
Abstract
The nuclear factor κB (NFκB) transcription factor plays critical roles in inflammation and immunity. The dysregulation of NFκB is associated with inflammatory and autoimmune diseases and cancer. NFκB activation is negatively regulated by the ubiquitin-dependent proteasomal degradation pathway. In the present review, we discuss recent advances in our understanding of how ubiquitin ligases regulate the NFκB degradation pathway.
Similar content being viewed by others


Main
Nuclear factor κB (NFκB) was identified approximately 20 years ago by Dr David Baltimore as a transcription factor that binds to the intronic enhancer of the kappa light chain gene (the κB site) in B cells.1 The NFκB family is composed of several members including p50, p52, p65/RelA, c-Rel and RelB and plays a central role in cell growth, inflammation, immunity and apoptosis. NFκB activation is dependent on the stability of the inhibitor IκBα. IκBα stabilizes the NFκB complex so that after its degradation the remaining subunits translocate from the cytoplasm to the nucleus.2 The phosphorylation of IκBα is catalyzed by IκB kinase (IKK), a complex composed of three subunits: IKKα/IKK1, IKKβ/IKK2, and IKKγ/NEMO. IKK1 and IKK2 are the catalytic subunits, whereas IKKγ serves as a non-enzymatic regulator.3,4,5 The IKKs/NFκB pathway is activated by extracellular stimuli such as cytokines, ultraviolet irradiation, free radicals, bacterial or viral antigens and oxidized low density lipoprotein.3,4 In the absence of persistent upstream stimuli, NFκB transcriptional activity is terminated by the NFκB/IκBα negative feedback loop.4
The activation of NFκB in immune or stromal cells causes a pro-inflammatory response, and persistent tissue inflammation has been linked to inflammation-associated cancer.6 NFκB activation is negatively regulated by ubiquitin-dependent proteasomal degradation.7,8,9 The ubiquitin-proteasome system comprises a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2) and a ubiquitin ligase (E3). Ubiquitin E3 ligases play a critical role in substrate recognition and polyubiquitination by recruiting E2 ubiquitin-conjugating enzymes to specific substrates.10 SOCS-1 (suppressor of cytokine signaling) is one of the components of the EC2S (Elongin BC-CUL2-SOCS-box protein) ubiquitin ligase complex that mediates JAK2 (Janus kinase 2) ubiquitination and degradation.11,12 Similar to JAK2, NFκB/p65 is also a substrate of the EC2S ubiquitin ligase complex in both mice and cancer cells.13 Pin1 increases NFκB/p65 stability and transactivation, while the loss of Pin1 leads to SOCS-1-mediated p65 ubiquitination and degradation.13 This offers a new mechanism for NFκB-mediated pathogenesis.
NFκB is also activated by viral infections. In immunodeficiency virus-1-infected CD4+ lymphocytes, NFκB activation is terminated by COMMD1 (MURR1), resulting in the inhibition of immunodeficiency virus-1 growth in unstimulated or cytokine-stimulated CD4+ T cells.14 This study demonstrated that COMMD1 decreases NFκB transcriptional activity in T cells by inducing the ubiquitin-dependent proteasomal degradation of p65, but did not identify the mechanism of p65 degradation.14 Marine et al.15 found that COMMD1 promoted the ubiquitination and degradation of nuclear NFκB/p65 in cancer cell lines through its interaction with the EC2S multisubunit ubiquitin ligase complex. As a component of the EC2S ubiquitin ligase complex, COMMD1 serves as a cofactor of the EC2S ubiquitin ligase complex and promotes the degradation of nuclear p65.15 The main controversy is that while COMMD1 is predominantly localized to the cytoplasm, it induces the ubiquitination and degradation of nuclear p65. It is unknown whether COMMD1 can affect the stability of cytoplasmic p65 protein. In addition, it is unclear whether COMMD1 induces NFκB/p65 degradation in CD4+ T cells by the same EC2S multisubunit ubiquitin ligase. Therefore, multiple issues remain to be resolved.
