

Abstract
Stearoyl-CoA desaturase (SCD) is conserved in all eukaryotes and introduces the first double bond into saturated fatty acyl-CoAs1,2,3,4. Because the monounsaturated products of SCD are key precursors of membrane phospholipids, cholesterol esters and triglycerides, SCD is pivotal in fatty acid metabolism. Humans have two SCD homologues (SCD1 and SCD5), while mice have four (SCD1–SCD4). SCD1-deficient mice do not become obese or diabetic when fed a high-fat diet because of improved lipid metabolic profiles and insulin sensitivity5,6. Thus, SCD1 is a pharmacological target in the treatment of obesity, diabetes and other metabolic diseases7. SCD1 is an integral membrane protein located in the endoplasmic reticulum, and catalyses the formation of a cis-double bond between the ninth and tenth carbons of stearoyl- or palmitoyl-CoA8,9. The reaction requires molecular oxygen, which is activated by a di-iron centre, and cytochrome b5, which regenerates the di-iron centre10. To understand better the structural basis of these characteristics of SCD function, here we crystallize and solve the structure of mouse SCD1 bound to stearoyl-CoA at 2.6 Å resolution. The structure shows a novel fold comprising four transmembrane helices capped by a cytosolic domain, and a plausible pathway for lateral substrate access and product egress. The acyl chain of the bound stearoyl-CoA is enclosed in a tunnel buried in the cytosolic domain, and the geometry of the tunnel and the conformation of the bound acyl chain provide a structural basis for the regioselectivity and stereospecificity of the desaturation reaction. The dimetal centre is coordinated by a unique spacial arrangement of nine conserved histidine residues that implies a potentially novel mechanism for oxygen activation. The structure also illustrates a possible route for electron transfer from cytochrome b5 to the di-iron centre.
Similar content being viewed by others


Main
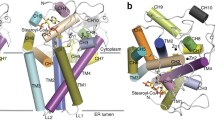
A Δ2–23 amino-terminal truncation of mouse SCD1 was crystallized in lipidic cubic phase11 and the structure was solved by single-wavelength anomalous dispersion (Extended Data Table 1). SCD1 has four transmembrane helices (TM1–TM4) arranged in a cone-like shape with TM4 sandwiched between TM1 and TM2 (Fig. 1a). Residues in the membrane-spanning region are largely hydrophobic, with the notable exception of a conserved arginine (Arg249; Extended Data Fig. 1) located on TM4 in the centre of the cone (Extended Data Fig. 2). Previous biochemical analysis12 determined that the amino and carboxy termini are on the cytosolic side of the membrane (Fig. 1b). On the cytosolic side, TM2 and TM4 protrude three helical turns out of the membrane and provide some of the coordinating residues for the dimetal active site. The cytosolic domain comprises 93 residues between TM2 and TM3 (C1) and the 90-residue C terminus (C2) (Fig. 2a). The C1 and C2 domains contain six and five α-helices, respectively. Three of the α-helices (AH1 on C1 and AH7 and AH9 on C2) are amphipathic and probably reside at the interface between the cytosolic domain and the lipid bilayer (Fig. 1a and Extended Data Fig. 3). These amphipathic helices and the locations of hydrophobic residues on the transmembrane helices indicate the approximate position of the lipid bilayer (Fig. 1a). In the crystal lattice, the interacting surfaces between neighbouring molecules are small (Extended Data Fig. 4), and size-exclusion chromatography (SEC) of the detergent solubilized protein indicated that mouse SCD1 was stable as a monomer. By contrast, previous in vivo studies have shown that SCDs are dimers in the cellular membrane13. Whether this difference is a consequence of isolation of the enzyme remains to be determined.

a, The crystal structure of mouse SCD1 is shown from two perpendicular orientations in the membrane, with the cytosolic side on top. Two zinc ions bound to the cytosolic domain are shown as grey spheres. b, Topology diagram of SCD1, with helices coloured by the same scheme as in a. Orange spheres represent conserved histidine residues involved in coordination of the dimetal centre.

a, Two views of the SCD1 structure with the C1 domain coloured teal and the C2 domain coloured magenta. The bound stearoyl-CoA is shown as yellow, red and blue sticks, and the two zinc ions as grey spheres. b, A close-up view of the CoA binding site shown as a surface representation (left), or with residues interacting with the CoA moiety shown as sticks (right). c, A close-up view of the substrate tunnel housing the acyl chain shown as a surface cross-section (top), or with residues forming the kink (Thr257, Gln143 and Trp149), hydrogen bonding to the acyl oxygen (Trp258), and capping the end of the substrate tunnel (Tyr104 and Ala108) shown as sticks (bottom). d, The locations of SCD3 mutations studied by yeast complementation experiments are mapped onto the SCD1 structure as green spheres. Residue positions are labelled according to the SCD3 sequence. e, Monounsaturated fatty acids in the total lipid of yeast L8–14C transformed with mouse SCD1, SCD3 or mutated SCD3 enzyme. SCD1 produces a mixture of 16:1 and 18:1 fatty acids, while SCD3 produces 16:1 nearly exclusively. The combination of Ile112Ala and Glu113Leu mutations converts SCD3 into an enzyme that yields a proportion of monounsaturated fatty acids indistinguishable from SCD1. The corresponding residues in SCD1, Ala108 and Leu109, lie at the end of the substrate binding tunnel. Error bars are s.d. of 3 technical replicates. f, Expression levels of the SCD3 mutants were similar as detected by western blotting. Uncropped gel is shown in Extended Data Fig. 6.
