicted by dashed red arrows).
Conclusions
The ABCG2 structure represents the first, to our knowledge, high-resolution structure of a human multidrug ABC transporter. The observed geometry and architecture of the hydrophobic substrate-binding cavity provide the structural basis of the polyspecific interaction with diverse hydrophobic compounds and offer an opportunity to model drugs and inhibitors into the multidrug-binding pocket, which may facilitate structure-based drug and inhibitor design. Our results also define the mechanism by which an inhibitory antibody, through long-range allosteric effects, precludes conformational changes that are essential for substrate transport. While probing of additional drug- and inhibitor-bound states is essential for a complete mechanistic understanding, our results can already be used for diagnostic and potential therapeutic approaches.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Expression and purification of human ABCG2
Human full-length ABCG2 (Uniprot: Q9UNQ0), containing an amino (N)-terminal Flag tag, was expressed in HEK293-EBNA (Thermo Fisher Scientific) cells as previously described49. The cells were treated with 5 μM kifunensine (Toronto Research Chemicals) for 30 min before transfection. After transfection, the cells were incubated for 2 days at 37 °C before harvesting.
Transfected cells were solubilized in 40 mM HEPES buffer pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 1 mM PMSF (phenylmethylsulfonyl fluoride), 2 μg ml−1 DNaseI (Roche), and protease inhibitor cocktail (Sigma) with 1% DDM (n-dodecyl-β-d-maltopyranoside) + 0.1% CHS (cholesteryl hemisuccinate) (w/v) (Anatrace) for 90 min at 4 °C. The solubilized lysate was ultracentrifuged at 100,000g and the supernatant was incubated with anti-Flag M2 affinity agarose gel (Sigma). The gel was washed extensively with purification buffer containing 40 mM HEPES pH 7.5, 150 mM NaCl, and 0.026% DDM + 0.0026% CHS (w/v). ABCG2 was eluted with Flag peptide (Sigma) and treated with EndoF before being loaded into a TSKgel G3000SWXL column (Tosoh Bioscience). The peak fractions were collected and reconstituted into nanodiscs or liposomes and used for either cryo-EM studies or functional assays.
ABCG2-liposome preparation for transport and ATPase assays
A brain polar lipid:cholesterol (BPL:chol) (Avanti Polar Lipids) mixture was prepared at a 4:1 (w/w) ratio as described previously50. Briefly, the BPL:chol mixture was extruded 11 times through a 400 nm filter and destabilized with 0.17% (v/v) Triton X-100. Detergent-purified ABCG2 was then mixed with BPL:chol at a 100:1 (w/w) lipid:protein ratio. Detergent was removed with Bio-Beads and proteoliposomes were spun at 100,000g, resuspended in 25 mM HEPES pH 7.5, 150 mM NaCl at a final lipid concentration of 10 mg ml−1, and the reconstitution efficiency was determined51.
ABCG2-nanodisc preparation for ATPase assays
Membrane scaffold protein (MSP) 1D1 was expressed and purified as described previously24. BPL:CHS (4:1) (w/w) was solubilized with a 3× molar excess of sodium cholate using a sonic bath. Solubilized BPL:CHS was mixed with MSP 1D1 and detergent-purified ABCG2 at a molar ratio of 100:5:0.2 (lipid:MSP:ABCG2). Bio-Beads were added and the sample was incubated at 4 °C overnight. Bio-Beads were removed and the sample was spun at 100,000g before being loaded into a Superdex 200 10/300 column (GE Healthcare) and used either for cryo-EM studies or ATPase assays.
Transport assays and E1S Km determination
Liposome-reconstituted ABCG2 was freeze-thawed in the presence or absence of a 3× molar excess of 5D3-Fab to get the 5D3-Fab inside the proteoliposomes. Freeze thawing was not detrimental to the function of ABCG2. Proteoliposomes were then extruded through a 400 nm polycarbonate filter and diluted to 4 mg ml−1 lipid in transport buffer (25 mM HEPES pH 7.5, 150 mM NaCl). MgCl2 (5 mM) and 3H-E1S (1–50 μM) were added in the presence or absence of 5D3-Fab or Ko143 and the samples incubated for 5 min at 30 °C. The transport reaction was initiated by the addition of 2 mM ATP. To stop the reaction, samples were removed, added to ice-cold transport buffer containing unlabelled E1S, filtered using a Multiscreen vacuum manifold (MSFBN6B filter plate, Millipore) and washed three times. Radioactivity trapped on the filters was measured using a Perkin Elmer 2450 Microbeta2 microplate scintillation counter. Data were analysed and curves were plotted using the nonlinear regression Michaelis–Menten analysis tool and initial rates were determined using linear regression in GraphPad Prism 7 (GraphPad Software, La Jolla, California, USA).
