
Abstract
Tyrosine kinase inhibitor (TKI) resistance and progression to blast crisis (BC), both related to persistent β-catenin activation, remain formidable challenges for chronic myeloid leukemia (CML). We observed overexpression of β-catenin in BC-CML stem/progenitor cells, particularly in granulocyte–macrophage progenitors, and highest among a novel CD34+CD38+CD123hiTim-3hi subset as determined by CyTOF analysis. Co-culture with mesenchymal stromal cells (MSCs) induced the expression of β-catenin and its target CD44 in CML cells. A novel Wnt/β-catenin signaling modulator, C82, and nilotinib synergistically killed KBM5T315I and TKI-resistant primary BC-CML cells with or without BCR–ABL kinase mutations even under leukemia/MSC co-culture conditions. Silencing of β-catenin by short interfering RNA restored sensitivity of primary BCR–ABLT315I/E255V BC-CML cells to nilotinib. Combining the C82 pro-drug, PRI-724, with nilotinib significantly prolonged the survival of NOD/SCID/IL2Rγ null mice injected with primary BCR–ABLT315I/E255V BC-CML cells. The combined treatment selectively targeted CML progenitors and inhibited CD44, c-Myc, survivin, p-CRKL and p-STAT5 expression. In addition, pretreating primary BC-CML cells with C82, or the combination, but not with nilotinib alone, significantly impaired their engraftment potential in NOD/SCID/IL2Rγ-null-3/GM/SF mice and significantly prolonged survival. Our data suggest potential benefit of concomitant β-catenin and Bcr–Abl inhibition to prevent or overcome Bcr–Abl kinase-dependent or -independent TKI resistance in BC-CML.
Similar content being viewed by others

Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasia, initiated by a reciprocal chromosomes 9 and 22 translocation resulting in the generation of a Bcr–Abl fusion protein.1, 2 The development of Bcr–Abl tyrosine kinase inhibitors (TKIs) is the most successful targeted cancer therapy to date.2, 3 Imatinib and other TKIs have revolutionized CML therapy and markedly improved treatment outcome for CML patients.2, 3, 4 Unfortunately, TKIs cannot eliminate the disease-initiating leukemic stem cell (LSC) in the bone marrow (BM) niche in majority of patients,2 supported by results from multiple prospective trials, including STIM, STOP 2G-TKI, ENESTFreedom and EURO-SKI, showing that 38–61% of patients who achieved and maintained a deep molecular response for at least 2 years after TKI discontinuation ultimately relapsed.5, 6 Furthermore, in the context of the development of TKI resistance and blast crisis (BC)-CML, formidable challenges remain.4, 5, 6, 7, 8 Approximately 10–15% of patients present beyond chronic phase and 7% of chronic-phase CML cases continue to progress to accelerated-phase or BC-CML even on TKI therapy. The frequency of transformation is recorded at 3–5% within the first few years of TKI therapy and drops to ~1% per year thereafter.8
The Wnt signaling cascade is known to be involved in virtually every aspect of development and homeostatic self-renewal of adult tissues. Its persistent activation is pivotal for tumorigenesis.9, 10, 11 Self-renewal is a key characteristic for both hematopoietic stem cells (HSCs) and LSCs.12, 13, 14, 15 Emerging evidence indicates that β-catenin, the canonical Wnt pathway’s central effector, is required for the development and maintenance of LSCs in both acute myeloid leukemia and CML.16, 17, 18, 19, 20, 21, 22, 23, 24, 25 Recent studies have demonstrated the impact of aberrant Wnt/β-catenin activity in the development of Bcr–Abl-induced myeloproliferative neoplasms in CML murine models.17, 18 Genetic and pharmacologic inhibition of β-catenin can target imatinib-resistant CML LSCs in mice. Importantly, β-catenin seems dispensable in fully developed adult HSCs.18 In BC-CML, activation of β-catenin in Lin−CD34+CD38+ granulocyte–macrophage progenitors (GMPs) can enhance their self-renewal capacity.21, 22, 23, 24, 25 Furthermore, anomalous Wnt/β-catenin signaling plays critical roles in Bcr–Abl kinase-independent TKI resistance and stromal-mediated microenvironmental protection.17, 21, 26, 27 Thus, activation of β-catenin pathway potentially represents a unique therapeutic target in BC-CML.
C82, an ICG-001-related compound but with higher specificity and potency, selectively binds to CREB-binding protein and prevents its interaction with β-catenin, thereby suppressing the expression of subsets of Wnt/β-catenin-driven genes that are required for cell proliferation and self-renewal.28, 29 We hypothesized that the combination of β-catenin and Bcr–Abl inhibition could serve as a novel approach to overcome TKI resistance and target BC-CML progenitors. We investigated, in TKI-resistant BC-CML, the activity of nilotinib alone and in combination with C82 in vitro, followed by investigation of combined C82 pro-drug PRI-724 with nilotinib in a murine CML xenograft model. Aided by a single-cell cytometry by time-of-flight (CyTOF) mass cytometry and subsequent data analysis with spanning-tree progression analysis of density-normalized events (SPADE),30, 31 we demonstrate that β-catenin inhibition reverses TKI resistance in BC-CML cells with or without BCR–ABL kinase domain mutations and synergizes with nilotinib in vitro and in vivo. Our data suggest that this combinatorial strategy may have broad clinical utility in the therapy of CML, particularly in TKI-resistant BC-CML.
