

Abstract
Haematopoietic stem cells (HSCs) are arguably the most extensively characterized tissue stem cells. Since the identification of HSCs by prospective isolation1, complex multi-parameter flow cytometric isolation of phenotypic subsets has facilitated studies on many aspects of HSC biology, including self-renewal2,3,4, differentiation, ageing, niche5, and diversity6,7,8. Here we demonstrate by unbiased multi-step screening, identification of a single gene, homeobox B5 (Hoxb5, also known as Hox-2.1), with expression in the bone marrow that is limited to long-term (LT)-HSCs in mice. Using a mouse single-colour tri-mCherry reporter driven by endogenous Hoxb5 regulation, we show that only the Hoxb5+ HSCs exhibit long-term reconstitution capacity after transplantation in primary transplant recipients and, notably, in secondary recipients. Only 7–35% of various previously defined immunophenotypic HSCs are LT-HSCs. Finally, by in situ imaging of mouse bone marrow, we show that >94% of LT-HSCs (Hoxb5+) are directly attached to VE-cadherin+ cells, implicating the perivascular space as a near-homogenous location of LT-HSCs.
Similar content being viewed by others


Main
Prospective isolation of HSCs requires that the isolated cells are capable of long-term production of all blood cell types in primary irradiated hosts, as well as self-renewal, such that the cells can be transplanted to secondary hosts to give rise to long-term multilineage repopulation. From the first enrichment and isolation of candidate HSCs1,9,10, this activity has been entirely contained in cell-surface-marker-defined cell populations, and more recently in fluorescent reporters11,12,13. However, the precise fraction of cells in those populations that are true LT-HSCs remains unknown.
To enable further purification of LT-HSCs, we sought to identify genes expressed exclusively in HSCs within cell populations resident in mouse bone marrow, detectable by flow cytometry and in situ fluorescence, and thus performed the following four-step screening (Fig. 1d).

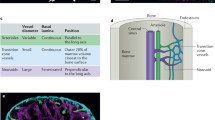
a, Microarray heat map depicting relative expression (pink, high; blue, low) of HSC-specific genes in haematopoietic and stromal populations. Each row represents a gene; each subcolumn a replicate microarray; each labelled column a category of cell populations. The 45 genes in the top panel displayed limited activity in all non-HSC populations examined. b, Transcriptional profiling by RNA-seq of the 45 genes from a. Three genes (top panel) exceeded the estimated threshold for detection (FPKM > 7.0) in HSCs while showing minimal expression (FPKM < 2.5) in MPPa and MPPb populations. c, Heterogeneity of expression for the three remaining candidate genes in HSCs as assessed by single-cell qPCR. d, Venn diagram reflecting the four-step screen that identified Hoxb5 as an ideal candidate in the HSC transcriptome. e, Targeting strategy to generate a triple-mCherry Hoxb5 knock-in mouse reporter line (Hoxb5–tri-mCherry). UTR, untranslated region; PGK, phosphoglycerate kinase I. f, Hoxb5 reporter expression (red) in pHSCs and MPPs compared to wild-type controls (blue). Values indicate the percentage of mCherry+ cells ± s.d. in each fraction for n = 3 mice.
First, we compared microarray gene expression assays among 28 distinct populations of the haematopoietic system (Extended Data Fig. 1a and Supplementary Table 1). Using the Gene Expression Commons platform14, we identified 118 candidate HSC-specific genes (Fig. 1a and Supplementary Table 2). Surprisingly, this list did not include all previously reported HSC-specific markers11,12,13 (Extended Data Fig. 1b and Supplementary Table 2). Second, to identify HSCs in situ, we excluded candidates that also label non-haematopoietic cells present in the bone marrow such as stromal and endothelial cells15,16. Consequently, we excluded genes expressed in eight distinct non-haematopoietic bone marrow populations, thereby narrowing the list to 45 candidate genes (Fig. 1a).
Next, to ensure that the expression of any candidates could be detected by both flow cytometry and in situ fluorescence, we used RNA-sequencing (RNA-seq) combined with a threshold gene standard to estimate the fragments per kilobase of transcript per million mapped reads (FPKM) value that could serve as a detection threshold. From the bone marrow of 12-week-old mice, we sorted and RNA-sequenced immunophenotypically defined (Lin−c-Kit+Sca-1+CD150+CD34−/loFlk2−) HSCs (hereafter referred to as pHSCs), multipotent progenitors subset A (MPPa; Lin−c-Kit+Sca-1+CD150+CD34+Flk2−), and multipotent progenitors subset B (MPPb; Lin−c-Kit+Sca-1+CD150−CD34+Flk2−) (Fig. 1b) to determine the FPKM value of candidate genes. On the basis of Bmi-1–eGFP knock-in reporter expression17, we found that a single copy of eGFP is detectable at an estimated FPKM value of ~20. However, this high threshold would have excluded all candidate genes. Therefore, we designed a targeting construct (Fig. 1e) with three copies of mCherry, bringing the theoretical detection limit to ~7 FPKM. Lastly, to minimize aberrant detection, we set threshold FPKM values for both the MPPa and MPPb fractions to 2.5. Only three genes, Hoxb5, Rnf208, and Smtnl1, met these criteria (Fig. 1b).