ATPase assays and stoichiometry
ATP hydrolysis activity was assessed as described previously52. All reactions were performed at 37 °C in the presence of 2 mM ATP and 10 mM MgCl2. E1S (50 μM), 100 μM sodium cholate, saturating amounts of Ko143, and a 3× molar excess of 5D3-Fab were used to assess their stimulating or inhibitory effects. The molar ratios of nanodisc-reconstituted ABCG2 and 5D3-Fab were varied to determine the functional stoichiometry. For ATPase assays in proteoliposomes, samples were freeze-thawed and extruded as described above. ATPase rates were determined using linear regression in GraphPad Prism 7.
Cryo-EM data acquisition
Cryo-EM grids of nanodisc-reconstituted ABCG2–5D3(Fab) were prepared using a Vitrobot Mark IV (FEI) with the environmental chamber set at 95% humidity and 10 °C. Aliquots (3 μl) of the complex, at a concentration of ~0.2 mg ml−1, were placed on glow-discharged Quantifoil carbon grids (300 mesh, copper). Grids were blotted for 1.5 s and flash-frozen in a mixture of liquid ethane and propane cooled by liquid nitrogen. Grids were imaged with a Titan Krios (FEI) electron microscope that operated at 300 keV equipped with a Quantum-LS energy filter and a K2 Summit electron counting direct detection camera (Gatan). Images were recorded using SerialEM53 in super-resolution mode using a 20 eV slit width of the GIF energy filter at a nominal magnification of 130,000×, resulting in a calibrated super-resolution pixel size of 0.5196 Å (physical pixel size 1.039 Å). Defocus was set to vary from −1.5 to −2 μm. The super-resolution micrographs were down-sampled twice by Fourier-cropping and drift-corrected and dose-weighted using MotionCor2 (ref. 54) through the Focus suite55. Each image was dose-fractionated to 40 frames (16 s exposure in total, 0.4 s per frame). The dose rate was set to ~4.5 electrons per pixel per second (~1.7 electrons per square ångström per frame), leading to a total dose of ~67 electrons per square ångström (Extended Data Table 1). Focus was used to remove bad micrographs manually from the data set.
Image processing
The drift-corrected, dose-weighted micrographs were imported in RELION-2.02-beta56,57,58, which was used for all data processing except where specified. Contrast transfer function (CTF) parameters were estimated with GCTF59. Micrographs had a calculated defocus of −1.1 to −2.5 μm. Particles were picked in template-free manner using Gautomatch60. A total of 92,326 particles were selected from 1,702 electron micrographs. After three rounds of two-dimensional classification and selection of particles belonging to ‘good’ classes, the data set was reduced to 32,806 particles. These particles were used to generate a model with three-dimensional auto-refine. The resolution of this initial map was 5.9 Å. This map was then filtered to 20 Å and projected in evenly spaced (15°) directions. Using these projections as templates, a total of 232,608 particles could be picked from the data set. After three steps of two-dimensional classification and selection, we obtained a final data set of 97,612 particles. Refinement of these particles against the same starting model as before using the three-dimensional auto-refine procedure resulted in a 4.2 Å map. After post-processing (including soft masking and B-factor sharpening with the automatically determined B-factor of −142.3 Å2) in RELION-1.4 (refs 56, 57), a map with a resolution of 3.8 Å (by FSC using the 0.143 Å cut-off criterion61,62,63) was obtained. To assist modelling, a local-resolution-filtered map was calculated with RELION-2.02-beta (using a B-factor of -142.3 Å2).