Materials and methods
Cells, cell culture and treatments
K562 was purchased from American Type Culture Collection (Rockville, MD, USA). KBM5 and KBM5T315I were established from a BC-CML patient sample as previously described.32, 33 Primary samples were acquired from patients and healthy donors using Institutional Review Board (IRB)-approved protocols after informed consent according to institutional guidelines. All patients (Table 1) were resistant to TKIs. Patients (nos 1–4) harbored BCR–ABL kinase domain mutations, including three with T315I/E255K or T315I/E255V compound mutations, whereas the others (nos 5–8) did not. Mononuclear cells from these samples were cultured in α-minimum essential medium supplemented with 10% heat-inactivated fetal bovine serum, 2 μM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. BM-derived mesenchymal stromal cells (MSCs) obtained from healthy donors as described previously34, 35 were cultured (5000–6000 cells per cm2) in the same medium. Proliferating and quiescent progenitors were identified by staining patient mononuclear cells with 5-(and 6-) carboxy-fluorescein diacetate succinimidyl ester (CFSE) and cultured with human BM-derived MSCs as described previously.36, 37 Cells were treated with C82, nilotinib or both with or without MSC co-culture (leukemia:MSC=4:1).
CyTOF
Primary BC-CML cells with or without in vitro treatments and BM cells from NOD/SCID/IL2Rγ-null mice (NSG, Jackson Laboratories, Bar Harbor, ME, USA) xenografted with cells from a BCR–ABLT315I/E255V BC-CML patient after 4-week treatment were stained with metal-labeled antibodies against cell surface markers, followed with antibodies against intracellular proteins (Table 2) and subjected to CyTOF analysis as previously described38, 39 with a CyTOF 2 mass cytometer (Fluidigm, San Francisco, CA, USA). The bead signature was routinely applied to normalize the raw CyTOF data before analysis using a previously reported method.31, 39 Protein expression determined by SPADE quantification was expressed as nonlinear ArcSinh units transformed from raw CyTOF counts or as percentage to the control.
Knockdown of β-catenin expression by short interfering RNA
Primary BC-CML cells with BCR–ABLT315I/E255V mutations (no. 2, Table 1) were electroporated with 750 nM CTNNB ON-TARGET plus short interfering RNA (Dharmacon, Lafayette, CO, USA) using an Amaxa apparatus (Solution V, program K-17; Lonza, Walkersville, MD, USA), following the manufacturer’s instructions. Twenty-four hours after short interfering RNA transfection, cells (0.5 × 106/ml) were treated with nilotinib for 48 h. Apoptosis and cell counts were determined by flow cytometry as described below.
Western blot
Protein levels were determined by western blot analysis as previously described.37 Antibodies against β-catenin (#8480), CD44 (#5640) and p-CRKL (#3181) were purchased from Cell Signaling Technology (Danvers, MA, USA), and survivin (AF886) from R&D Systems (Minneapolis, MN, USA). β-Actin was used as a loading control. Proteins were visualized with Odyssey infrared imaging system and quantitated using the Odyssey software program (version 3.0; LI-COR Biotechnology, Lincoln, NE, USA).
Cell viability assay
Apoptosis was determined by flow cytometry in cell lines, CD45+ leukemia bulk, CD34+, CD34+CD38+, and CD34+CD38− BC-CML cells and CD34+ healthy donor BM cells after staining cells with annexin V (BD Biosciences, San Jose, CA, USA), and in CD34+ proliferating (CD34+CFSEdim) and quiescent (CD34+CFSEbright) BC-CML cells after annexin V staining in the presence of 7-amino-actinomycin D (Sigma-Aldrich, St Louis, MO, USA) using LSR II (BD Biosciences). For co-cultures, leukemia cells were collected by combining cells in the supernatant and after two washes with phosphate-buffered saline. Apoptotic cells were defined as annexin V-positive CD45+ cells. The extent of drug-induced specific apoptosis was assessed by the formula:

Viable cell counts were determined by flow cytometry in the presence of 7-amino-actinomycin D and CountBright absolute counting beads (ThermoFisher, Waltham, MA, USA).
In vivo study
In vivo experiments were conducted following the Institution Animal Care and Use Committee-approved protocols. Female NSG mice (8-week-old) were irradiated (250 cGy) and injected with cells (2 × 106 per mouse) from a primary BC-CML patient (no. 2, Table 1). After engraftment was confirmed by flow cytometry, mice were randomized and treated with vehicle control, PRI-724 (continuous administration, 30 mg/kg per day) by osmotic pump (ALZET, model 1004, Cupertino, CA, USA), nilotinib (oral gavage, 50 mg/kg per day), or the combination (n=11 per group). After a 4-week treatment, leukemia burden and progenitor cells were determined by flow cytometry of mouse peripheral blood. Three mice per group were killed. BM cells were obtained for CyTOF and spleen cells for western blot analysis. Survival was followed for the residual mice.