Given previous reports of heterogeneity within pHSCs7,18,19,20, we analysed single cells to determine whether the remaining candidate genes were heterogenously expressed among pHSCs. We reasoned that an ideal pan-HSC candidate gene would label the majority of pHSCs, with quantitative differences potentially reflecting HSC heterogeneity/diversity. We thus performed single-cell quantitative PCR (qPCR) analysis of pHSCs, and evaluated expression of Hoxb5, Rnf208, and Smtnl1. Only Hoxb5 exhibited bimodal expression, in comparison to the unimodality of Rnf208 and Smtnl1 (Fig. 1c). Therefore, from the entire HSC transcriptome, only Hoxb5 satisfied the criteria of our extensive unbiased screening (Fig. 1d).
We next sought to generate a Hoxb5 reporter with minimal disruption of endogenous Hoxb5 function. Thus we designed our targeting construct and CRISPR guide sequences to facilitate an in-frame knock-in to the endogenous Hoxb5 gene locus immediately 5′ of the only endogenous stop codon. We used three tandem mCherry cassettes separated by porcine teschovirus-1 2A (P2A) sequences, with the terminal mCherry carrying a CAAX membrane localization sequence (Hoxb5–tri-mCherry) (Fig. 1e).
To evaluate the specificity of this reporter, we isolated whole bone marrow cells from 12-week-old reporter mice and measured mCherry+ cells in the following immunophenotypic populations: pHSC, MPPa, MPPb, Flk2+ multipotent progenitor, megakaryocyte erythrocyte progenitor, granulocyte monocyte progenitor, common myeloid progenitor, common lymphoid progenitor fractions; differentiated cell populations (B cell, T cell, natural killer (NK) cell, neutrophil, eosinophil, monocyte, macrophage, dendritic cell, red blood cell, and megakaryocyte); and in CD45− stromal fractions (Fig. 1f, Extended Data Fig. 2a, b, Extended Data Fig. 3, and unpublished data). Consistent with our initial screen (Fig. 1a–d), and using wild-type mice as a fluorescence minus one (FMO) threshold21, mCherry-labelled cells were highly enriched in the pHSC fraction (21.8% ± 0.90%), had a low frequency in the MPPa fraction (1.88% ± 0.64%), and background frequencies in the remaining fractions (Fig. 1f, Extended Data Figs 2b and 3 and unpublished data). Interestingly, as only a minority of pHSCs were mCherry+, this suggested that either our reporter labelled only a subfraction of HSCs or only a subfraction of pHSCs were indeed HSCs.
To distinguish between these two possibilities and to determine whether Hoxb5 is a reporter of LT-HSCs, we characterized Hoxb5-expressing cells by transplantation. In order to be inclusive of all events in the pHSC gate, we used wild-type FMO to define Hoxb5 negativity (Hoxb5neg or sometimes referred to as Hox5−) and divided the positive fraction (sometimes referred to as Hox5+) into Hoxb5hi (top 5th percentile) and Hoxb5lo (14.1 ± 7.7%) (Extended Data Fig. 4). Ten-cell and three-cell grafts of Hoxb5hi, Hoxb5lo, or Hoxb5neg pHSCs were transplanted with supporting bone marrow cells into irradiated mouse recipients. We used CD45.2 expression to assess donor HSC contribution to haematopoietic lineages at 4-week intervals (Fig. 2a). Analysis of peripheral blood 16 weeks after transplantation of ten cells demonstrated that multilineage reconstitution was present in 78% of Hoxb5hi, 70% of Hoxb5lo, and 44% Hoxb5neg pHSC recipients (Fig. 2b and Extended Data Fig. 5a). Three-cell transplants exhibited similar kinetics (Extended Data Fig. 5c). Notably, the chimaerism of the Hoxb5neg pHSCs decreased over time, in particular between 4 to 8 weeks and shifted towards a predominantly lymphoid chimaerism (Fig. 2b, d and Extended Data Fig. 5a), suggesting that either the Hoxb5neg fraction comprised of lymphoid-biased HSCs or, more likely, transiently self-renewing short-term (ST)-HSCs and/or MPPs that had given rise to long-lived lymphocytes.

a, Experimental schematic for long-term haematopoietic reconstitution assays. CD45.1+ recipient mice were lethally irradiated and competitively transplanted with ten or three Hoxb5–tri-mCherry HSCs and 2 × 105 CD45.1+/CD45.2+ supporting cells. For secondary transplants, 1 × 107 whole bone marrow (WBM) cells or 100 sorted LSK cells were transferred from primary recipient mice. PB, peripheral blood. b, Percentage chimaerism at 16 weeks in primary recipients receiving ten Hoxb5neg (n = 9 mice), Hoxb5lo (n = 13 mice), or Hoxb5hi (n = 18 mice) pHSCs. Each column represents an individual mouse. c, Percentage chimaerism at 16 weeks following whole bone marrow secondary transplant. d, Average donor lineage contribution in ten-cell primary transplants. Error bars denote s.d. e, Individual donor chimaerism by lineage in whole bone marrow secondary recipients. Each line represents an individual mouse (n = 6 mice for Hoxb5neg; n = 5 mice for Hoxb5lo; and n = 8 mice for Hoxb5hi).