The cytosolic domain contains a substantial non-protein density consistent with an 18-carbon acyl-CoA molecule (Fig. 2a and Extended Data Fig. 5a). We modelled a stearoyl-CoA molecule into this density, although we were unable to distinguish between oleoyl-CoA and stearoyl-CoA solely from the crystallographic maps. The electron density for the CoA moiety is well resolved, and the CoA group interacts primarily with hydrophilic and charged residues on the outer surface of the C1 domain (Fig. 2b). The residues that form polar interactions with the CoA group in the mouse SCD1 structure are strongly conserved among known stearoyl-CoA desaturases, including human SCD1, but not among stearoyl-lipid desaturases (Extended Data Fig. 1).
The acyl chain is enclosed in a long, narrow tunnel extending approximately 24 Å into the mostly hydrophobic interior of the protein. This tunnel is sharply kinked where it binds to C9 and C10 on stearoyl-CoA, the atoms involved in formation of the cis-double bond (Fig. 2c). The positioning of C9 and C10 by the kink is enforced by the shape complementarity of this substrate tunnel, by the location of the CoA binding site, and by a hydrogen bond between the Trp258 side chain and the acyl carbonyl (Fig. 2c). The kink in the tunnel is created by the side chains of two conserved residues, Trp149 and Thr257, which are stabilized by hydrogen bonds with conserved Gln143 (Fig. 2c). We note that the narrow and kinked tunnel precisely positions the acyl chain for Δ9-regioselective desaturation, an idea that was envisioned by Bloch over four decades ago8.
Previous studies showed that rat SCD1 was effective on acyl chains containing between 14 and 19 carbons, and had the highest activity with substrates 17 to 19 carbons in length9. Regardless of the acyl chain length, the double bond is exclusively placed between C9 and C10, as enforced by the substrate–enzyme interactions described above. Residues that probably have a role in determining substrate length are found at the end of the substrate tunnel, which is capped by Tyr104 on TM2 (Fig. 2c). There is a distance of 4.1 Å between the end of the 18:0 acyl chain and the tyrosine hydroxyl oxygen, which agrees well with the observed preference of the enzyme for 18:0 (ref. 9). Tyr104 is highly conserved in animal SCD1. However, an atypical acyl-CoA desaturase (ChDes1) from the marine copepod Calanus hyperboreus has a threonine at the position corresponding to Tyr104 in mouse SCD1 (ref. 14). ChDes1 preferentially acts on very long-chain fatty acyl-CoAs (22:0–26:0), but when this threonine was mutated to tyrosine, desaturation of 26:0 was lost while desaturation of 18:0 was retained14. Another conserved residue, Ala108, is located one helical turn above Tyr104 facing the substrate tunnel (Fig. 2c). Desat2 from Drosophila melanogaster has a methionine at this position, and can only accept acyl substrates up to 14 carbons long15. Combined, these observations suggest that the tunnel-facing residues 104 and 108 on TM2 are critical determinants of the substrate chain length.
To explore the relationship between the structure of the substrate tunnel in mouse SCD1 and acyl chain selectivity further, we transformed yeast monounsaturated fatty acid auxotroph L8–14C with either mouse SCD1 or SCD3, which allowed growth in media lacking unsaturated fatty acids. Although SCD1 and SCD3 share 89% primary sequence identity, they yield remarkably different total fatty acid profiles in the yeast host cells, probably reflecting differences in their preferences for reaction with 16:0 and 18:0 (Fig. 2e and ref. 16). In SCD1, Ala108, Leu109, Ala288 and Val289 line the distal end of the substrate binding channel, Ala115 is near the position of double bond formation, and Gln277 and Ser278 are on the cytoplasmic surface opposite to the CoA binding site. The corresponding residues in SCD3 are Ile112, Glu113, Ser292 and Met293, Val119, and Asp281 and Pro282, respectively (Fig. 2d). The stacked mutations Ile112Ala/Glu113Leu were able to convert SCD3 from exclusively a 16:0 desaturase into a predominantly 18:0 desaturase (Fig. 2e, f and Extended Data Fig. 6). The stacked mutations Val119Ala/Asp281Gln/Pro282Ser, which are located away from the end of the substrate tunnel, caused no change in the reaction specificity.