Model building and refinement
For model building, we used mainly the post-processed map, but also the local-resolution-filtered map and the original refinement map. The Coot program64 was used for all modelling steps. 5D3-Fab was placed as a rigid body followed by separate fitting of the variable domain and constant domain. A half-transporter atomic model for ABCG2 was generated using the HHpred server with MODELLER65, utilizing the ABCG5 X-ray structure18 (chain A of Protein Data Bank (PDB) accession number 5DO7). The TMD and NBD domains were fitted separately in the electrostatic potential map. The half-transporter atomic coordinates were then duplicated and rigid-body docked into the region of the map corresponding to the other half of ABCG2 to complete the full transporter model.
The post-processed map was of excellent quality and allowed de novo model building (Extended Data Fig. 4) of the ABCG2 TMD and the variable domain of the 5D3-Fab. For the constant domain of 5D3-Fab, we were especially careful when adapting the model to the map and took advantage of the local-resolution filtered map. In the region of the ABCG2 NBDs, the post-processed map was of lower quality than in the regions of ABCG2 TMD and 5D3-Fab. A similar reduced resolution of NBDs was recently observed in the cryo-EM structures of the zebra fish CFTR protein and human TAP66,67. Given the lower resolution of the density covering the NBDs, we generated an NBD model based on the crystal structure of ABCG5/G8, which shares ~40% sequence identity in the NBD region. Upon docking this model into the density, we could adjust the position of the secondary structure elements by fitting them into the density. We used the local-resolution filtered map and the original non-post-processed map for very conservative adjustment of the model, guided by the structures of ABCG5 and ABCG8 (chains A and B, respectively, of PDB accession number 5DO7), the HHpred models using these structures as a template, as well as HHpred models based on the structures of Thermotoga maritima TM_0544 (PDB accession number 1VPL), Alicyclobacillus acidocaldarius CysA68 (chain A of PDB accession number 1Z47), T. maritima TM_1403 (chain A of PDB accession number 4YER), Geobacillus stearothermophilus ArtP in complex with ADP/Mg2+ (chain A of PDB accession number 2OLJ), and Thermoanaerobacter tengcongensis CbiO69 (chain B of PDB accession number 4MKI). We then placed two cholesterol molecules in cavity 1 of the ABCG2 TMD in the orientation that resulted in the best visual fit to the map. Finally, the complete ABCG2–5D3(Fab) atomic model was refined against the local-resolution-filtered map (using a resolution limit of 3.8 Å) with phenix.real_space_refine70. For the final round of model refinement, we performed global real-space refinement with standard geometry restraints as well as rotamer, Ramachandran plot, C-beta, non-crystallographic symmetry, and secondary structure restraints, coupled to reciprocal-space refinement of the B-factors. Refinement was performed including all side chains and with both cholesterol molecules. Refinement statistics of that model are given in Extended Data Table 1.
For validation of the refinement, random shifts (up to 0.3 Å) were introduced into the coordinates of the final refined model, followed by refinement with phenix.real_space_refine (using the same parameters as described before) against the first unfiltered half-map (half-map 1). The small ‘gap’ between the FSC curve of the model with random displacements refined against half-map 1 versus half-map 1 and the FSC curve of the same model versus half-map 2 (against which it was not refined) indicated no over-refinement took place (Extended Data Fig. 2g)71,72.
Orientation determination of yellow fluorescent protein–3C–ABCG2 in proteoliposomes
ABCG2 was N-terminally tagged with yellow fluorescent protein (YFP) and 3C protease (YFP–3C–ABCG2), and was expressed, purified, and reconstituted into liposomes as described above. The sample was divided into two (disrupted and non-disrupted). Triton X-100 (1% v/v) was added to the ‘disrupted’ sample while the equivalent volume of transport buffer was added to the ‘non-disrupted’ sample, and both were incubated for 30 min at room temperature. 3C was added to both samples to cleave the YFP and the samples were centrifuged at 100,000g. Supernatants were collected and analysed by reducing SDS–PAGE in-gel fluorescence using ImageJ.