To evaluate these two possibilities, we carried out a secondary transplantation of whole bone marrow from primary Hoxb5hi, Hoxb5lo, or Hoxb5neg pHSC recipients into lethally irradiated secondary recipients (Fig. 2a). Sixteen weeks after secondary transplantation, peripheral blood analyses revealed robust multilineage chimaerism from all Hoxb5hi and Hoxb5lo transplant recipients, with minimal chimaerism from the Hoxb5neg fraction (Fig. 2c, e and Extended Data Fig. 5b). Furthermore, bone marrow analysis of primary recipients for donor pHSCs revealed that the Hoxb5hi pHSC recipients contained Hoxb5hi, Hoxb5lo, and Hoxb5neg cells (100%, n = 10 mice), whereas the recipients of Hoxb5neg cells were only Hoxb5neg (36%, n = 4 mice) or devoid of donor cells (64%, n = 7 mice). These results further suggested that the Hoxb5neg pHSCs were in fact transiently self-renewing ST-HSCs/MPPs. To determine if Hoxb5 expression could also distinguish LT-HSCs in a primary transplant, we normalized the number of donor cells used for secondary transplant by sorting 100 Lin-c-Kit+Sca-1+ (LSK) donor cells from the primary Hoxb5hi recipients and transplanted them into irradiated secondary recipients (n = 24 mice) (Fig. 2a). As with the primary bone marrow transplantations, the chimaerism was minimal in Hoxb5− compared to Hoxb5+ recipients (Extended Data Fig. 5d). Limiting dilution analysis revealed that the frequency of long-term plus short-term HSCs in primary hosts at 16 weeks was 1 in 2.1 for Hoxb5hi, 1 in 2.4 for Hoxb5lo, and 1 in 16.1 for Hoxb5neg recipients (Extended Data Fig. 6). Taken together, these results demonstrate that Hoxb5 labels functional LT-HSCs.
Given that Hoxb5− cells are non-LT-HSCs, we re-examined the specificity of past definitions (Fig. 3a, f), including previously reported refinements to the LT-HSC immunophenotype18,19,20 and the most widely used in situ definition over the past decade22. We found that 78.5% ± 2.6% of CD11a− HSCs19, 63.9% ± 3% of the HSC-1 (Lin−c-Kit+Sca-1+CD48−CD150+CD229−CD244−)20, and 82.5% ± 0.4% of fraction I HSCs (Lin−c-Kit+Sca-1+ CD34−/loCD150+CD41−)18 were Hoxb5– (Fig. 3b, c, and d, respectively). Surprisingly, 91.3% ± 0.4% of Lin−CD48−CD41−CD150+ (ref. 22) cells were Hoxb5− (Fig. 3e). As this subset was initially used for localization of HSCs in situ22, we re-examined the in situ location of HSCs using Hoxb5 expression.

Flow cytometry plots of bone marrow from 12-week-old mice depicting Hoxb5–tri-mCherry reporter activity in previously reported HSC immunophenotypes. a, pHSC (LSK CD150+CD34−/loFlk2−) Hoxb5+ (red) and Hoxb5− (blue). b, CD11a− (LSK CD150+CD34−/loCD11a−)19. c, HSC-1 (LSK CD150+CD48−/loCD229−/loCD244−)20. d, Fraction I (LSK CD150+CD34−/loCD41−)18. e, CD150+CD48−CD41− cells, currently used to identify HSCs in situ22. Wild-type FMO used to define Hoxb5 negativity for each panel. f, Summary percentage of Hoxb5+ and Hoxb5− negative cells in characterized HSC subfractions. Error bars denote s.d.; n = 5 mice (see Extended Data Fig. 7a–d for gating scheme).
Visualizing LT-HSCs in bone marrow and identifying the cellular constituents and structures of the HSC niche remains challenging. Despite this, multiple constituent cell types have been proposed, including mesenchymal stromata, endosteal osteoblasts, glia, endothelia, and pericytes5. In situ studies are made difficult by several technical limitations, including the number of fluorescent colours, difficulty in identifying HSCs surrounded by non-HSCs, and difficulty in translating the same fluorescently defined positive and negative thresholds from flow cytometry to tissue sections.
To address these issues, we used the Hoxb5–tri-mCherry reporter. Using flow cytometry, we determined the utility of Hoxb5 alone in identifying LT-HSCs. After logical exclusion of autofluorescence by comparison to wild-type control mice, we found that all Hoxb5+ events are within the c-Kit+ fraction and 62.0 ± 12.8% of all Hoxb5+ events are located in the pHSC gate (Extended Data Fig. 8), representing an eight- to nine-fold enrichment compared to previous in situ labelling of HSCs (Lin−CD48−CD41−CD150+)22 (Fig. 3e).
To reveal the three-dimensional (3D) HSC niche architecture, we applied the CUBIC technique (clear, unobstructed brain imaging cocktails and computational analysis23) to tibial bone marrow, facilitating depletion of autofluorescent cellular components (Fig. 4a). Given that fluorescence intensity is preserved by CUBIC23, the specificity of LT-HSCs in situ correlates with Hoxb5 intensity determined by flow cytometry. Therefore, on the basis of the number of Hoxb5+ LT-HSCs detected in one tibia (Extended Data Fig. 9a, b), we objectively gated the same number of Hoxb5+ cells by intensity as in situ Hoxb5+ cells. We observed a uniform distribution of in situ Hoxb5+ cells along the longitudinal axis of the tibia (Fig. 4e and Extended Data Fig. 10a–c). The average percentage of in situ Hoxb5+ cells was 30.8 ± 5.9 in the proximal epiphysis, 39.0 ± 2.4 in the diaphysis, and 30.1 ± 5.4 in the distal epiphysis per field (1.25 × 1.25 × 0.4 mm3) (Fig. 4e).

