
Abstract
In April 2013 our group published a review on predictive molecular pathology in this journal. Although only 2 years have passed many new facts and stimulating developments have happened in diagnostic molecular pathology rendering it worthwhile to present an up-date on this topic. A major technical improvement is certainly given by the introduction of next-generation sequencing (NGS; amplicon, whole exome, whole genome) and its application to formalin-fixed paraffin-embedded (FFPE) tissue in routine diagnostics. Based on this ‘revolution’ the analyses of numerous genetic alterations in parallel has become a routine approach opening the chance to characterize patients’ malignant tumors much more deeply without increasing turn-around time and costs. In the near future this will open new strategies to apply ‘off-label’ targeted therapies, e.g. for rare tumors, otherwise resistant tumors etc. The clinically relevant genetic aberrations described in this review include mutation analyses of RAS (KRAS and NRAS), BRAF and PI3K in colorectal cancer, KIT or PDGFR alpha as well as BRAF, NRAS and KIT in malignant melanoma. Moreover, we present several recent advances in the molecular characterization of malignant lymphoma. Beside the well-known mutations in NSCLC (EGFR, ALK) a number of chromosomal aberrations (KRAS, ROS1, MET) have become relevant. Only very recently has the clinical need for analysis of BRCA1/2 come up and proven as a true challenge for routine diagnostics because of the genes’ special structure and hot-spot-free mutational distribution. The genetic alterations are discussed in connection with their increasingly important role in companion diagnostics to apply targeted drugs as efficient as possible. As another aspect of the increasing number of druggable mutations, we discuss the challenges personalized therapies pose for the design of clinical studies to prove optimal efficacy particularly with respect to combination therapies of multiple targeted drugs and conventional chemotherapy. Such combinations would lead to an extremely high complexity that would hardly be manageable by applying conventional study designs for approval, e.g. by the FDA or EMA. Up-coming challenges such as the application of methylation assays and proteomic analyses on FFPE tissue will also be discussed briefly to open the door towards the ultimate goal of reading a patients' tissue as ‘deeply’ as possible. Although it is yet to be shown, which levels of biological information are most informative for predictive pathology, an integrated molecular characterization of tumors will likely offer the most comprehensive view for individualized therapy approaches. To optimize cancer treatment we need to understand tumor biology in much more detail on morphological, genetic, proteomic as well as epigenetic grounds. Finally, the complex challenges on the level of drug design, molecular diagnostics, and clinical trials make necessary a close collaboration among academic institutions, regulatory authorities and pharmaceutical companies.
Similar content being viewed by others

Introduction
Targeted drugs, in particular therapeutic antibodies, kinase inhibitors and PARP inhibitors, have started to transform current treatment strategies in oncology and new organizational structures, which we have started to discuss previously and which we now review in more detail.1 Prior to application almost all of them require a so called pre-therapeutic companion diagnostic test to identify certain molecular alterations. More precisely, a well-defined biomarker, most often a characteristic genetic alteration or particular protein (over-)expression, that indicates the efficacy of the respective drug has to be identified in the tumor tissue of individual patients. The tissue-based analysis is mostly done by predictive molecular pathology applying conventional or high-throughput techniques on FFPE tissue. This is the basis of personalized, individualized or precision medicine.
This new and extremely exciting field of medicine creates a number of challenges which can be managed only by new ways of thinking and new organizational structures. Some examples are
-
1
The scope of the interdisciplinary tumor board that consists of radiologists, pathologists, oncologists, surgeons and organ specialists, needs to be extended to account for the increasingly available clinically relevant molecular profiling information by including specially trained molecular pathologists.
-
2
It is pivotal to further integrate ‘molecular knowledge’ into diagnostic pathology training.
-
3
Due to the complexity of new combination treatment new designs of clinical studies have to be defined and approved by the regulatory authorities.
-
4
The classification of malignant tumors may change considerably by introducing a molecular classification of malignant tumors. One example is the molecular subtyping of breast carcinomas. The up-coming edition of the WHO Blue Series on brain tumors will include new entities, which are defined mainly or only by molecular characteristics.
Despite the increasing importance of molecular information for tumor classifications, examples such as the different druggability of BRAF V600E mutations in melanoma compared to colorectal cancer indicate that for the foreseeable future the classical morphology-based WHO classification will remain in place but, will certainly have to be supplemented by genetic, proteomic metabolomic and other data so that finally the combination of all the techniques and the provided data will lead to an optimal strategy for patient care.