Pull-down assays of human and rat ABCG2 using 5D3-Fab
Expression and solubilization of YFP-tagged human ABCG2 and of YFP-tagged rat ABCG2 was performed as described above but without kifunensine treatment. After ultracentrifugation, the supernatants were incubated with 5D3-monoclonal antibody-coupled sepharose resin. The resin was washed extensively with purification buffer and analysed by reducing SDS–PAGE in-gel fluorescence.
Expression, purification, crystallization, and structure determination of the 5D3-Fab
The 5D3 hybridoma cells, producing the 5D3 monoclonal antibody, were obtained from B. Sorrentino16 and cultured in WHEATON CELLine Bioreactors according to the manufacturer’s protocol. 5D3-Fab was purified from the supernatant as described in the Fab Preparation Kit protocol (Thermo Fisher Scientific). The purity and yield of 5D3-Fab were evaluated by reducing and non-reducing SDS–PAGE, and the purified protein was crystallized in 100 mM Tris pH 8.5 and 16% PEG 6000 at 20 °C. A single 5D3-Fab crystal was mounted at 100 K on the goniometer of beamline PXI at the Swiss Light Source and a complete data set was collected to a maximum resolution of 1.5 Å (Extended Data Table 2). The 5D3-Fab X-ray structure was determined by molecular replacement in Phaser73 using the Fab of PDB accession number 4CAD as the search model74. Iterative model building was performed in Coot75 and the X-ray structure was refined in PHENIX70.
Figure preparation
Figures were prepared using the programs PyMOL (The PyMOL Molecular Graphics System, DeLano Scientific) and Chimera76.
Data availability
Atomic coordinates of the ABCG2–5D3(Fab) model after truncating the side chains of the NBD and of the 5D3-Fab constant domain to alanine and removing the two cholesterol molecules have been deposited in the PDB under accession number 5NJ3. The de novo built structure of the ABCG2 TMD in complex with 5D3-Fab variable domain was separately deposited in the Protein Data Bank (PDB) under accession number 5NJG. Atomic coordinates of the X-ray structure of 5D3-Fab have been deposited in the PDB under accession number 5NIV, and the three-dimensional cryo-EM density post-processed, masked map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession number EMD-3654. All other data are available from the corresponding authors upon reasonable request.
References
Moitra, K. & Dean, M. Evolution of ABC transporters by gene duplication and their role in human disease. Biol. Chem. 392, 29–37 (2011)
Dean, M., Rzhetsky, A. & Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11, 1156–1166 (2001)
Cuperus, F. J., Claudel, T., Gautherot, J., Halilbasic, E. & Trauner, M. The role of canalicular ABC transporters in cholestasis. Drug Metab. Dispos. 42, 546–560 (2014)
Fetsch, P. A. et al. Localization of the ABCG2 mitoxantrone resistance-associated protein in normal tissues. Cancer Lett. 235, 84–92 (2006)
Diestra, J. E. et al. Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J. Pathol. 198, 213–219 (2002)
Robey, R. W. et al. ABCG2: a perspective. Adv. Drug Deliv. Rev. 61, 3–13 (2009)
Ishikawa, T ., Aw, W. & Kaneko, K. Metabolic interactions of purine derivatives with human ABC transporter ABCG2: genetic testing to assess gout risk. Pharmaceuticals (Basel) 6, 1347–1360 (2013)
Hillgren, K. M. et al. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin. Pharmacol. Ther. 94, 52–63 (2013)
Sarkadi, B., Homolya, L., Szakács, G. & Váradi, A. Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol. Rev. 86, 1179–1236 (2006)
Gottesman, M. M., Fojo, T. & Bates, S. E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58 (2002)
Sharom, F. J. The P-glycoprotein multidrug transporter. Essays Biochem. 50, 161–178 (2011)
Gillet, J. P. & Gottesman, M. M. Advances in the molecular detection of ABC transporters involved in multidrug resistance in cancer. Curr. Pharm. Biotechnol. 12, 686–692 (2011)
Mo, W. & Zhang, J. T. Human ABCG2: structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 3, 1–27 (2012)
Robey, R. W., Ierano, C., Zhan, Z. & Bates, S. E. The challenge of exploiting ABCG2 in the clinic. Curr. Pharm. Biotechnol. 12, 595–608 (2011)
Tamaki, A., Ierano, C., Szakács, G., Robey, R. W. & Bates, S. E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 50, 209–232 (2011)