a, Tissue preparation and representative images of tibial bone marrow plug after paraformaldehyde fixation (day 0) and treatment with reagent-1 (see ref. 23) (day 7, day 14). b, Localization of Hoxb5+ cells (red and arrows) and VE-cadherin+ cells (green) in 3D-reconstructed images. Scale bar, 30 μm. c, Representative 2D images of direct (top panel) and non-direct (bottom panel) association of Hoxb5+ cells (red) with VE-cadherin+ cells (green). Scale bar, 10 μm. d, Frequency of Hoxb5+ cells (n = 287 cells, from n = 3 mice) and random spots (n = 600 spots, from n = 3 mice) plotted against proximity to VE-cadherin+ cells. ***P < 0.0001. e, Average number of Hoxb5+ cells in proximal, medial, and distal regions of tibia (n = 3 mice). NS, not significant. Unpaired Student’s t-test (d, e).
To investigate the association of LT-HSCs with vasculature, we injected anti-mouse VE-cadherin antibody into reporter mice and analysed the bone marrow with CUBIC23. We found that 94.1 ± 1.9% of in situ Hoxb5+ cells were directly attached to the abluminal surface of VE-cadherin+ endothelial cells. In contrast, only 52.8 ± 2.3% of the random (Hoxb5−) spots were directly associated with VE-cadherin+ cells. This implied a near-homogenous perivascular location for the LT-HSC niche (Fig. 4b–d).
Following the first successful enrichment of HSCs in 1988 (ref. 1), many groups have attempted to identify surface markers to isolate LT-HSCs18,19,20,22,24,25. Identification of CD150, CD34, and CD48 enabled isolation of LT/ST-HSCs from MPPs. However, complete separation of LT-HSCs from ST-HSCs has never been fully accomplished. Our study demonstrates that Hoxb5 expression is specifically limited to LT-HSCs in adult mouse bone marrow. Limiting dilution assay shows that at least 1 in 2.1 of Hoxb5hi cells are LT-HSCs. However, this assay underestimates the functional potential of candidate HSCs. Transplantation, although the gold standard for prospective isolation of HSCs, may not take into account differences between sessile and mobile HSCs26,27, cell cycle status28,29, expression of CD47 (an anti-phagocytic molecule expressed highly on mobilized HSCs but at low levels on sessile bone marrow HSCs30), irradiation, and their influence on engraftment efficiency.
Our results demonstrate that the LT-HSCs compartment is near-homogenously perivascular. Although other compartments have been implicated5, including less homogenous association of candidate HSCs with the vasculature11, it is likely that our results differ owing to the fraction of assayed populations that are LT-HSCs (Extended Data Fig. 1b and Supplementary Table 3). These results suggest that the model of Hoxb5–tri-mCherry is the most specific available, to our knowledge, for identification of LT-HSCs, and could facilitate in situ lineage tracing to define their role in haematopoiesis in the absence of transplantation into irradiated hosts.
Methods
Data Reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to outcome assessment.
Mice
Eight-to-twelve-week-old C57BL/6J male mice (Jackson Laboratory) were used as wild-type controls. Eight-to-twelve-week-old male B6.SJL- Ptprca Pepcb/BoyJ mice (Jackson Laboratory) were used as recipients for transplantation assay. Supporting cells for competitive reconstitution assays were collected from B6.SJL-Ptprca Pepcb/BoyJ × C57BL/6J (F1 mice CD45.1+/CD45.2+). Mice were bred at our animal facility according to NIH guidelines. All animal protocols were approved by the Stanford University Administrative Panel on Laboratory Animal Care. Hoxb5–tri-mCherry (C57BL/6J background) mice were used as donor cells for transplantation as well as for analysis. Please see Gene targeting for mouse derivation.
Microarray data
All microarray data employed in this study are available at Gene Expression Commons (http://gexc.stanford.edu) and GEO GSE77078. Two to four microarray replicates were assessed for each distinct cell population14,15,16. The immunophenotype definition of each fraction is included in Supplementary Table 1.
RNA sequencing
RNA sequencing was performed as previously described31. In brief, total RNA was isolated with trizol, treated with RQ1 RNase free DNase (Promega) to remove minute quantities of genomic DNA if present, and cleaned up using RNeasy minelute columns (QIAGEN). cDNA libraries were prepared for pHSC, MPPa, and MPPb populations using Ovation RNA-Seq System V2 (NuGen) and sequenced separately using HiSeq 2500 (Illumina) to obtain 2 × 150 base pair (bp) paired-end reads. Raw transcriptome sequence data were mapped to Mus musculus reference mRNAs using OLego32 to produce a reference-guided transcript assembly. Four replicates were sequenced for each population. Data are accessible at NCBI SRP068593.