In addition to the bound stearoyl-CoA molecule, SCD1 also contains two metal ions. The metal ions in the current structure were identified as zinc by X-ray fluorescence, and by diffraction data collected at a wavelength near the zinc absorption edge that yielded two prominent anomalous difference peaks in each protein (Extended Data Fig. 5b–e). Incorporation of zinc instead of iron into the protein was probably an artefact of protein overexpression, and zinc remained the predominant metal species even when the growth media and purification solutions were supplemented with iron.
The dimetal cluster sits at the kink in the substrate tunnel adjacent to C9 and C10 on the substrate, where the double bond is introduced. Zinc 1 (M1) is positioned 5.2 Å from C9, while zinc 2 (M2) is 4.7 Å from C10 (Fig. 3a). M1 and M2 are coordinated by four and five histidine residues, respectively, provided by the helices TM2, TM4, H2 and H8 (Fig. 3b and Extended Data Fig. 7a). The coordination of both zinc ions is consistent with an octahedral geometry with one missing ligand. The nine histidines are highly conserved (Extended Data Fig. 1), and eight of them belong to three histidine-containing motifs (two HXXHH motifs and one HX4H motif in SCD1) that are characteristic of integral membrane desaturases, alkane hydroxylases and xylene monooxygenases3,17. The predominance of histidine ligands in SCD1 is consistent with the assignment of nitrogen-rich ligation of the di-iron centre in alkane hydroxylase from Mössbauer isomer shifts18. Mutation of any of these eight histidines into an alanine in rat SCD1 led to a nonfunctional enzyme17. The ninth histidine (His265) is conserved in other SCDs but had not been previously identified. The water molecule coordinating M1 is hydrogen bonded to an asparagine residue, Asn261 on TM4. Interestingly, this asparagine and His265 belong to a NX3H motif that is symmetrically equivalent to the HX4H motif interacting with M2 (Fig. 3c). Likewise, the two HXXHH motifs have symmetrical interactions with M1 and M2 (Fig. 3c).

a, b, Two views of the dimetal centre and coordinating residues, marked with distances between the zinc ions and C9 and C10 on the substrate (a), and coordination distances (b). The zinc ions and an ordered water molecule are shown as grey and red spheres, respectively. See Extended Data Fig. 7 for a stereo view. c, Schematic showing the locations of the coordinating His and Asn residues in four conserved motifs on TM2, TM4, H2 and H8.
One notable aspect of the structure is that the two metal ions are separated by 6.4 Å (Fig. 3a). This is longer than in any previously solved structures of soluble di-iron enzymes19,20,21, including the soluble plant acyl-ACP desaturases that catalyse the same reaction as SCD1. In these soluble enzymes, the two iron ions are bridged by a glutamate residue with bidentate coordination (Extended Data Fig. 7b) that constrains the inter-iron distance to roughly 3–4 Å (refs 19, 20, 21), and permits formation of reaction intermediates such as cis-μ-1,2 peroxo22,23 and diferryl24. In SCD1, there is no carboxylate coordination between the metal ions. This is probably not an artefact caused by Zn2+, as the closest glutamate or aspartate residues are over 6 Å away from the metal centres and provide hydrogen-bonding interactions to the metal-bound His residues (Extended Data Fig. 7a). Since Zn2+ has an ionic radius of 0.88 Å, it may have served as a reasonable substitute for Fe2+ (ionic radius 0.92 Å) in terms of size and charge during heterologous expression. Notably, the bound Zn2+, which often have tetrahedral coordination, have octahedral coordination in the SCD1 structure as typical for iron ions. Given the similar B-values for residues around the observed metal sites, the presence of stearoyl-CoA bound in an appropriately kinked configuration relative to the metals, and the absence of reasonably positioned carboxylate residues that might serve as bridging ligands, we propose that the unfortunate presence of Zn2+ has not significantly altered the structure and that the SCD1 structure indicates a new avenue for activation of O2 in biological oxidation reactions.
Two known aspects of the desaturase reaction are compatible with this active site. Removal of the pro-R hydrogen atom from C9 proceeds with kH/kD of ∼6–7 (refs 9, 25), indicating that this step is rate-limiting. The distance from the pro-R C9 hydrogen atom to the water bound to M1 is 3.5 Å, possibly corresponding to the direction through which the desaturation reaction will initiate. Furthermore, no oxygen atom transfer to carbon is anticipated during the desaturation reaction26,27. The enforcement of a long distance between the acyl chain and metals would be consistent with promotion of an electron transfer mechanism in which O atoms are retained on an oxidizing metal centre as electrons and protons are extracted from the bound and sterically configured acyl chain.