Zhou, S. et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 7, 1028–1034 (2001)
Robey, R. W., Massey, P. R., Amiri-Kordestani, L. & Bates, S. E. ABC transporters: unvalidated therapeutic targets in cancer and the CNS. Anticancer. Agents Med. Chem. 10, 625–633 (2010)
Lee, J. Y. et al. Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature 533, 561–564 (2016)
Rosenberg, M. F. et al. Three-dimensional structure of the human breast cancer resistance protein (BCRP/ABCG2) in an inward-facing conformation. Acta Crystallogr. D 71, 1725–1735 (2015)
László, L., Sarkadi, B. & Hegedüs, T. Jump into a new fold-a homology based model for the ABCG2/BCRP multidrug transporter. PLoS ONE 11, e0164426 (2016)
Suzuki, M., Suzuki, H., Sugimoto, Y. & Sugiyama, Y. ABCG2 transports sulfated conjugates of steroids and xenobiotics. J. Biol. Chem. 278, 22644–22649 (2003)
Karlsson, J. E. et al. High-activity P-glycoprotein, multidrug resistance protein 2, and breast cancer resistance protein membrane vesicles prepared from transiently transfected human embryonic kidney 293-epstein-barr virus nuclear antigen cells. Drug Metab. Dispos. 38, 705–714 (2010)
Allen, J. D. et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 1, 417–425 (2002)
Ritchie, T. K. et al. Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 (2009)
Nath, A., Atkins, W. M. & Sligar, S. G. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry 46, 2059–2069 (2007)
Dawson, R. J. & Locher, K. P. Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 (2006)
Aller, S. G. et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 (2009)
Li, J., Jaimes, K. F. & Aller, S. G. Refined structures of mouse P-glycoprotein. Protein Sci. 23, 34–46 (2014)
Locher, K. P., Lee, A. T. & Rees, D. C. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science 296, 1091–1098 (2002)
Woo, J. S., Zeltina, A., Goetz, B. A. & Locher, K. P. X-ray structure of the Yersinia pestis heme transporter HmuUV. Nat. Struct. Mol. Biol. 19, 1310–1315 (2012)
Polgar, O. et al. Mutational analysis of ABCG2: role of the GXXXG motif. Biochemistry 43, 9448–9456 (2004)
Frank, G. A. et al. Cryo-EM Analysis of the conformational landscape of human p-glycoprotein (ABCB1) during its catalytic cycle. Mol. Pharmacol. 90, 35–41 (2016)
Shintre, C. A. et al. Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl Acad. Sci. USA 110, 9710–9715 (2013)
Perez, C. et al. Structure and mechanism of an active lipid-linked oligosaccharide flippase. Nature 524, 433–438 (2015)
Macalou, S. et al. The linker region of breast cancer resistance protein ABCG2 is critical for coupling of ATP-dependent drug transport. Cell. Mol. Life Sci. 73, 1927–1937 (2016)
Henriksen, U., Fog, J. U., Litman, T. & Gether, U. Identification of intra- and intermolecular disulfide bridges in the multidrug resistance transporter ABCG2. J. Biol. Chem. 280, 36926–36934 (2005)
Wakabayashi, K. et al. Identification of cysteine residues critically involved in homodimer formation and protein expression of human ATP-binding cassette transporter ABCG2: a new approach using the flp recombinase system. J. Exp. Ther. Oncol. 5, 205–222 (2006)
Kage, K., Fujita, T. & Sugimoto, Y. Role of Cys-603 in dimer/oligomer formation of the breast cancer resistance protein BCRP/ABCG2. Cancer Sci. 96, 866–872 (2005)
Nakagawa, H. et al. Disruption of N-linked glycosylation enhances ubiquitin-mediated proteasomal degradation of the human ATP-binding cassette transporter ABCG2. FEBS J. 276, 7237–7252 (2009)
Özvegy-Laczka, C. et al. Function-dependent conformational changes of the ABCG2 multidrug transporter modify its interaction with a monoclonal antibody on the cell surface. J. Biol. Chem. 280, 4219–4227 (2005)
Telbisz, Á., Hegedüs, C., Váradi, A., Sarkadi, B. & Özvegy-Laczka, C. Regulation of the function of the human ABCG2 multidrug transporter by cholesterol and bile acids: effects of mutations in potential substrate and steroid binding sites. Drug Metab. Dispos. 42, 575–585 (2014)
Tarling, E. J. & Edwards, P. A. ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc. Natl Acad. Sci. USA 108, 19719–19724 (2011)
Tarr, P. T. & Edwards, P. A. ABCG1 and ABCG4 are coexpressed in neurons and astrocytes of the CNS and regulate cholesterol homeostasis through SREBP-2. J. Lipid Res. 49, 169–182 (2008)