To evaluate treatments on BC-CML stem/progenitor cell function, female NOD/SCID/IL2Rγ-null-3/GM/SF (7–9-week-old; NSGS, Jackson Laboratories) mice were irradiated (250 cGy), randomized based on age, and injected via tail vein with 2 × 106 cells from a BCR–ABLT315/E255V BC-CML patient sample (no. 2, Table 1) untreated or after ex vivo treatment with C82 (1.25 μM), nilotinib (0.5 μM) or both for 48 h (n=7 per group). Leukemia engraftment and BC-CML stem/progenitor cells were assessed by flow cytometry 4–5 weeks after transplant and survival was monitored.
Statistical analyses
Experiments using cell lines were conducted in triplicates. Results were expressed as the mean±s.e.m. Student’s t-tests were performed using GraphPad Prism version 7.01 (GraphPad Software, Inc., La Jolla, CA, USA). All tests were two-sided. Statistical significance was set at P<0.05. The combination index (CI), based on the Chou–Talalay method40 and determined by CalcuSyn software (BIOSOFT, Cambridge, UK), was expressed as the mean of the CI values obtained at the effective doses of 50, 75 and 90%. CI<1 was considered synergistic, CI=1 additive and CI>1 antagonistic. CyTOF data were analyzed and quantified by SPADE.38, 39 Mouse survival data were analyzed using the log-rank test. Sample sizes for mouse experiments were sufficient for statistical analyses.
Results
β-Catenin is overexpressed in primary BC-CML progenitors and induced by BM MSCs
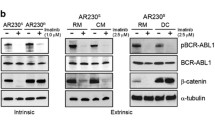
To characterize β-catenin expression and survival/proliferative signaling pathways in phenotypically defined BC-CML progenitors, cells from BC-CML patients (nos 1, 2 and 7; Table 1) were stained with metal-tagged antibodies for myeloid cell surface markers and intracellular proteins (Table 2) and subjected to CyTOF analysis with data interpretation by SPADE. Phenotypically defined myeloid cell populations were identified and the expression of proteins of interest was determined (Figure 1a). Notably, Tim-3 was highly expressed in a small subset of CD34+CD38+CD123hi progenitors (CD34+CD38+CD123hiTim-3hi) (Figure 1a, boxed). SPADE quantification analysis showed overexpression of β-catenin in BC-CML CD34+ (P=0.0316), CD34+CD38+ (P=0.0041) and CD34+CD38− (P=0.0383) progenitors compared to CD45+ bulk leukemia (Figure 1b), consistent with previous reports.21, 22, 23, 24, 25 Interestingly, among CD34+CD38+ GMP BC-CML progenitors, β-catenin expression was even higher in the CD34+CD38+CD123hi subset (P=0.0432) and highest in CD34+CD38+CD123hiTim-3hi (Tim-3hiGMP) BC-CML stem/progenitor cells (P=0.009; Figure 1b). High β-catenin-expressing cells also expressed high levels of CD44 and c-Myc, known β-catenin targets. High expression of β-catenin in BC-CML progenitors was associated with high expression of p-CRKL, p-STAT5, p-STAT3, p-Tyr and p-AKT (Figure 1b), suggesting high Bcr–Abl signaling in these cells. To mimic the BM microenvironment, we co-cultured BC-CML cells with MSCs. Co-culture induced β-catenin and CD44 expression in the leukemia cells (Figure 1c). These data suggest that β-catenin is overexpressed in BC-CML stem/progenitor cells, and can be induced by BM stromal cells.

BC-CML stem/progenitor cells overexpress β-catenin and survival/proliferating signaling molecules, and β-catenin expression is induced by MSCs. (a) SPADE tree map, generated using all surface markers of a representative BC-CML sample (no. 2, Table 1) showing cell populations, and the expression of β-catenin and survival/proliferating signaling proteins in various cell populations. The boxed notes were identified by the expression of CD34, CD38, CD123 and Tim-3 markers. ArcSinh-transformed counts for each protein were exported and quantified in each cell population. (b) Left: BC-CML populations and stem/progenitor subsets (CD45+, CD34+, CD34+CD38+, CD34+CD38−, CD34+CD38+CD123hi and CD34+CD38+CD123hiTim-3hi) were located in SPADE tree according to its colored versions based on (bars highlighted by by red boxes) surface markers. Right: SPADE quantification analysis demonstrating that CD34+CD38+CD123hiTim-3hi BC-CML stem/progenitor subset has the highest expression of β-catenin and proteins for survival/proliferating pathways, including CD44, c-Myc, p-CRKL, p-STAT5, p-STAT3, p-Tyr and p-AKT. Data are presented as ArcSinh expression (n=3; nos 1, 2 and 7; Table 1). (c) Western blot showing that MSC co-culture induced β-catenin and CD44 expression in KBM5T315I cells.