Release of a desaturated acyl chain from the active site merits additional consideration. Given the kink and the narrow aperture of the substrate tunnel, it seems unlikely that substrate entrance or product release can occur by simple linear diffusion in and out of the tunnel. However, a break in the hydrogen bond between Gln143 and Thr257 below the kink in the substrate tunnel would create a fenestration into the hydrophobic core of the membrane (Fig. 2c), allowing lateral transfer of substrates and products into and out of the well-formed substrate tunnel. A separation between TM4 and the loop between helices H1 and H2 could break this bond (Extended Data Fig. 8).
In SCD1, the electrons needed for the desaturation reaction are obtained from cytochrome b5 (cytb5), which in turn obtains electrons from NAD(P)H via cytochrome b5 reductase9. Although it is known that cytb5 consists of an N-terminal haem-binding domain and a C-terminal membrane anchor domain28, and must be membrane-anchored to function in the desaturation reaction1, the cytb5 binding site on SCD is unknown. Examination of the mouse SCD1 structure shows that the dimetal centre could be accessible from the cytoplasm via a groove formed between the soluble domains C1 and C2. In the crystal structure, the N terminus of the protein lies along this groove (Extended Data Fig. 9); however, it forms few interactions with the cytoplasmic domain and potentially could be displaced by cytb5. Electrostatic calculations on the two proteins demonstrate that the predominantly positive surface of SCD1 is well complemented by the mostly negative surface of cytb5 (Fig. 4a). Placement of the cytb5 functional domain along this groove would place the electron donor groups within 14 Å of the dimetal centre, an acceptable distance for electron transfer between biological redox centres29 (Fig. 4b). His157 (H2) and His298 (H8) sit directly above the two metal ions and are potential candidates to form an electron transfer interface. This proposed complex also places negatively charged residues on cytb5 demonstrated to be necessary for complex formation28 close to positively charged residues on H4 of SCD. The structure of mouse SCD1 enables us to address questions related to basic mechanisms of the desaturation reaction more precisely.

a, Electrostatic surfaces of cytb5 (left) and mouse SCD1 (right) oriented with their proposed interaction surfaces towards the viewer. b, A proposed model of the complex between cytb5 and SCD1. Cytb5 is coloured green; domains C1 and C2 on SCD1 are teal and magenta, respectively. The inset shows a closer view of the proposed electron transfer pathway, with the haem domain shown as blue sticks and the metal ions as grey spheres. The last residue resolved in the cytb5 structure is approximately 35 Å from the predicted position of the bilayer. The 18-residue linker from this residue to the transmembrane helix of cytb5 should therefore be long enough to allow for the haem-containing domain to bind in this orientation.
PDB 4YMK
Methods
No statistical methods were used to predetermine sample size.
Desaturase expression and purification
To identify an optimal SCD candidate for crystallization, we tested 24 eukaryotic homologues for overexpression in High Five (Trichoplusia ni) insect cells using the Bac-to-Bac system (Invitrogen) to generate virus. During the process of identifying suitable homologs, we found that the InterPro fatty acid desaturase family 1 motif30, G-E-X-[FYN]-H-N-[FY]-H-H-X-F-P-X-D-Y, is found in more than 7,000 eukaryotic sequences (fatty acid desaturase type 1, InterPro accession IPR005804) from animals, plants and fungi. The three His residues from this motif are now known to provide ligands to both metal ions, and so provide an excellent indicator for the presence of a mouse SCD1-like protein fold. Inclusion of bacterial genes having a related three-His motif expands to more than 22,000 sequences with 90 distinct domain architectures where a mouse SCD1-like fold is fused to domains with a redox centre such as cytb5, plant-type and Rieske-type ferredoxins, rubredoxins and thioredoxins, as well as domains for peptidases, hydrolases, kinases, phosphatases, and many others. This diversity of domain architectures suggests a considerably greater versatility for the SCD1 fold than the originally anticipated desaturase and alkane hydroxylase activities17.
The pFastBac vector was modified to include a tobacco etch virus (TEV) protease cleavage site before the C-terminal polyhistidine tag. The majority of the 24 homologues expressed, but the mouse SCD1 clone (GI:13938635, Life Technologies Open Biosystems) gave the highest yield and stability. Since full-length mouse SCD1 was prone to aggregation and precipitation, we designed five more mouse SCD1 constructs of various N-terminal truncations based on secondary structure prediction and sequence conservation. The construct containing residues 24–355 was stable and yielded diffracting crystals.
Cells were infected with baculovirus at a density of ∼3 × 106 cells ml−1 and were grown at 27 °C for 48–56 h before being collected by centrifugation at 2,000g for 20 min. Cell membranes were isolated from cell pellets following a published protocol31. In brief, cell pellet from 1 l of culture was lysed in 50 ml hypotonic 10 mM HEPES, pH 7.5, containing 10 mM NaCl, 5 mM MgCl2 and 25 μg ml−1 DNase I. After centrifugation at 55,000g for 45 min, cell membranes were washed with 50 ml high-osmotic buffer containing 25 mM HEPES, pH 7.5 and 1 M NaCl. Purified membranes were re-suspended in a low-osmotic buffer containing 25 mM HEPES, pH 7.5, 150 mM NaCl and 40% (v/v) glycerol, flash frozen with liquid N2, and stored at −80 °C.