Single-cell qPCR
Single cells for qPCR were processed using the Single Cell-to-CT qRT–PCR Kit (Life Technologies) as per the manufacturer’s instructions. Cells were sorted directly into lysis solution in a 96-well plate, subjected to a reverse transcription reaction for cDNA synthesis, amplified for 14 cycles with pooled TaqMan Gene Expression Assays, and diluted with 1× TE buffer (pH 8.0). For real-time PCR, samples were amplified for 50 cycles of 3 s at 95 °C followed by 30 s at 60 °C using the following TaqMan probes: Mm00657672_m1 for Hoxb5; Mm03039759_s1 for Rnf208; Mm00470338_m1 for Smtnl1; and Mm99999915_g1 for Gapdh. All thermocycling was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems). Presence of a single cell was validated by a Ct value of 30 or less for Gapdh.
Gene targeting
The targeting construct was cloned into pJYC (derived from pUC19, golden gate insert of TALE backbone vector33, and removal of LacZ sequence). Right and left homology arms of ~700 bp were cloned by PCR from C57BL/6J genomic DNA, and the entire construct was sequence validated. BRUCE-4 embryonic stem (ES) cells (Millipore) derived from C57BL/6J mice were transfected with the targeting construct as well as bicistronic CRISPR vector PX330 expressing Cas9 and a Hoxb5-specific single guide RNA (5′-GGCUCCUCCGGAUGGGCUCA-3′)34. Following homologous recombination, recombinant clones were positively selected for one week with neomycin (100 μg ml−1), transfected with an EF1α-Cre-puromycin vector, and selected for a second week with puromycin (1 μg ml−1). Serially diluted ES colonies were then individually picked, expanded, and screened by PCR and qPCR for site-specific integration, exclusion of off-target effects, and correct copy number. After sequence validation of the targeted site, successfully targeted ES clones were used to establish chimaeras. Chimaeric mice were crossed with C57BL/6 − Tyrc-2j/J female mice to establish germline transmission.
Flow cytometry and cell sorting
Flow cytometry and cell sorting were performed on a FACS Aria II cell sorter (BD Biosciences) and analysed using FlowJo software (Tree Star). Bone marrow cells were collected from bilateral tibias, femurs, humeri, and pelvises by crushing (unless otherwise specified) using a mortar and pestle in Ca2+- and Mg2+-free PBS supplemented with 2% heat-inactivated bovine serum (Gibco) and 2 mM EDTA. Cells were passed through 100 μm, 70 μm, and 40 μm strainers before analysis and sorting. To enrich HSCs and progenitor populations, cells were stained with APC-conjugated anti-c-Kit (2B8) and fractionated using anti-APC magnetic beads and LS columns (both Miltenyi Biotec). c-Kit+ cells were then stained with combinations of antibodies against the following surface markers: Sca-1, Flk2, CD150, CD34, IL-7R, CD16/32, and the lineage markers Ter-119, B220, CD2, CD3, CD4, CD5, CD8a, Gr-1, CD11a, CD11b, CD41, CD48, CD229, and CD244. For lymphoid populations, bone marrow cells were stained with antibodies against CD3, CD4, CD8a, CD11b, CD11c, Gr-1, NK-1.1, Ter-119, B220, c-Kit, and F4/80. For myeloid populations, cells were stained with antibodies against CD3, CD3-, CD11b, CD11c, CD19, Gr-1, NK-1.1, Ter-119, and F4/80. Antibody staining was performed at 4 °C and cells were incubated for 30 min. Cells stained with CD34 were incubated for 90 min. Before sorting or analysis, cells were stained with SYTOX Red Dead Cell Stain (Life Technologies) to assess viability as per the manufacturer’s recommendations. Transplanted cells were double-sorted for purity. Further details regarding flow cytometry reagents are provided in Supplementary Table 4.
Transplantations and peripheral blood analyses
B6.SJL-Ptprca Pepcb/BoyJ (Jackson Laboratory) recipient mice were lethally irradiated at a single dose of 9.1 Gy. For reconstitution assays, donor cells were first combined with 2 × 105 whole bone marrow supporting cells (B6.SJL- Ptprca Pepcb/BoyJ × C57BL/6J F1 mice CD45.1+/CD45.2+) in 200 μl of PBS with 2% FBS, then injected into the retro-orbital venous plexus. Peripheral blood analyses were performed at 4, 8, 12, and 16 weeks after primary and secondary transplantations. At each time point, 50 μl of blood was collected from the tail vein and added to 100 μl of PBS with 2 mM EDTA. Red blood cells were subsequently lysed using BD Pharm Lyse Buffer (BD Pharmingen), as per the manufacturer’s protocol, for 3 min on ice, followed by blocking with 5 μg ml−1 rat IgG. Leukocytes were stained with antibodies against (refer to Supplementary Table 4 for specific clones and colours): CD45.1 (FITC), CD45.2 (PE), CD11b (BUV395), Gr-1 (Alexa-Flour700), B220 (BV786), CD3 (BV421), TCRβ (BV421), and NK-1.1 (PerCP-cy5.5). For each mouse, the percentage of donor chimaerism in the peripheral blood was defined as the percentage of CD45.1−CD45.2+ cells among total CD45.1−CD45.2+ and CD45.1+CD45.2+ cells. To control for variability in host response to lethal irradiation, mice with host chimaerism of 50% or higher 16 weeks after transplantation were excluded from our analyses. The frequency of chimaerism in peripheral blood was analysed as follows. For evaluation of donor (CD45.1−CD45.2+) chimaerism kinetics, after exclusion of recipient (CD45.1+CD45.2−) fraction, the frequency of the donor fraction was calculated. Within the whole donor fraction, the frequency of each lineage (NK cell, B cell, T cell, granulocyte, and monocyte) was determined. For evaluation of lineage contribution kinetics, after gating each lineage, the frequency of the donor fraction (CD45.1−CD45.2+) was calculated. Any recipients that exhibited lower than 1% of chimaerism were treated as negative to exclude ambiguous cases.