β-Catenin contributes to TKI resistance in BCR–ABLT315I BC-CML cells
To assess the effect of β-catenin inhibition in BC-CML, we first treated CML cell lines KBM5, KBM5T315I and K562 with C82, nilotinib and both. The combination synergistically induced apoptosis and reduced viable cells not only in TKI-sensitive KBM5 and K562, but also TKI-resistant KBM5T315I cells, regardless of MSC co-culture (Supplementary Figure 1a) and decreased β-catenin targets CD44 and survivin, and Bcr–Abl target p-CRKL (Supplementary Figure 1b). It was reported that β-catenin is required for Bcr–Abl-independent TKI resistance.21, 26, 41 These data suggested that β-catenin also contributed to Bcr–Abl-dependent TKI resistance in CML.
To further investigate the role of β-catenin in TKI resistance, especially in BC-CML cells with mutations in the BCR–ABL gene, we knocked down β-catenin in primary BCR–ABLT315I/E255V BC-CML cells (no. 2, Table 1). After 24 h short interfering RNA transfection, cells were treated with nilotinib for 48 h. Interestingly, β-catenin knockdown significantly re-sensitized primary BCR–ABLT315I/E255V BC-CML cells to nilotinib regardless of MSC co-culture, not only in CD45+ leukemia bulk cells but also in CD34+CD38+ and CD34+CD38− cell populations (Figure 2), implicating that β-catenin contributes, at least in part, to Bcr–Abl-dependent TKI resistance in BC-CML cells.

Inhibition of β-catenin with short interfering RNA (siRNA) restores sensitivity of BCR–ABLT315I BC-CML primary cells to nilotinib in vitro. β-Catenin siRNA silencing significantly increased sensitivity of TKI-resistant BCR–ABLT315I/E255V BC-CML CD45+ cells, CD34+CD38+ and CD34+CD38− subsets to nilotinib (***P<0.001). Western blot confirmed β-catenin knockdown by siRNA at 24 h. Solid lines, without MSC (alone); dashed line, with MSC co-culture (cocox). NIL, nilotinib.
Co-inhibition of β-catenin and Bcr–Abl synergistically targets TKI-resistant BC-CML progenitors, including CD34+ quiescent progenitors in vitro
We next determined the effects of C82, nilotinib and the combination in samples from extensively treated and resistant primary BC-CML patients (Table 1). The combination synergistically induced apoptosis not only in CD45+ bulk leukemia cells but also in CD34+CD38− and CD34+CD38+ stem/progenitor subsets (Figure 3a, n=5). This was also observed in the CD34+CFSEdim-proliferating population, and more importantly in the CD34+CFSEbright quiescent population (Figure 3b, n=6). The synergy occurred in TKI-resistant BC-CML cells without or with BCR–ABL kinase domain mutations (Figures 3a and b, Table 1). These data indicated that β-catenin inhibition could reverse both BCR–ABL kinase-dependent and -independent TKI resistance. Similar results were obtained when cells were co-cultured with MSCs (Figures 3a and b), implying that the combination could overcome MSC-mediated CML microenvironmental protection. No cytotoxic effect was observed in normal CD34+ progenitor cells exposed to the same agents (Supplementary Figure 2).

Combination of C82 and nilotinib synergistically decreases viability of TKI-resistant BC-CML cells with or without BCR–ABL kinase mutations in vitro. Cells from primary TKI-resistant BC-CML patients were treated with C82, nilotinib or both for 48 h with or without MSC co-culture. The combination synergistically decreased viability in (a) leukemia bulk and stem/progenitor subsets (n=5), and (b) primitive proliferating (CD34+CFSEdim) and quiescent stem/progenitor cells (CD34+CFSEbright; n=6). (c) CyTOF/SPADE analysis showing that the combination significantly eliminated BC-CML stem/progenitor subsets (n=3, Table 1). CON, control; COM, combination; M, million.
To determine responses in phenotypically defined subpopulations, we treated primary BC-CML samples (n=3, Table 1) with C82, nilotinib or the combination for 48 h, stained cells with the same panel of antibodies for cell surface markers as described above (Table 2), and performed CyTOF/SPADE analysis. As expected, nilotinib alone had no effect. C82 alone inhibited various subpopulations and the combination significantly decreased various BC-CML progenitor subpopulations, including the β-catenin highest CD34+CD38+CD123hiTim-3hi subset compared to control or nilotinib alone (Figure 3c). The combination reduced CD44, p-CRKL and survivin expression in these samples (Supplementary Figure 3). These results indicated that co-inhibition of β-catenin and Bcr–Abl could synergistically target TKI-resistant BC-CML stem/progenitor cells.