Purified membranes were thawed and dounced in (10 ml per gram membrane) 20 mM HEPES, pH 7.5, 150 mM NaCl and 2 mM β-mercaptoethanol, and solubilized with 2% (w/v) n-decyl-β-d-maltoside (Anatrace) at 4 °C for 2 h. After centrifugation (55,000g, 45 min, 4 °C), the desaturase was purified from the supernatant using a cobalt-based affinity resin (Talon, Clontech) and the His-tag was cleaved by TEV protease (leaving an extra ENLYFQ peptide at the C terminus). Purified desaturase was collected and concentrated to 5 mg ml−1 (Amicon 50 kDa cutoff, Millipore) and loaded onto a size-exclusion column (Superdex 200 10/300 GL, GE Health Sciences) equilibrated with 25 mM HEPES, pH 7.5, 150 mM NaCl, 0.18% (w/v) n-decyl-β-d-maltoside and 5 mM β-mercaptoethanol. The peak fractions containing desaturase were pooled and immediately used for crystallization.
Lipidic cubic phase crystallization
Crystallization trials with detergent-solubilized protein and the bicelle method32 failed to yield crystals, whereas the in meso method33 succeeded. For lipidic cubic phase crystallization, the purified desaturase was concentrated to 50 mg ml−1 and two volumes of protein solution and three volumes of molten monoolein (Sigma) were mixed with a coupled syringe device. Crystallization trials were performed with 96-well glass sandwich plates (Molecular Dimensions) and a Gryphon crystallization robot using 50 nl protein-lipid mixture overlaid with 800 nl precipitant solution in each well. The initial crystal hits were systematically optimized by screening against salt and PEG concentrations, pH values, and different lipids. The best crystals grew to a final size of ∼50 × 50 × 20 μm within 5 days in 100 mM MES buffer, pH 6.7–7.1, containing 33–37% (v/v) PEG 400, 200 mM NaCl, 4% (v/v) ethylene glycol. Crystals were collected directly from the protein-lipid mixture using 50 μm MiTeGen MicroMounts and immediately flash frozen in liquid nitrogen.
X-ray data collection and processing
X-ray diffraction data were collected at beamline 24ID-C (NE-CAT) at the Advanced Photon Source at Argonne National Laboratory. A data set collected from a single crystal at a wavelength of 1.254 Å with a resolution of 2.8 Å was used for phasing (Extended Data Table 1). The phasing data were processed with XDS34 and scaled with AIMLESS35. The presence of zinc as the predominant ion was confirmed via fluorescence emission spectra using an Amptek SDD fluorescence detector, and analysis of the anomalous signal. A second data set with a resolution of 2.6 Å was collected on a second crystal at a wavelength of 0.9795 Å for use in molecular replacement. The higher resolution data were indexed, integrated, and scaled using HKL2000 (ref. 36). The crystals belonged to the space group P212121 with unit cell dimensions of a = 77.06 Å, b = 113.77 Å and c = 141.70 Å.
Structure determination and refinement
SHELXD/SHELXE37 found four anomalous scatterers per asymmetric unit and the resulting density modified map was used to build an all-helical partial model of one protein molecule. The phased translation function from MOLREP38 was then used to locate a second protein molecule. This partial model and the heavy atom sites were input into PHASER-EP39, which was run in MR-SAD phasing mode. Further density modification was carried out with RESOLVE40. The structure model was then further built through successive rounds with COOT41 and refinement with phenix.refine42. This model was then used as the input model for molecular replacement using PHASER with the 2.6 Å diffraction data. The model was completed through successive rounds of model building with COOT and refinement with phenix.refine. The crystallographic map was easily interpretable. In the final stages of refinement, TLS groups were determined using TLSMD43 and protein geometry was validated with MolProbity44. Figures were produced with PyMOL (Schrödinger LLC.). Electrostatic surfaces were generated with Chimera45. For Fig. 4, coordinates were used from rat cytochrome b5 (PDB accession 1BFX; ref. 46), which is 100% identical to mouse cytochrome b5 over the depicted residue range.
Functional studies of mouse SCDs
For expression using the galactose-inducible yeast expression vector pYES-DEST52 (Invitrogen,)47, mouse SCD1 and SCD3 genes were cloned with an 81 nucleotide leader sequence encoding the N terminus of Saccharomyces cerevisiae desaturase (ole1) appended to the 5′ end. Mutations of mouse SCD3 were made using QuikChange (Stratagene). Expression plasmids were transformed into L8–14C (ref. 48), an ole1− yeast strain, and cultured on agar plates containing 0.5 mM each of oleic acid and palmitoleic acid17. SCD expression was induced on agar plates containing 2% galactose. SCD expression was detected by western blotting using a His-Tag monoclonal antibody (Novagen, 70796-3), goat anti-mouse IgG AP conjugate secondary antibody (Novagen, 69266-3), and alkaline phosphatase reagent (Novagen). Total fatty acids were determined as methyl esters using gas chromatography and mass spectroscopy49.