Limiting dilution analysis
The frequency of long-term and short-term HSCs was calculated using the transplantation data of ten- and three-cell Hoxb5hi, Hoxb5lo, or Hoxb5neg pHSC transplants. Any mice showing long-term (>16 week) multilineage reconstitution (>1% in each lineage) were counted as positive recipients. A nonlinear regression semi-log best fit line was used to calculate the frequency of LT/ST-HSCs at F0 = 0.368 (GraphPad Prism 6).
CUBIC bone marrow imaging
Bone clearing protocol was modified from the original CUBIC protocol23. Specifically, tibias were collected and fixed in 4% PFA solution for two days, after which bone marrow plugs were extracted from the distal end by flushing method with a 25-gauge syringe. For nuclear staining, bone marrow plugs were immersed in DAPI/PBS solution at 37 °C for three days with gentle shaking. For clearing, marrow plugs were immersed in ScaleCUBIC-1 (Reagent-1) at 37 °C for two weeks with gentle shaking. The solution was changed every 48 h. To visualize vasculature, 20 μg of Alexa488-conjugated anti-mouse VE-cadherin antibody (BV13) was administrated intravenously (retro-orbital) with tibias collected 30 min later. Processed plugs were embedded in 4-mm-diameter glass capillaries with 2% agarose for imaging. Images were acquired using a Zeiss Z1 Lightsheet microscope (Zeiss) and reconstituted into 3D images using Zen software (Zeiss). Acquired 3D images were analysed with Imaris software (Bitplane). After exclusion of outliers including events of extraordinary size (>30 μm) or intensity, all other mCherry+ (Hoxb5+) cells were analysed in the tibia (n = 287 cells in total from n = 3 mice). The mCherry-negative threshold was determined on the basis of the intensity level of the wild-type control tibial plugs. These mCherry− events with intensities ranging from 0.002–75.998 (75.998 representing the upper bound of mCherry intensity) were transformed into an integer list of 75,000 values (75,000 representing the length of the list), and 600 spots were then randomly selected from this integer list using the ‘randbetween’ (1,75,000) function in Excel 2015 (Microsoft). Using the list of random spots identified by intensity, the location and distance to VE-cadherin+ cells was measured using the Imaris software. All CUBIC imaging experiments were performed in biological triplicates from three mice.
Genotyping
Genomic DNA from Hoxb5–tri-mCherry mice was isolated from tail biopsies using QuickExtract solution (EpiCentre). PCR amplification was performed using the same forward primer (5′-GACGTATCGAGATCGCCCAC-3′) with two reverse primers to distinguish between the Hoxb5–tri-mCherry (5′-CCTTGGTCACCTTCAGCTTGG-3′) and wild-type (5′-AGATTGGAAGGGTCGAGCTG-3′) alleles.
Statistics
All comparative analyses were performed using unpaired Student’s t tests. Pearson’s chi-square test was performed using online software (http://vassarstats.net/).
References
Spangrude, G. J., Heimfeld, S. & Weissman, I. L. Purification and characterization of mouse hematopoietic stem cells. Science 241, 58–62 (1988)
Guo, W. et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 453, 529–533 (2008)
Yilmaz, O. H. et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482 (2006)
Ito, K. et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431, 997–1002 (2004)
Morrison, S. J. & Scadden, D. T. The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014)
Dykstra, B. et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 1, 218–229 (2007)
Beerman, I. et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl Acad. Sci. USA 107, 5465–5470 (2010)
Lu, R., Neff, N. F., Quake, S. R. & Weissman, I. L. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nature Biotechnol. 29, 928–933 (2011)
Uchida, N. & Weissman, I. L. Searching for hematopoietic stem cells: evidence that Thy-1.1lo Lin− Sca-1+ cells are the only stem cells in C57BL/Ka-Thy-1.1 bone marrow. J. Exp. Med. 175, 175–184 (1992)
Morrison, S. J. & Weissman, I. L. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1, 661–673 (1994)
Acar, M. et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 526, 126–130 (2015)
Gazit, R. et al. Fgd5 identifies hematopoietic stem cells in the murine bone marrow. J. Exp. Med. 211, 1315–1331 (2014)
Hills, D. et al. Hoxb4-YFP reporter mouse model: a novel tool for tracking HSC development and studying the role of Hoxb4 in hematopoiesis. Blood 117, 3521–3528 (2011)
Seita, J. et al. Gene expression commons: an open platform for absolute gene expression profiling. PLoS ONE 7, e40321 (2012)
Chan, C. K. F. et al. Identification and specification of the mouse skeletal stem cell. Cell 160, 285–298 (2015)
Chan, C. K. F. et al. Clonal precursor of bone, cartilage, and hematopoietic niche stromal cells. Proc. Natl Acad. Sci. USA 110, 12643–12648 (2013)
Hosen, N. et al. Bmi-1-green fluorescent protein-knock-in mice reveal the dynamic regulation of bmi-1 expression in normal and leukemic hematopoietic cells. Stem Cells 25, 1635–1644 (2007)
Yamamoto, R. et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126 (2013)
Fathman, J. W. et al. Upregulation of CD11A on hematopoietic stem cells denotes the loss of long-term reconstitution potential. Stem Cell Reports 3, 707–715 (2014)