Combined inhibition of β-catenin and Bcr–Abl synergistically targets TKI-resistant BC-CML progenitors in vivo and prolongs survival of BC-CML-bearing NSG mice
We next examined the effect of PRI-724, a C82 pro-drug, and nilotinib combination in NSG mice engrafted with cells from a BCR–ABLT315I/E255V BC-CML patient sample (no. 2, Table 1; Figure 4a). We collected BM cells (n=3 per group) after 4-week treatment and performed CyTOF/SPADE analysis. We discovered that while nilotinib alone was inactive, PRI-724 and its combination with nilotinib significantly decreased various BC-CML stem/progenitor subsets, particularly CD34+CD38+CD123hi and most effectively Tim-3hi GMPs (Figure 4b), which were further confirmed by conventional flow cytometry in blood samples (Supplementary Figure 4), indicating that the combination also synergistically targeted BC-CML progenitor cells with mutations in BCR–ABL gene in vivo. As shown in Figure 4c, PRI-724 alone (median, 38.5 days) significantly improved the overall survival of the leukemia-bearing NSG mice compared to controls (median 34.5 days, P=0.0281). The combination further prolonged survival (median, 43.5 days) compared to PRI-724 (P=0.0368), nilotinib (median, 35.5 days; P=0.0044) and control (P=0.0009) groups. No apparent drug-related toxicities were observed in mice with single agent or combination treatment.

Co-inhibition of β-catenin by PRI-724 and Bcr–Abl by nilotinib targets BC-CML stem/progenitor cells and significantly improves survival of BC-CML-bearing NSG mice. (a) Schematic diagram of the in vivo study. Engraftment was confirmed by flow cytometry and defined as huCD45+ cells >1% in mouse blood (n=11 per group). (b) CyTOF/SPADE analysis revealed that 4-week combination treatment significantly reduced BC-CML stem/progenitor subsets in mouse BM (n=3). (c) Survival curves (n=8 per group). PRI, PRI-724; hu, human.
Combination of PRI-724 and nilotinib inhibits CD44, c-Myc, p-CRKL, p-STAT5 and survivin expression in BC-CML progenitors in vivo
To further identify mechanisms of synergy for the PRI-724 and nilotinib combination in BC-CML stem/progenitor cells, we determined the expression of CD44 and intracellular signaling proteins by CyTOF in various cell populations of BM cells from the treated NSG mice. SPADE analysis demonstrated that PRI-724 alone and its combination with nilotinib significantly decreased the expression of β-catenin targets, CD44 and c-Myc in BM BC-CML progenitor subsets after 4 weeks in vivo treatment (Figure 5a). CD44 is well known to play a critical role in mediating LSC-BM niche interactions.42, 43, 44 Furthermore, PRI-724 and its combination with nilotinib decreased p-CRKL and p-STAT5 levels in high β-catenin-expressing stem/progenitor subsets (Figure 5b) and significantly inhibited survivin expression in spleen cells obtained from the treated NSG mice (Figure 5c), implicating potential roles of these proteins in TKI resistance.

Combination of PRI-724 and nilotinib inhibits the expression of CD44, c-Myc, p-STAT5 and p-CRKL in BC-CML stem/progenitor cells and survivin in BC-CML cells in vivo. PRI-724 and the combination reduced the expression of CD44 and c-Myc (a), and p-CRKL and p-STAT5 (b) in various BC-CML stem/progenitor subsets in BM cells by CyTOF/SPADE analysis and survivin by western blot (the right panel, quantification) in spleen cells (c) from human BC-CML xenograft NSG mice after 4 weeks in vivo treatment (n=3 per group). *P<0.05, **P<0.01 and ***P<0.001.
Prior exposure to C82 or the combination with nilotinib impairs engraftment potential of BC-CML cells and prolongs survival in NSGS mice
After ex vivo treatment with C82, nilotinib or both for 48 h, BC-CML patient cells from untreated and each treatment group were injected into NSGS mice. Although not affected by nilotinib as expected, engraftment abilities of C82- and the combination-treated cells were significantly abolished (4 weeks after transplant, Figure 6a). Furthermore, mice transplanted with C82- or the combination-treated cells had significantly less BC-CML stem/progenitor subsets, including CD34+CD38+CD123+ cells expressing Tim-3 (5 weeks after transplant, Figure 6b). Importantly, mice transplanted with C82-treated cells survived markedly longer (median, 47 days) than with nilotinib-treated cells (median, 41 days; **P=0.0028) and the untreated controls (median, 41 days; **P=0.0054), and mice injected with the combination-treated cells had significantly longer survival (median, 63 days) than with C82-treated cells (*P=0.026; Figure 6c). Collectively, these data indicate that inhibition of β-catenin targets BC-CML stem/progenitor cells and impairs leukemia stem cell function, and this effect is further enhanced by TKIs.

Pretreatment with C82 or the combination significantly impairs BC-CML cell engraftment potential and prolongs mouse survival. BC-CML cells (no. 2, Table 1) were untreated, treated with nilotinib, C82 or both for 48 h, then injected into irradiated NSGS mice. Flow cytometry analysis of (a) engraftment (4 weeks) and (b) BC-CML cell populations (5 weeks) after transplant. (c) Survival curves.