Interestingly, the histone acetyltransferase GCN5 serves as a cofactor for COMMD1 to promote NFκB/p65 ubiquitination and degradation in cancer cell lines, independent of its enzymatic activity.16 GCN5 induces p65 ubiquitination depending on the phosphorylation state of the Ser 468 residue of p65.16 The interaction between GCN5, COMMD1 and the other components of the EC2S ubiquitin ligase promotes NFκB/p65 degradation.16 These observations leave some unanswered questions: do other GCN5 cofactors induce NFκB/p65 degradation by the GCN5/COMMD1/EC2S complex; and do any other proteins hijack the EC2S ubiquitin ligase to induce p65 ubiquitination and degradation?
NFκB plays a crucial role in the innate immune response against microbial and viral infections. The inhibition of NFκB activation has been observed in various viral infections. The lymphotropic gammaherpesvirus MuHV-4 (murid herpesvirus-4) ORF73 protein inhibits host NFκB transcriptional activity by inducing NFκB/p65 ubiquitination and degradation.17 The SOCS-box motif of ORF73 acts by forming one component of the EC5S (ElonginC/Cullin5/SOCS) ubiquitin ligase complex. The genetic deletion of the SOCS-box of ORF73 suppresses MuHV-4 expansion in germinal center B cells and prevents persistent MuHV-4 infection in mice.17 This study suggests that the virus escapes host immune surveillance by using the ubiquitin-dependent proteasomal degradation pathway. In addition to degrading NFκB/p65 via the EC2/5S ubiquitin ligase complex, PDLIM2 alone can act as a ubiquitin ligase to induce the ubiquitination and degradation of nuclear p65 in T cells and macrophages.18 PDLIM2 is associated with several malignancies including breast cancer and adult T-cell leukemia. As a nuclear protein, PDLIM2 contains a LIM domain similar to those of the RING family of ubiquitin ligases.18 However, there is no direct evidence demonstrating PDLIM2 ubiquitin ligase activity in in vitro ubiquitination assays.18 As stated above, COMMD1 and PDLIM2 induce the ubiquitination and degradation of nuclear p65, although it is unknown whether they act in the same cell types and in response to the same stimuli.
In the ubiquitin–proteasome pathway, ubiquitin is attached to the lysine residue of a substrate. The ubiquitin-tagged proteins are recognized and degraded by the proteasome pathway. Although PDLIM2 and COMMD1 induce p65 ubiquitination and degradation, the identity of the ubiquitin-modified lysine residue of p65 is still unclear.14,18 Saccani et al.7 were the first to identify that NFκB/p65 undergoes ubiquitination and degradation in cancer cells in response to TNF-α. Consistent with these findings, Fan et al.19 found that the lysine-195 residue of p65 was critical for TNF-α-induced ubiquitination by an unknown ubiquitin E3 ligase. Because GCN5 and COMMD1 can induce p65 degradation in response to TNF-α, the possibility that they are responsible for the TNF-α-mediated p65 ubiquitination and degradation needs to be further studied.
Like PDLIM2 and COMMD1,14,18 peroxisome proliferator-activated receptor gamma (PPAR-γ or PPARG) acts as a ubiquitin E3 ligase to induce the ubiquitination and degradation of both cytoplasmic and nuclear p65.8 This proteasome-mediated degradation requires the lysine-28 residue of p65.8 PPARγ contains two zinc-finger domains and does not have a typical RING domain. The two zinc-finger domains of PPARγ cooperate in its enzymatic activity by interacting with UbcH3 (an ubiquitin-conjugating enzyme). This is an important prerequisite for the ubiquitin ligase to transfer activated ubiquitin from E2 to the substrate.8 The glitazone receptor PPARγ is implicated in numerous diseases including obesity, diabetes, atherosclerosis, and cancer.20,21,22,23 PPARγ inhibits NFκB activation in response to bacterial stimuli,24 oxidized low density lipoprotein25 and TNF-α.26
Recently, a new mechanism for the PPARγ-mediated inhibition of NFκB signaling has been revealed,8 and another ubiquitin E3 ligase involved in NFκB degradation was identified.9 Inhibitor of growth 4 (ING4) has ubiquitin E3 ligase activity but lacks the typical RING domain.9 ING4 belongs to the ING family and inhibits tumor growth via the suppression of NFκB activation.27,28 ING4 is present in plant and animal transcriptional regulatory pathways, and has a highly conserved PHD (plant homeodomain) zinc finger domain that can act as a ubiquitin E3 ligase.27,28,29,30 Consistent with this, the PHD of ING4 functions as a ubiquitin ligase and induces the degradation of cytoplasmic and nuclear p65. Importantly, the lysine-62 residue of p65 is essential for proteasomal degradation.9 These observations indicate that either PPARγ or ING4 promote p65 degradation in the cytoplasm and nucleus,8,9 distinct from PDLIM2 and COMMD1-mediated nuclear p65 degradation.14,18 It is unclear whether other PPAR family members (PPARα or PPARβ) or INGs1,2,3,4,5 have E3 ligase activity.