References
Goren, M. A. & Fox, B. G. Wheat germ cell-free translation, purification, and assembly of a functional human stearoyl-CoA desaturase complex. Protein Expr. Purif. 62, 171–178 (2008)
Paton, C. M. & Ntambi, J. M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. Endocrinol. Metab. 297, E28–E37 (2009)
Sperling, P., Ternes, P., Zank, T. K. & Heinz, E. The evolution of desaturases. Prostaglandins Leukot. Essent. Fatty Acids 68, 73–95 (2003)
Strittmatter, P. et al. Purification and properties of rat liver microsomal stearyl coenzyme A desaturase. Proc. Natl Acad. Sci. USA 71, 4565–4569 (1974)
Gutiérrez-Juárez, R. et al. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J. Clin. Invest. 116, 1686–1695 (2006)
Ntambi, J. M. et al. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl Acad. Sci. USA 99, 11482–11486 (2002)
Zhang, Z., Dales, N. A. & Winther, M. D. Opportunities and challenges in developing stearoyl-coenzyme A desaturase-1 inhibitors as novel therapeutics for human disease. J. Med. Chem. 57, 5039–5056 (2014)
Bloch, K. Enzymatic synthesis of monounsaturated fatty acids. Acc. Chem. Res. 2, 193–202 (1969)
Enoch, H. G., Catala, A. & Strittmatter, P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J. Biol. Chem. 251, 5095–5103 (1976)
Behrouzian, B. & Buist, P. H. Fatty acid desaturation: variations on an oxidative theme. Curr. Opin. Chem. Biol. 6, 577–582 (2002)
Pebay-Peyroula, E., Rummel, G., Rosenbusch, J. P. & Landau, E. M. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 277, 1676–1681 (1997)
Man, W. C., Miyazaki, M., Chu, K. & Ntambi, J. M. Membrane topology of mouse stearoyl-CoA desaturase 1. J. Biol. Chem. 281, 1251–1260 (2006)
Lou, Y. & Shanklin, J. Evidence that the yeast desaturase Ole1p exists as a dimer in vivo . J. Biol. Chem. 285, 19384–19390 (2010)
Meesapyodsuk, D. & Qiu, X. Structure determinants for the substrate specificity of acyl-CoA Δ9 desaturases from a marine copepod. ACS Chem. Biol. 9, 922–934 (2014)
Dallerac, R. et al. A Δ9 desaturase gene with a different substrate specificity is responsible for the cuticular diene hydrocarbon polymorphism in Drosophila melanogaster . Proc. Natl Acad. Sci. USA 97, 9449–9454 (2000)
Miyazaki, M., Bruggink, S. M. & Ntambi, J. M. Identification of mouse palmitoyl-coenzyme A Δ9-desaturase. J. Lipid Res. 47, 700–704 (2006)
Shanklin, J., Whittle, E. & Fox, B. G. Eight histidine residues are catalytically essential in a membrane-associated iron enzyme, stearoyl-CoA desaturase, and are conserved in alkane hydroxylase and xylene monooxygenase. Biochemistry 33, 12787–12794 (1994)
Shanklin, J., Achim, C., Schmidt, H., Fox, B. G. & Munck, E. Mossbauer studies of alkane omega-hydroxylase: evidence for a diiron cluster in an integral-membrane enzyme. Proc. Natl Acad. Sci. USA 94, 2981–2986 (1997)
Högbom, M., Huque, Y., Sjoberg, B. M. & Nordlund, P. Crystal structure of the di-iron/radical protein of ribonucleotide reductase from Corynebacterium ammoniagenes. Biochemistry 41, 1381–1389 (2002)
Lindqvist, Y., Huang, W., Schneider, G. & Shanklin, J. Crystal structure of delta9 stearoyl-acyl carrier protein desaturase from castor seed and its relationship to other di-iron proteins. EMBO J. 15, 4081–4092 (1996)
Sazinsky, M. H. & Lippard, S. J. Correlating structure with function in bacterial multicomponent monooxygenases and related diiron proteins. Acc. Chem. Res. 39, 558–566 (2006)
Broadwater, J. A., Ai, J., Loehr, T. M., Sanders-Loehr, J. & Fox, B. G. Peroxodiferric intermediate of stearoyl-acyl carrier protein Δ9 desaturase: oxidase reactivity during single turnover and implications for the mechanism of desaturation. Biochemistry 37, 14664–14671 (1998)
Moënne-Loccoz, P., Baldwin, J., Ley, B. A., Loehr, T. M. & Bollinger, J. M., Jr O2 activation by non-heme diiron proteins: identification of a symmetric μ-1,2-peroxide in a mutant of ribonucleotide reductase. Biochemistry 37, 14659–14663 (1998)
Banerjee, R., Proshlyakov, Y., Lipscomb, J. D. & Proshlyakov, D. A. Structure of the key species in the enzymatic oxidation of methane to methanol. Nature 518, 431–434 (2015)