Discussion
TKI resistance and progression to BC are major challenges in CML therapy in the TKI era.2, 3, 4, 5, 6, 7, 8 Although the mechanism is not well understood, the self-renewal capacity of LSC has been implicated.21, 22, 45, 46, 47 β-Catenin is involved in several critical aspects of CML biology, including promoting disease progression, contributing to BCR–ABL kinase-independent resistance and LSC self-renewal.15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 41
TKI resistance consists of Bcr–Abl kinase-dependent and -independent mechanisms in CML.2, 5, 6, 41 Bcr–Abl kinase-independent resistance may develop intrinsically by the activation of alternative signaling pathways or through extrinsic BM microenvironmental factors.2, 25, 26, 41 Bcr–Abl-dependent resistance occurs through point mutations in BCR–ABL kinase domain that could impair or even totally abrogate TKI binding, thus allow the reactivation of kinase activity.2, 41, 46 Using a stochastic mathematical model, scientists presumed that progenitors, which acquire a mutation conferring self-renewal, are the most likely cells of origin of myeloid malignancies.48 Furthermore, it was reported that T315I-mutated BCR–ABL is functional at the stem cell level, conferring leukemic cells self-renewal capacity in murine embryonic stem cell model.49 As a critical signaling in the regulation of LSC self-renewal, β-catenin is required for Bcr–Abl-independent TKI resistance.26, 41 However, its role in BCR–ABL gene mutation-mediated TKI resistance is not yet understood.
We investigated the effect of combined inhibition of β-catenin and Bcr–Abl on BC-CML cells. Synergy was observed in TKI sensitive, surprisingly, also in TKI-resistant KBM5T315I cells. This effect was further confirmed by β-catenin silencing in TKI-resistant primary BCR–ABLT315I/E255V cells and by combination treatment of primary TKI-resistant BC-CML cells with BCR–ABL kinase domain mutations in vitro and in vivo. These data provide evidence that β-catenin inhibition restores sensitivity to nilotinib in otherwise TKI-resistant BCR–ABL gene-mutated CML cells. We also show, for the first time, that β-catenin contributes to BCR–ABL kinase-dependent TKI resistance. The mechanism of this regulation is currently unclear, and the regulation between β-catenin and Bcr–Abl remains controversial. Reduced Bcr–Abl protein levels were found in β-catenin-deficient CML mice, implicating β-catenin upstream of BCR–ABL.17 Conversely, Bcr–Abl was reported to regulate β-catenin in CML by phosphorylating and stabilizing it.50 We found that inhibition of β-catenin and more so the combination reduced Bcr–Abl targets p-CRKL and p-STAT5 expression and synergistically induced apoptosis in BCR–ABLT315I/E255V BC-CML, suggesting that β-catenin can regulate the kinase function. Attempt to measure Bcr–Abl protein level in β-catenin knockdown cells failed due to very low basal Bcr–Abl level in the cells.
The activation of β-catenin was reported to represent a ‘second hit’ for CML GMPs that confers the regaining of self-renewal capacity during disease progression to BC.20, 21, 22, 23, 24, 25, 26, 51, 52, 53, 54 Aided by CyTOF, a single-cell proteomics system that has been increasingly applied to analyze cell signaling and phenotypic heterogeneity of leukemia,30, 31, 39, 55 we demonstrated overexpression of β-catenin in BC-CML CD34+ progenitors, consistent with previous reports.22, 23 Furthermore, we identified a novel CD34+CD38+CD123hiTim-3hi/Tim-3hi GMP subset that expresses the highest level of β-catenin and intracellular survival and proliferative signaling proteins. As a newly-recognized LSC surface marker, Tim-3 was reported to drive acute myeloid leukemia LSC self-renewal and leukemic progression via a Tim-3/Galectin-9 autocrine stimulatory loop.56, 57 The role of Tim-3 in CML LSC has not been well investigated. We suggest that Tim-3 is an important factor also in BC-CML LSC function, and that it might play a critical role in CML disease progression via activation of β-catenin in LSC and/or mediating cross-talk between LSC and BM microenvironment, such as through the Tim-3/Galectin-9 and Tim-3/HMGB1 signaling pathways,58 which can be further evaluated in the future with the development of Tim-3 specific antibodies/inhibitors.
Using the second-generation β-catenin/CREB-binding protein antagonist, C82, we demonstrated that the inhibition of β-catenin could target β-cateninhigh progenitor subsets, and importantly, remarkably impaired the engraftment potential of BC-CML stem/progenitor cells. The treatment spared normal human CD34+ cells, consistent with previous report.59 Compared to the genetic or pharmacologic inhibition of β-catenin by cyclooxygenase inhibitors in chronic-phase CML mice models,17, 18 PRI-724 has higher specificity and potency and has entered clinical trials for advanced myeloid leukemias (NCT01606579). PRI-724 is well tolerated in the clinic. Although it has limited single agent activity, our studies provide evidence that the combination with TKIs could be explored to test whether the earlier progenitors may be eliminated in CML patients, or whether it can improve outcome for patients in advanced stages. There is little overlapping toxicity of TKIs with PRI-724, which may make these combinations feasible. Particularly, our data strongly support that combined inhibition of β-catenin and Bcr–Abl synergistically targets TKI-resistant BC-CML progenitors in vitro and in vivo.
Collectively, we demonstrate that combined inhibition of β-catenin and Bcr–Abl tyrosine kinase overcomes Bcr–Abl-dependent and -independent TKI resistance, targets BC-CML progenitors and BM niche components, and impairs engraftment potential of LSC. Thus, the aforementioned combination represents a novel strategy for treating TKI-resistant CML and improving clinical outcomes of BC-CML patients and warrants clinical evaluation.