Oguro, H., Ding, L. & Morrison, S. J. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13, 102–116 (2013)
Herzenberg, L. A., Tung, J., Moore, W. A., Herzenberg, L. A. & Parks, D. R. Interpreting flow cytometry data: a guide for the perplexed. Nature Immunol. 7, 681–685 (2006)
Kiel, M. J. et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121 (2005)
Susaki, E. A. et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739 (2014)
Christensen, J. L. & Weissman, I. L. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proc. Natl Acad. Sci. USA 98, 14541–14546 (2001)
Osawa, M., Hanada, K., Hamada, H. & Nakauchi, H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 273, 242–245 (1996)
Wright, D. E., Wagers, A. J., Gulati, A. P., Johnson, F. L. & Weissman, I. L. Physiological migration of hematopoietic stem and progenitor cells. Science 294, 1933–1936 (2001)
Forsberg, E. C. et al. Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS ONE 5, e8785 (2010)
Fleming, W. H. et al. Functional heterogeneity is associated with the cell cycle status of murine hematopoietic stem cells. J. Cell Biol. 122, 897–902 (1993)
Passegué, E., Wagers, A. J., Giuriato, S., Anderson, W. C. & Weissman, I. L. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 202, 1599–1611 (2005)
Jaiswal, S. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138, 271–285 (2009)
Moraga, I. et al. Tuning cytokine receptor signaling by re-orienting dimer geometry with surrogate ligands. Cell 160, 1196–1208 (2015)
Wu, J., Anczuków, O., Krainer, A. R., Zhang, M. Q. & Zhang, C. OLego: fast and sensitive mapping of spliced mRNA-Seq reads using small seeds. Nucleic Acids Res. 41, 5149–5163 (2013)
Sanjana, N. E. et al. A transcription activator-like effector toolbox for genome engineering. Nature Protocols 7, 171–192 (2012)
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013)
Acknowledgements
We would like to acknowledge N. Neff and G. Mantalas for advice regarding RNA sequencing; B. Yu and A. Beel for providing critical input on imaging data; H. Nishikii for advice regarding imaging data; S. Karten for help in editing the manuscript; L. Jerabek and T. Storm for laboratory management; A. McCarty and C. Wang for animal care; P. Lovelace and J. Ho for FACS facility management; H. Zeng, Y. Li, and C. Wang for collaboration in generating the mouse model; and Y. Sato for technical assistance in Imaris software analysis. The authors would like to acknowledge ongoing support for this work: NCI and NHLBI of the NIH under award numbers R01 CA086065, U01 HL099999, and R01 HL058770, and by the Virginia and D. K. Ludwig Fund for Cancer Research (I.L.W.); Stanford University Medical Scientist Training Program (T32 GM007365) and NHLBI Ruth L. Kirschstein National Research Service Award (F30-HL122096) (J.Y.C.); and Human Frontier Science Program Long-Term Fellowships, the Uehara Memorial Foundation Research Fellowship, Toyobo Biotechnology Foundation Research Fellowship, and Kanzawa Medical Research Foundation Overseas study grants (M.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH.
Author information
Authors and Affiliations
Contributions
J.Y.C. and M.M. contributed equally to this work, and either has the right to list himself first in bibliographic documents. M.M. and J.Y.C. conceived, performed, analysed, and oversaw the experiments, with suggestions from I.L.W. M.M. and J.Y.C. identified Hoxb5 as a LT-HSC marker, and made and characterized the Hoxb5–tri-mCherry mouse. S.K.W. and K.S.K. performed experiments and prepared figures under the supervision of M.M. and J.Y.C. S.Y. generated CUBIC data and evaluated the association with VE-cadherin vasculature. R.S. designed and performed RNA-seq and associated data analysis. J.S. and D.S. designed the gene expression commons for microarray analysis. D.S. provided critical advice regarding combined analysis of microarray and RNA-seq data. M.M., J.Y.C., S.K.W., K.S.K., and I.L.W. wrote the manuscript. H.N. and R.S. provided comments on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 GEXC expression of previously reported HSC markers in mouse bone marrow.
a, Ideal expression pattern of HSC-specific genes (pink represents increased expression, blue represents decreased expression). b, Relative expression of Hoxb5 (top left), α-catulin/Ctnnal1 (top middle), Fgd5 (top right), CD150/Slamf1 (bottom left), Hoxb4 (bottom middle), Gfi-1 (bottom right) in haematopoietic and stromal populations as determined by microarray analysis.
Extended Data Figure 2 Gating scheme for HSC and progenitors.
a, Representative flow cytometry gating to isolate pHSCs, MPPs, and oligopotent progenitors from mouse bone marrow. Panels gated as shown after exclusion of doublets and dead cells. b, Hoxb5 reporter expression (red) in Flk2+ MPPs, megakaryocyte erythrocyte progenitor (MEP), granulocyte monocyte progenitor (GMP), common myeloid progenitor (CMP), and common lymphoid progenitor (CLP) populations compared to wild-type controls (blue). Values indicate the percentage of mCherry+ cells ± s.d. in each fraction for n = 3 mice.