Basseville, A. & Bates, S. E. Gout, genetics and ABC transporters. F1000 Biol. Rep. 3, 23 (2011)
Furukawa, T. et al. Major SNP (Q141K) variant of human ABC transporter ABCG2 undergoes lysosomal and proteasomal degradations. Pharm. Res. 26, 469–479 (2009)
Honjo, Y. et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 61, 6635–6639 (2001)
Ejendal, K. F., Diop, N. K., Schweiger, L. C. & Hrycyna, C. A. The nature of amino acid 482 of human ABCG2 affects substrate transport and ATP hydrolysis but not substrate binding. Protein Sci. 15, 1597–1607 (2006)
Locher, K. P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 23, 487–493 (2016)
Geisse, S., Jordan, M. & Wurm, F. M. Large-scale transient expression of therapeutic proteins in mammalian cells. Methods Mol. Biol. 308, 87–98 (2005)
Geertsma, E. R., Nik Mahmood, N. A., Schuurman-Wolters, G. K. & Poolman, B. Membrane reconstitution of ABC transporters and assays of translocator function. Nat. Protocols 3, 256–266 (2008)
Schaffner, W. & Weissmann, C. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal. Biochem. 56, 502–514 (1973)
Chifflet, S., Torriglia, A., Chiesa, R. & Tolosa, S. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application to lens ATPases. Anal. Biochem. 168, 1–4 (1988)
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005)
Li, X. et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013)
Biyani, N. et al. Focus: the interface between data collection and data processing in cryo-EM. J. Struct. Biol. 198, 124–133 (2017)
Scheres, S. H. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415, 406–418 (2012)
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012)
Kimanius, D., Forsberg, B. O., Scheres, S. H. & Lindahl, E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5, e18722 (2016)
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016)
Urnavicius, L. et al. The structure of the dynactin complex and its interaction with dynein. Science 347, 1441–1446 (2015)
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003)
Scheres, S. H. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012)
Chen, S. et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013)
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010)
Webb, B. & Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 1137, 1–15 (2014)
Zhang, Z. & Chen, J. Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell 167, 1586–1597 (2016)
Oldham, M. L., Grigorieff, N. & Chen, J. Structure of the transporter associated with antigen processing trapped by herpes simplex virus. eLife 5, e21829 (2016)
Scheffel, F. et al. Structure of the ATPase subunit CysA of the putative sulfate ATP-binding cassette (ABC) transporter from Alicyclobacillus acidocaldarius. FEBS Lett. 579, 2953–2958 (2005)
Chai, C. et al. Structural basis for a homodimeric ATPase subunit of an ECF transporter. Protein Cell 4, 793–801 (2013)
Adams, P. D . et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010)
Brown, A. et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr. D 71, 136–153 (2015)
Fischer, N. et al. The pathway to GTPase activation of elongation factor SelB on the ribosome. Nature 540, 80–85 (2016)
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007)
Manolaridis, I. et al. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature 504, 301–305 (2013)
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004)
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004)
Acknowledgements
This research was supported by the Swiss National Science Foundation through the National Centre of Competence in Research (NCCR) TransCure, and by a Swiss Federal Institute of Technology Zürich (ETH Zürich) research grant ETH-22-14-1. We thank the staff of the X06SA beamline of the Swiss Light Source for support during data collection. We thank K. Goldie, A. Fecteau-LeFebre, and R. McLeod for support during electron microscope use, and R. Adaixo, L. Muckenfuss, N. Biyani, and R. Righetto for support and discussions during EM data analysis. We thank L. Lancien and B. Prinz for help with protein expression and cell culture work. We thank B. Sorrentino for providing the 5D3-producing hybridoma cell line, B. Stieger for discussions about transport assays, and J.-I. Kim for initial ABCG2 expression and purification experiments.