Data availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information files.
References
De Klein A, van Kessel AG, Grosveld G, Bartram CR, Hagemeijer A, Bootsma D et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature 1982; 300: 765–767.
Apperley JF . Chronic myeloid leukaemia. Lancet 2015; 385: 1447–1459.
Goldman JM, Melo JV . Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001; 344: 1084–1086.
Hochhaus A, Kantarjian HM, Baccarani M, Lipton JH, Apperley JF, Druker BJ et al. Dasatinib induces notable hematologic and cytogenetic responses in chronic-phase chronic myeloid leukemia after failure of imatinib therapy. Blood 2007; 109: 2303–2309.
Mahon FX, Réa D, Guilhot J, Guilhot F, Huguet F, Nicolini F et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010; 11: 1029–1035.
Saußele S, Richter J, Hochhaus A, Mahon FX . The concept of treatment-free remission in chronic myeloid leukemia. Leukemia 2016; 30: 1638–1647.
Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355: 2408–2417.
Schemionek M, Elling C, Steidl U, Bäumer N, Hamilton A, Spieker T et al. BCR-ABL enhances differentiation of long-term repopulating hematopoietic stem cells. Blood 2010; 115: 3185–3195.
Clevers H . Wnt/beta-catenin signaling in development and disease. Cell 2006; 127: 469–480.
MacDonald BT, Tamai K, He X . Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17: 9–26.
Moon RT, Bowerman B, Boutros M, Perrimon N . The promise and perils of Wnt signaling through beta-catenin. Science 2002; 296: 1644–1646.
Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003; 423: 409–414.
Scheller M, Huelsken J, Rosenbauer F, Taketo MM, Birchmeier W, Tenen DG et al. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol 2006; 7: 1037–1047.
Jeannet G, Scheller M, Scarpellino L, Duboux S, Gardiol N, Back J et al. Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood 2008; 111: 142–149.
Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton IG et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci USA 2009; 106: 3925–3929.
Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010; 327: 1650–1653.
Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007; 12: 528–541.
Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA, Stein L et al. Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell 2012; 10: 412–424.
Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012; 119: 1501–1510.
Kavalerchik E, Goff D, Jamieson CH . Chronic myeloid leukemia stem cells. J Clin Oncol 2008; 26: 2911–2915.
Perrotti D, Jamieson C, Goldman J, Skorski T . Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest 2010; 120: 2254–2264.
Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 2004; 351: 657–667.
Minami Y, Stuart SA, Ikawa T, Jiang Y, Banno A, Hunton IC et al. BCR-ABL-transformed GMP as myeloid leukemic stem cells. Proc Natl Acad Sci USA 2008; 105: 17967–17972.
Giotopoulos G, van der Weyden L, Osaki H, Rust AG, Gallipoli P, Meduri E et al. A novel mouse model identifies cooperating mutations and therapeutic targets critical for chronic myeloid leukemia progression. J Exp Med 2015; 212: 1551–1569.
Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest 2013; 123: 4144–4157.
Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 2013; 121: 1824–1838.
Kleppe M, Levine RL . Targeting beta-catenin in CML: leukemia stem cells beware!. Cell Stem Cell 2012; 10: 351–353.
El-Khoueiry AB, Ning Y, Yang D, Cole S, Kahn M, Zoghbi M et al. A phase I first-in-human study of PRI-724 in patients with advanced solid tumors. J Clin Oncol 2013; 31 (Suppl); Abstract 2501).
Eguchi M, Nguyen C, Lee SC, Kahn M . ICG-001, a novel small molecule regulator of TCF/beta-catenin transcription. Med Chem 2005; 1: 467–472.
Bendall SC, Simonds EF, Qiu P, Amir el-AD, Krutzik PO, Finck R et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011; 332: 687–696.
Qiu P, Simonds EF, Bendall SC, Gibbs Jr KD, Bruggner RV, Linderman MD et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol 2011; 29: 886–891.
Beran M, Pisa P, O’Brien S, Kurzrock R, Siciliano M, Cork A et al. Biological properties and growth in SCID mice of a new myelogenous leukemia cell line (KBM-5) derived from chronic myelogenous leukemia cells in the blastic phase. Cancer Res 1993; 53: 3603–3610.
Ricci C, Scappini B, Divoky V, Gatto S, Onida F, Verstovsek S et al. Mutation in the ATP-binding pocket of the ABL kinase domain in an STI571-resistant BCR/ABL-positive cell line. Cancer Res 2002; 62: 5995–5998.
Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M . Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res 2002; 62: 3603–3608.
Konopleva M, Konoplev S, Hu W, Zaritskey AY, Afanasiev BV, Andreeff M . Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 2002; 16: 1713–1724.
Holtz MS, Slovak ML, Zhang F, Sawyers CL, Forman SJ, Bhatia R . Imatinib mesylate (STI571) inhibits growth of primitive malignant progenitors in chronic myelogenous leukemia through reversal of abnormally increased proliferation. Blood 2002; 99: 3792–3800.