Extended Data Figure 3 Hoxb5 is not expressed in CD45− bone marrow.
Hoxb5 reporter expression in the CD45− compartment within bone marrow of wild-type (red) and three Hoxb5–tri-mCherry mice (blue, orange, and green, n = 3 mice).
Extended Data Figure 4 FMO gating for Hoxb5+ signal.
Representative flow cytometry gating to separate mCherry (Hoxb5)-high, -low, and -negative populations in both wild-type and Hoxb5–tri-mCherry mice.
Extended Data Figure 5 Hoxb5 distinguishes between LT-HSCs and non-LT-HSCs.
a, Reconstitution kinetics in primary recipients 4, 8, and 12 weeks after receiving ten Hoxb5neg (n = 9 mice), Hoxb5lo (n = 13 mice), or Hoxb5hi (n = 18 mice) pHSCs. Each column represents an individual mouse. b, Reconstitution kinetics 4, 8, and 12 weeks after whole bone marrow secondary transplant. c, Reconstitution kinetics in primary recipients receiving three Hoxb5neg (n = 11 mice), Hoxb5lo (n = 12 mice), or Hoxb5hi (n = 14 mice) pHSCs. Each column represents an individual mouse. d, Reconstitution kinetics following secondary transplant of 100 sorted LSK Hoxb5− (n = 14 mice) or Hoxb5+ (n = 9 mice) cells and 2 × 105 supporting cells.
Extended Data Figure 6 Limiting dilution analysis of Hoxb5+ and Hoxb5− pHSCs.
Limiting dilution results of ten- and three-cell transplants of Hoxb5hi (red, n = 18 mice for ten-cell and n = 14 mice for three-cell), Hoxb5lo (green, n = 13 mice for ten-cell and n = 12 mice for three-cell), and Hoxb5neg (blue, n = 9 mice for ten-cell and n = 11 mice for three-cell). Frequency of LT/ST-HSCs by limiting dilution analysis is 1 in 2.1 for Hoxb5hi, 1 in 2.4 for Hoxb5lo, and 1 in 16.1 for Hoxb5neg cells.
Extended Data Figure 8 Specificity of Hoxb5 as a single marker for LT-HSCs.
a, Flow cytometry plots of wild type (top row) and Hoxb5–tri-mCherry (bottom row) after excluding doublets, dead cells, autofluorescence, and gating on Hoxb5+ events. Frequencies shown are percentage in gate ± s.d. in each fraction (n = 3 mice).
Extended Data Figure 9 Comparison of processing methods on pHSC and Hoxb5+ LT-HSC yield.
a, b, Relative frequency of pHSCs (a) and Hoxb5+ LT-HSCs (b) in tibial plugs (flushed) (n = 6 mice) compared to tibial plugs plus bones (crushed) (n = 6 mice).
Extended Data Figure 10 Hoxb5+ HSCs are evenly distributed in the tibia.
a, Distribution of Hoxb5+ cells (red and arrows) in bone marrow in 3D-reconstructed images. Nuclei are counterstained with DAPI (blue) wild-type (top panel) Hoxb5–tri-mCherry (middle and bottom panel). Scale bar, 100 μm. b, Cartoon representing the location of the proximal, medial, and distal sampling. c, Representative 3D-reconstructed images of Hoxb5+ cells (red) in proximal (left column), medial (middle column), and distal (right column) regions of the tibia. Scale bar, 150 μm. Nuclei are counterstained with DAPI (blue); n = 3 mice.
Supplementary information
Supplementary Table 1
The file shows the marker definitions of the cell populations used in gene microarray profiling. (XLSX 30 kb)
Supplementary Table 2
This file contains a list of HSC-specific genes found by screening the haematopoietic sub-fractions in the adult mouse bone marrow. (XLSX 33 kb)
Supplementary Table 3
This file contains the RNA expression of α-catulin in Hoxb5 subsets within the pHSC gate. (XLSX 9 kb)
Supplementary Table 4
Tis file contains a list of flow cytometry reagents used in this study, including, antigen, clone, fluorophore, and manufacturer. (XLSX 10 kb)
Rights and permissions
About this article

Cite this article
Chen, J., Miyanishi, M., Wang, S. et al. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature 530, 223–227 (2016). https://doi.org/10.1038/nature16943
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature16943
- Springer Nature Limited
This article is cited by
-
Depleting myeloid-biased haematopoietic stem cells rejuvenates aged immunity
Nature (2024)
-
A conserved ZFX/WNT3 axis modulates the growth and imatinib response of chronic myeloid leukemia stem/progenitor cells
Cellular & Molecular Biology Letters (2023)
-
Functions and regulatory mechanisms of resting hematopoietic stem cells: a promising targeted therapeutic strategy
Stem Cell Research & Therapy (2023)
-
Cell-intrinsic factors governing quiescence vis-à-vis activation of adult hematopoietic stem cells
Molecular and Cellular Biochemistry (2023)
-
Multiphoton intravital microscopy of rodents
Nature Reviews Methods Primers (2022)