Author information
Authors and Affiliations
Contributions
I.M. expressed human and rat ABCG2. I.M. and S.M.J. purified and reconstituted ABCG2 in liposomes and nanodiscs. S.M.J. performed functional assays. J.K. performed initial negative-stain and cryo-EM analyses and prepared all grids. N.M.I.T. and H.S. performed cryo-EM data collection. N.M.I.T. performed ABCG2 structure determination and built the ABCG2 model. I.M., K.P.L. and N.M.I.T. revised the model, and N.M.I.T. performed model refinement. I.M. determined the 5D3-Fab crystal structure. K.P.L., I.M., S.M.J., and H.S. conceived the project and planned the experiments. I.M., S.M.J. and K.P.L. wrote the manuscript; all authors contributed to its revision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks A. Ward and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Functional studies of liposome-reconstituted ABCG2.
a, Transport curves showing E1S uptake by ABCG2 in the presence and absence of ATP. b, The rate of E1S transport was measured at different E1S concentrations and the Michaelis constant was determined. c, ATP-dependent transport curves showing E1S uptake in the presence and absence of Ko143 or 5D3-Fab. d, The 30 s to 2 min linear portion of the curves from c used to determine the initial rates of E1S transport. e, Representative SDS–PAGE to determine the orientation of liposome-reconstituted YFP–3C–ABCG2 using in-gel fluorescence, comparing the intensity of free YFP after the addition of 3C protease in disrupted and non-disrupted samples; 54 ± 2% of ABCG2 was oriented with the NBDs on the outside of the proteoliposomes and all assays were subsequently corrected for this value; error, s.d. (n = 3). Non-disrupted supernatant (+3C) (lane 1); disrupted supernatant (+3C) (lane 2); non-disrupted supernatant (−3C) (lane 3); disrupted supernatant (−3C) (lane 4). f, The basal ATPase activity of nanodisc-reconstituted ABCG2 is equivalent to the E1S-stimulated activity of liposome-reconstituted ABCG2 and cannot be stimulated further. ABCG2 is not responsive to cholate in the presence or absence of E1S. The curves have been normalized to the basal ATPase activity of liposome-reconstituted ABCG2. In a–d and f, two separate experiments were performed in triplicate (error bars, s.d.).
Extended Data Figure 2 Cryo-EM map generation and atomic model refinement.
a, An example micrograph (drift-corrected, dose-weighted, and low-pass filtered to 20 Å) of the nanodisc-reconstituted ABCG2–5D3(Fab) data set. White scale bar, 1,000 Å. b, Averages of the 28 most prevalent two-dimensional class averages of the final round of two-dimensional classification, sorted in decreasing order by the number of particles assigned to each class. c, Map obtained from the RELION three-dimensional auto-refine procedure shown at a lower (white, transparent) and higher (blue, non-transparent) threshold. Density corresponding to the nanodisc is indicated with an arrow. d, Same as c but rotated. e, Angular distribution plot for the final reconstruction. The refinement map is shown at two different thresholds as in c, in the same orientation. f, FSC from the RELION auto-refine procedure of the unmasked half-maps (yellow), the random-phase corrected half-maps (green), the half-maps after masking (blue), and the half-maps after masking and correction for the influence of the mask (red). A horizontal line (black) is drawn for the FSC = 0.143 criterion. For both the unmasked and the corrected FSC curves, their intersection with the FSC = 0.143 line is indicated by an arrow, and the resolution at this point is indicated. g, FSC of the final model versus the map calculated with all data after local-resolution filtering, against which it was refined, (dark blue) and of the final model with introduced shifts (up to 0.3 Å) refined against the first unfiltered half-map (half-map 1) versus the same unfiltered half-map (violet) and versus the other unfiltered half-map (half-map 2) against which it was not refined (grey).