Mak DH, Schober WD, Chen W, Konopleva M, Cortes J, Kantarjian HM et al. Triptolide induces cell death independent of cellular responses to imatinib in blast crisis chronic myelogenous leukemia cells including quiescent CD34+ primitive progenitor cells. Mol Cancer Ther 2009; 8: 2509–2516.
Han L, Qiu P, Zeng Z, Jorgensen JL, Mak DH, Burks JK et al. Single-cell mass cytometry reveals intracellular survival/proliferative signaling in FLT3-ITD-mutated AML stem/progenitor cells. Cytometry A 2015; 87: 346–356.
Carter BZ, Mak PY, Mu H, Zhou HS, Mak DH, Schober W et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci Transl Med 2016; 8: 355ra117.
Chou TC, Talalay P . Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55.
Eiring AM, Khorashad JS, Anderson DJ, Yu F, Redwine HM, Mason CC et al. β-Catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia 2015; 29: 2328–2337.
Krause DS, Lazarides K, von Andrian UH, Van Etten RA . Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med 2006; 12: 1175–1180.
Holm FL, Hellqvist E, Mason C . The niche specific role of CD44 splice isoform expression in blast crisis leukemia stem cell generation. Cancer Res 2014; 74 (19 Suppl); Abstract 4798).
Konopleva MY, Jordan CT . Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol 2011; 29: 591–599.
Carter BZ, Mak DH, Cortes J, Andreeff M . The elusive chronic myeloid leukemia stem cell: does it matter and how do we eliminate it? Semin Hematol 2010; 47: 362–370.
Burchert A . Roots of imatinib resistance: a question of self-renewal? Drug Resist Updat 2007; 10: 152–161.
Savona M, Talpaz M . Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer 2008; 8: 341–350.
Haeno H, Levine RL, Gilliland DG, Michor F . A progenitor cell origin of myeloid malignancies. Proc Natl Acad Sci USA 2009; 106: 16616–16621.
Melkus M, Bennaceur-Griscelli A, Valogne Y, Flamant S, Chomel JC, Sorel N et al. Biological effects of T315I-mutated BCR-ABL in an embryonic stem cell-derived hematopoiesis model. Exp Hematol 2013; 4: 335–345.
Coluccia AM, Vacca A, Duñach M, Mologni L, Redaelli S, Bustos VH et al. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J 2007; 26: 1456–1466.
McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, Montalto G et al. Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia 2014; 28: 15–33.
Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010; 464: 852–857.
Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 2014; 506: 240–244.
Eaves CJ, Humphries RK . Acute myeloid leukemia and the Wnt pathway. N Engl J Med 2010; 362: 2326–2327.
Ornatsky O, Bandura D, Baranov V, Nitz M, Winnik MA, Tanner S . Highly multiparametric analysis by mass cytometry. J Immunol Methods 2010; 361: 1–20.
Kikushige Y, Yuda J, Shima T, Miyamoto T, Akashi K . TIM-3 and its ligand, Galectin-9 constitute a pan-myeloid autocrine loop to develop and maintain malignant stem cells in most human myeloid leukemia. Blood 2014; 124: 835.
Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S et al. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell 2015; 17: 341–352.
Tang D, Lotze MT . Tumor immunity times out: TIM-3 and HMGB1. Nat Immunol 2012; 13: 808–810.
Kabiri Z, Numata A, Kawasaki A, Edison, Tenen DG, Virshup DM . Wnts are dispensable for differentiation and self-renewal of adult murine hematopoietic stem cells. Blood 2015; 126: 1086–1094.
Acknowledgements
We thank Drs Michael Kahn for helping with technical questions and Numsen Hail for editing the manuscript. This work was supported in part by the research funding from PRISM Pharma/Eisai (to BZC) and grants from the National Institutes of Health (P01CA49639 and MD Anderson’s Cancer Center Support Grant CA016672), Cancer Prevention Research Institute of Texas (CPRIT, RP121010), MDACC moonshot program, and the Paul and Mary Haas Chair in Genetics (to MA) and grant from Science and Technology Planning Project of Guangdong Province, China (2016A020215112 to HSZ).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhou, H., Mak, P., Mu, H. et al. Combined inhibition of β-catenin and Bcr–Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 31, 2065–2074 (2017). https://doi.org/10.1038/leu.2017.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.87
- Springer Nature Limited
This article is cited by
-
circCRKL, a circRNA derived from CRKL, regulates BCR-ABL via sponging miR-877-5p to promote chronic myeloid leukemia cell proliferation
Journal of Translational Medicine (2022)
-
Chronic myeloid leukemia stem cells: targeting therapeutic implications
Stem Cell Research & Therapy (2021)
-
Third-line therapy for chronic myeloid leukemia: current status and future directions
Journal of Hematology & Oncology (2021)
-
Depleting long noncoding RNA HOTAIR attenuates chronic myelocytic leukemia progression by binding to DNA methyltransferase 1 and inhibiting PTEN gene promoter methylation
Cell Death & Disease (2021)
-
Overcoming Wnt–β-catenin dependent anticancer therapy resistance in leukaemia stem cells
Nature Cell Biology (2020)