In summary, NFκB activation is negatively regulated by the ubiquitin-dependent proteasomal degradation pathway (Figure 1), which can modulate inappropriate or excessive activity during NFκB-mediated inflammatory disease and in cancer. The NFκB degradation pathway provides a novel therapeutic target in these diseases.

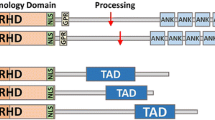
NFκB/p65 ubiquitination and degradation signaling. NFκB contains RHD and TAD domains. NFκB/p65 undergoes ubiquitination and degradation by the ubiquitin ligases EC2S, EC5S, PDLIM2, PPARγ or ING4. GCN5 or COMMD1 serves as a cofactor for EC2S ubiquitin ligase-mediated p65 degradation. Ubiquitin is attached to the lys-195, lys-28 or lys-62 residue on p65 in response to ubiquitin ligase. NFκB, nuclear factor κB.
References
Sen R, Baltimore D . Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986; 46: 705–716.
Chen LF, Greene WC . Shaping the nuclear action of NFkappaB. Nat Rev Mol Cell Biol 2004; 5: 392–401.
Hou Y, Mortimer L, Chadee K . Entamoeba histolytica cysteine proteinase 5 binds integrin on colonic cells and stimulates NFkappaB-mediated pro-inflammatory responses. J Biol Chem 2010; 285: 35497–35504.
Baldwin ASJ . The transcription factor NFκB and human disease. J Clin Invest 2001; 107: 3–6.
Yamamoto Y, Gaynor RB . IkappaB kinases: key regulators of the NFkappaB pathway. Trends Biochem Sci 2004; 29, 72–79.
Solinas G, Germano G, Mantovani A, Allavena P . Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 2009; 86: 1065–1073.
Saccani S, Marazzi I, Beg AA, Natoli G . Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor KB response. J Exp Med 2004; 200: 107–113.
Hou Y, Moreau F, Chadee K . PPARg is an E3 ligase that induces the degradation of NFkB/p65. Nat Commun 2012; 3: 1300.
Hou Y, Zhang Z, Xu Q, Wang H, Xu Y, Chen KP . Inhibitor of growth 4 induces NF kB/p65 ubiquitin-dependent degradation. Oncogene 2014; 33: 1997–2003.
Deshaies RJ, Joazeiro CA . RING domain E3 ubiquitin ligases. Annu Rev Biochem 2009; 78: 399–434.
Kamizono S, Hanada T, Yasukawa H, Minoguchi S, Kato R, Minoguchi M et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem 2001; 276: 12530–12538.
Frantsve J, Schwaller J, Sternberg DW, Kutok J, Gilliland DG . Socs-1 inhibits TEL-JAK2-mediated transformation of hematopoietic cells through inhibition of JAK2 kinase activity and induction of proteasome-mediated degradation. Mol Cell Biol 2001; 21: 3547–3557.
Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G et al. Regulation of NFkappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell 2003; 12: 1413–1426
Ganesh L, Burstein E, Guha-Niyogi A, Louder MK, Mascola JR, Klomp LW et al. The gene product Murr1restricts HIV-1 replication in resting CD4+ lympho-cytes. Nature 2003; 426: 853–857.
Marine GN, Mao X, Komarck CM, Burstein E . COMMD1 promotes the ubiquitination of NF-κB subunits through a cullin-containing ubiquitin ligase. EMBO J 2007; 26: 436–447.
Mao X, Gluck N, Li D, Maine GN, Li H, Zaidi IW et al. GCN5 is a required cofactor for a ubiquitin ligase that targets NFkB/RelA. Genes Dev 2009; 23: 849–861.
Rodrigues L, Filipe J, Seldon MP, Fonseca L, Anrather J, Soares MP et al. Termination of NFκB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J 2009; 28: 1283–1295.
Tanaka T, Grusby MJ, Kaisho T . PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol 2007; 8: 584–591.
Fan Y, Mao R, Zhao Y, Yu Y, Sun W, Song P et al. Tumor necrosis factor-α induces RelA degradation via ubiquitination at lysine 195 to prevent excessive nuclear factor-kB activation. J Biol Chem 2009; 284: 29290–28297.
Greene ME, Blumberg B, McBride OW, Yi HF, Kronquist K, Kwan K et al. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: expression in hematopoietic cells and chromosomal mapping. Gene Express 1995; 4: 281–299.
Girnun GD, Naseri E, Vafai SB, Qu L, Szwaya JD, Bronson R et al. Synergy between PPARgamma ligands and platinum-based drugs in cancer. Cancer Cell 2007; 11: 395–406.
Zhang Z, Xu Y, Xu Q, Hou Y . PPARγ against tumors by different signaling pathways. Onkologie 2013; 36: 598–601.
Hou Y, Gao J, Xu H, Xu Y, Zhang Z, Xu Q et al. PPARγ E3 ubiquitin ligase regulates MUC1-C oncoprotein stability. Oncogene 2013; in press.
Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol 2004; 5: 104–112.
Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G et al. Oxidized low-density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J Biol Chem 2000; 275: 32681–3267.
Ruan H, Pownall HJ, Lodish HF . Troglitazone antagonizes tumor necrosis factor-alpha-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of NF-kappaB. J Biol Chem 2003; 278: 28181–28192.
Garkavtsev I, Kozin SV, Chernova O, Xu L, Winkler F, Brown E et al. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature 2004; 428: 328–332.
Coles AH, Gannon H, Cerny A, Kurt-Jones E, Jones SN . Inhibitor of growth-4 promotes IkappaB promoter activation to suppress NF-kappaB signaling and innate immunity. Proc Natl Acad Sci USA 2010; 107: 11423–11428.
Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell 2007; 28: 823–837.
Coscoy L, Ganem D . PHD domains and E3 ubiquitin ligases: viruses make the connection. Trends Cell Biol 2003; 13: 7–12.
Acknowledgements
This work was supported by the Jiangsu Province Natural Science Foundation (SBK201320232); the National Science Foundation of China (31271272, 31071030); and the Scientific Research Foundation for Returned Overseas Chinese Scholars, State Education Ministry.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Xu, H., You, M., Shi, H. et al. Ubiquitin-mediated NFκB degradation pathway. Cell Mol Immunol 12, 653–655 (2015). https://doi.org/10.1038/cmi.2014.99
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2014.99
- Springer Nature Limited
Keywords
This article is cited by
-
Dual function of activated PPARγ by ligands on tumor growth and immunotherapy
Medical Oncology (2024)
-
TRIMs: selective recruitment at different steps of the NF-κB pathway—determinant of activation or resolution of inflammation
Cellular and Molecular Life Sciences (2021)
-
The ubiquitin–proteasome system in kidney physiology and disease
Nature Reviews Nephrology (2019)
-
STAT3 and NF-κB are Simultaneously Suppressed in Dendritic Cells in Lung Cancer
Scientific Reports (2017)
-
Cellular and molecular regulation of innate inflammatory responses
Cellular & Molecular Immunology (2016)