Extended Data Figure 3 Local resolution.
a, Full view of the RELION local-resolution-filtered map coloured by local resolution as calculated by ResMap. b, Same as a but only showing the posterior half (front clipping plane in the middle of the molecule). c, Same as a but only showing a region up to 3 Å around one chain of ABCG2 (including the putative cholesterol molecule and side chains of the NBD). d, Same as c but rotated. e, Same as a but only showing a region up to 3 Å around one 5D3-Fab. f, Same as e but rotated.
Extended Data Figure 4 Fit of the model to the density.
a, Fit of fragments of the final model of the ABCG2 TMD to the post-processed and masked map from RELION. A region of up to 2 Å around the atoms is shown. The residue numbers are indicated, as are helices that are contained in these fragments and the EL3. b, Same as a but showing regions of 5D3-Fab. Residue numbers and whether the fragment belongs to the heavy or the light chain is indicated. c, Same as b but showing regions of the 5D3-Fab constant domain. d, Fit of the ABCG2 NBD model to the local-resolution-filtered map. A region of up to 3 Å around one ABCG2 NBD (including side chains) is shown. e, Same as a but showing the cholesterol (CHL) molecule. f, Fit of fragments of ABCG2 TM2, TM5a, and cholesterol to the post-processed and masked map from RELION. A region of up to 2 Å around the atoms is shown. g, Same as f but rotated by 180°.
Extended Data Figure 5 The X-ray structure and electrostatic surface potential of 5D3-Fab.
a, Cartoon representation of the X-ray structure of 5D3-Fab. The light and heavy chain CDR1s are coloured red, CDR2s are blue and CDR3s are purple. b, The molecular surface of 5D3-Fab, colour-coded by electrostatic potential ranging from blue (most positive) to red (most negative) to white (uncharged).
Extended Data Figure 6 Electrostatic surface potential of ABCG2 and comparison of ABCG2 and ABCG5/G8.
a, The molecular surface of ABCG2, colour-coded by electrostatic potential ranging from blue (most positive) to red (most negative) to white (uncharged). The lipid membrane (40 Å) is indicated by the black lines on the basis of the distribution of non-polar residues and transmembrane helices, and the 5D3-Fab binding surface is boxed. b, Overlay of the ABCG2 and ABCG5/G8 TMDs with EL3 boxed. Panels a and b are coloured as in Fig. 2a and ABCG5/G8 is coloured green. c, EL3 sequence alignment of the G-family of ABC transporters with the cysteines involved in disulfide bond formation indicated by red asterisks and with EL3 boxed.
Extended Data Figure 7 Purification of human ABCG2 and pull-down assays of human and rat ABCG2 using the 5D3 monoclonal antibody.
a, Representative SDS–PAGE of purified ABCG2. The same sample was applied after (lane 1) or before (lane 2) disulfide reduction using DTT, demonstrating disulfide-dependent dimerization of ABCG2 (black arrows). M indicates marker proteins, with selected masses indicated on the left. b, Pull-down assay showing that the sepharose-immobilized 5D3-monoclonal antibody does not bind to rat YFP–ABCG2. Human YFP–ABCG2 cell lysate (lane 1), flow-through (lane 2), and elution (lane 3). Rat YFP–ABCG2 cell lysate (lane 4), flow-through (lane 5), and elution (lane 6).
Rights and permissions
About this article

Cite this article
Taylor, N., Manolaridis, I., Jackson, S. et al. Structure of the human multidrug transporter ABCG2. Nature 546, 504–509 (2017). https://doi.org/10.1038/nature22345
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature22345
- Springer Nature Limited
This article is cited by
-
Impacts of ABCG2 loss of function variant (p. Gln141Lys, c.421 C > A, rs2231142) on lipid levels and statin efficiency: a systematic review and meta-analysis
BMC Cardiovascular Disorders (2024)
-
Inhibition and transport mechanisms of the ABC transporter hMRP5
Nature Communications (2024)
-
Cadmium transport by mammalian ATP-binding cassette transporters
BioMetals (2024)
-
The net electrostatic potential and hydration of ABCG2 affect substrate transport
Nature Communications (2023)
-
Advances in the structure, mechanism and targeting of chemoresistance-linked ABC transporters
Nature Reviews Cancer (2023)