Behrouzian, B. et al. Mechanism of fatty acid desaturation in the green alga Chlorella vulgaris . Eur. J. Biochem. 268, 3545–3549 (2001)
Buist, P. H., Behrouzian, B., Kostas, A. A., Dawson, B. & Black, B. Fluorinated fatty acids: new mechanistic probes for desaturases. Chem. Commun. 23, 2671–2672 (1996)
Light, R. J., Lennarz, W. J. & Bloch, K. The metabolism of hydroxystearic acids in yeast. J. Biol. Chem. 237, 1793–1800 (1962)
Dailey, H. A. & Strittmatter, P. Characterization of the interaction of amphipathic cytochrome b5 with stearyl coenzyme A desaturase and NADPH:cytochrome P-450 reductase. J. Biol. Chem. 255, 5184–5189 (1980)
Page, C. C., Moser, C. C., Chen, X. & Dutton, P. L. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402, 47–52 (1999)
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 (2015)
Jaakola, V. P. et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217 (2008)
Ujwal, R. & Bowie, J. U. Crystallizing membrane proteins using lipidic bicelles. Methods 55, 337–341 (2011)
Caffrey, M. & Cherezov, V. Crystallizing membrane proteins using lipidic mesophases. Nature Protocols 4, 706–731 (2009)
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010)
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D 69, 1204–1214 (2013)
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data. Methods Enzymol. 276, 307–326 (1997)
Sheldrick, G. M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D 66, 479–485 (2010)
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D 66, 22–25 (2010)
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007)
Wang, J. W. et al. SAD phasing by combination of direct methods with the SOLVE/RESOLVE procedure. Acta Crystallogr. D 60, 1244–1253 (2004)
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004)
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 68, 352–367 (2012)
Painter, J. & Merritt, E. A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D 62, 439–450 (2006)
Davis, I. W. et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007)
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004)
Arnesano, F., Banci, L., Bertini, I., Felli, I. C. & Koulougliotis, D. Solution structure of the B form of oxidized rat microsomal cytochrome b 5 and backbone dynamics via 15N rotating-frame NMR-relaxation measurements. Biological implications. Eur. J. Biochem. 260, 347–354 (1999)
West, R. W., Jr, Yocum, R. R. & Ptashne, M. Saccharomyces cerevisiae GAL1–GAL10 divergent promoter region: location and function of the upstream activating sequence UASG. Mol. Cell. Biol. 4, 2467–2478 (1984)
Stukey, J. E., McDonough, V. M. & Martin, C. E. Isolation and characterization of OLE1, a gene affecting fatty acid desaturation from Saccharomyces cerevisiae . J. Biol. Chem. 264, 16537–16544 (1989)
Gomez, F. E. et al. Molecular differences caused by differentiation of 3T3–L1 preadipocytes in the presence of either dehydroepiandrosterone (DHEA) or 7-oxo-DHEA. Biochemistry 41, 5473–5482 (2002)
Acknowledgements
This work was supported by the US National Institutes of Health (R01DK088057, R01GM098878, R01HL086392, U54GM095315, U54GM094584 and R01GM050853), the American Heart Association (12EIA8850017), and the Cancer Prevention and Research Institute of Texas (R12MZ). Final data were collected at Northeastern Collaborative Access Team (NE-CAT) beamlines, which are supported by a grant from the National Institute of General Medical Sciences (P41GM103403). Crystals were screened at beamline 17-ID at the Advanced Photon Source, beamlines 8.2.2 and 5.0.2 at Berkeley Center for Structural Biology at the Lawrence Berkeley Laboratory.
Author information
Authors and Affiliations
Contributions
M.Z., B.G.F. and Y.B. conceived the project. Y.B., J.G.M. and E.J.L. expressed, purified and crystallized mouse SCD1, and solved and refined the structure. P.S. and B.G.F. designed and performed mutagenesis and yeast complementation experiments. K.R.R. advised on data collection and structure determination. All authors analysed data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Sequence alignment of mouse SCD1 with other integral membrane desaturases.
The N terminus of mouse SCD1 is not shown. For all the other sequences, only the region aligning to mouse SCD1 is included. Secondary structure elements from the mouse SCD1 crystal structure are labelled. Residues discussed in the text are highlighted in red (histidines in the primary coordination sphere of the dimetal unit), purple (carboxylates in the secondary coordination sphere of the dimetal unit), blue (acyl-chain binding site), yellow (CoA-binding site), green (residues that may determine the length of bound acyl chains), black (mutations that change the substrate specificity in mouse SCD3) and grey (Arg249 in the transmembrane region). The accession numbers for sequences included in the alignment are: mouse SCD1 (GI: 31543675), mouse SCD3 (GI: 13277368), human SCD1 (GI: 53759151), zebrafish SCD1 (GI: 28394115), D. melanogaster desat2 (GI: 24646295), C. hyperboreus ChDes9-1 (GI: 589834955), C. elegans FAT5 (GI: 544604099), delta-9 desaturase from Synechocystis sp. PCC 6803 (GI: 339274799), delta-9 desaturase from A. thaliana (GI: 18402641), and yeast OLE1 (GI: 1322552).
Extended Data Figure 2 Structural role of Arg249.
The conserved arginine residue Arg249, located on TM4 within the transmembrane region of the protein, forms a hydrogen bond with the carbonyl oxygen of Cys222 on TM3. This interaction may help stabilize the kink in TM3 caused by Pro226 on the following turn.
Extended Data Figure 3 Structure of the SCD1 cytoplasmic domain.
Four views of the cytoplasmic domain. The proposed amphipathic helices are coloured blue, while the other helices forming the cytoplasmic domain are green.
Extended Data Figure 4 The mouse SCD1 crystal lattice.
Cross-sections of the crystal lattice for the P212121 mouse SCD1 lipidic cubic phase crystals, viewed from two perpendicular directions. One asymmetric unit is coloured blue. Within an individual asymmetric unit, interactions between the two chains are mediated by residues from a C-terminal cloning artefact. All interactions with chains in neighbouring asymmetric units involve antiparallel orientations of the interacting monomers and have small interface areas.
Extended Data Figure 5 Electron density maps for acyl-CoA and the dimetal centre.
a, Stearoyl-CoA bound to SCD1 is superposed with the weighted 2Fo − Fc electron density contoured at 1.5σ (left) or Fo − Fc electron density calculated with the substrate molecule omitted and contoured at 2.3σ (right). b, Stereoview of the dimetal centre and coordinating histidines, shown with the weighted 2Fo − Fc density contoured at 2σ. c–e, The dimetal centre superposed with the anomalous difference map, contoured at 5σ (c), the Fo − Fc density calculated with the zinc atoms omitted, contoured at 3σ (d), and the Fo − Fc density calculated with the ordered water molecule between M1 and Asn261 omitted, contoured at 3σ (e).
Extended Data Figure 6 Western blot analysis of SCD expression.
a, b, Analysis of two separate yeast expression trials after introduction of mutations to mouse SCD3 to impart catalytic specificity of mouse SCD1. Contents of lanes are as indicated in the gel. The position of SCD is indicated by a black star. Additional bands are other proteins detected by the polyclonal antibody. Dotted line in a shows the portion of the complete gel image included in Fig. 2f; dotted line in b shows the corresponding expression trials from the second experiment. a, Expression trial 1, with gel artefacts in lanes 2 and 3. b, Expression trial 2, with gel artefacts in lanes 4, 6 and 7.
Extended Data Figure 7 Coordination in diiron-containing desaturases.
a, Stereoview of residues forming both the first and second coordination shell around the dimetal centre in mouse SCD1. b, Stereoview of the coordination of the dimetal centre in the stearoyl-acyl carrier protein desaturase from the castor bean (PDB accession 1AFR).
Extended Data Figure 8 The Thr257–Gln143 hydrogen bond blocks product egress.
The surface of the substrate tunnel housing the acyl chain is shown, with the structural elements AH1, H2 and TM4, and the hydrogen-bonded residues Thr257 and Gln143 highlighted in the inset. The proximity of these two residues creates the kinked shape of the substrate tunnel, and their separation would result in a larger opening capable of releasing the product into the bilayer.
Extended Data Figure 9 The SCD1 N terminus.
Two perpendicular views of mouse SCD1, from within the plane of the membrane and from the cytoplasmic side, showing the interaction between the N terminus (red ribbon) and the cytosolic domain (beige surface). The dashed yellow circles indicate the approximate location of the metal atoms.
Rights and permissions
About this article

Cite this article
Bai, Y., McCoy, J., Levin, E. et al. X-ray structure of a mammalian stearoyl-CoA desaturase. Nature 524, 252–256 (2015). https://doi.org/10.1038/nature14549
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14549
- Springer Nature Limited
This article is cited by
-
Integrated multi-omics analysis reveals variation in intramuscular fat among muscle locations of Qinchuan cattle
BMC Genomics (2023)
-
Lipid saturation controls nuclear envelope function
Nature Cell Biology (2023)
-
High-level production of nervonic acid in the oleaginous yeast Yarrowia lipolytica by systematic metabolic engineering
Communications Biology (2023)
-
The pyridazine heterocycle in molecular recognition and drug discovery
Medicinal Chemistry Research (2023)
-
Advances in regulation and function of stearoyl-CoA desaturase 1 in cancer, from bench to bed
Science China Life Sciences (2023)