Clinical trial design—the combinatorial complexity challenge in precision oncology and predictive pathology
Actionable mutations indicating a response to targeted therapy are often only present in a minority of tumors. EML4-ALK translocations, for instance, are found in only about 4% of all NSCLC, which poses a major challenge for patient recruitment in clinical trials. The clinical study that demonstrated the efficacy of Crizotinib in EML4-ALK-positive NSCLC comprised 105 sites in 21 countries only to recruit 347 patients.2 This comes as no surprise as the low frequency of EML4-ALK required about 10,000 NSCLC patients to be screened. To address this challenge, alternative trial designs have been proposed, such as the so called ‘basket’ or ‘umbrella’ trials, which recruit patients with different cancers but identical mutations or vice versa.2, 3 While this partially solves the low frequency problem in a monotherapy setting, the situation becomes more difficult when it comes to combination therapies, which will likely become necessary because of frequent resistance development against single drug therapies. Although only a relatively small proportion out of the hundreds of genetic aberrations found in most cancers (over 800 in an average squamous cell lung cancer4 are currently therapeutically targetable, the list of actionable mutations and corresponding drugs is constantly growing). Imagine a rather conservative scenario with only 10 mutations: already here, 45 alternative 2-drug combination therapies exist. In only moderately more complex situations, such as selecting a triple-therapy against 3 out of 20 mutations, over 1,000 alternatives exist, a number that increases to nearly 10,000 for 40 possible targets. Although the number of possible combination therapies may be reduced to some extent based on knowledge on pathway biology and clinical experience, multiple drug combinations that would have to be compared in clinical trials remain. This complexity is practically not addressable with existing clinical trial strategies especially when seen in conjunction with the low frequency challenge described above. As an example, when a triple-therapy against 3 actionable mutations that each occur in 20% of the tumors is to be evaluated in a clinical trial, 25,000 patients would have to be screened to recruit just 200 patients.
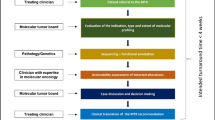
A solution to these challenges requires novel, more patient-centric clinical trial design strategies, similar to approaches implemented in the so called N-of-1 trials.5 Such trials would among other things rely on a—compared to static genomic profiling—more functionally oriented systems medicine approach based on proteogenomic analyses and bioinformatic modeling (Figure 1). Predicting the functional relevance and efficacy of a therapy for individual patients would help avoid systematic or random testing of alternatives. Novel patient-centric clinical trials would then not just evaluate a particular therapy for a given mutational signature, but assess the capability of the predictive molecular pathology approach to design a custom-tailored therapy for an individual patient6, 7 for a more detailed discussion.

Proposal for a systems medicine approach to patient-centric precision oncology trials. Conventional clinical trial design is incompatible with the high combinatorial complexity of targeted combination therapy selection even for moderate numbers of actionable mutations found with next-generation sequencing. However, additional proteomic profiling enhanced by computational modeling facilitates the functional evaluation of molecular aberrations and may be used to predict personalized therapies for individual patients. The proposed trial design no longer tests a particular drug for a defined cancer in a homogeneous cohort of patients, but evaluates the method used to predict the custom-tailored therapies for individual patients.
Methodolocigal considerations
Since formalin fixation and paraffin embedding (FFPE) is and will be the by far the most widely used method for tissue fixation in the routine diagnostic setting the high-through-put technologies have been adapted to the particular quality of DNA and RNA in FFPE tissue. This opens the door to extended molecular analyses of almost all tissues. It has to be emphasized that carefully controlled pre-analytic steps, standard operating procedures (SOPs) and bio-mathematic knowledge are of absolute importance. In addition the labs working in clinical diagnostics should constantly perform external quality controls.
Next-generation sequencing and liquid biopsy
Next-generation sequencing (NGS) has recently been adapted to diagnostic requirements and a growing number of diagnostic laboratories and companies offer a variety of NGS-based services. However, different NGS-based approaches ranging from amplicon-based (targeted) NGS, whole exome (WGS) or whole genome NGS (WES) exist and are, as yet, suited for different applications. With respect to tumor diagnostics in molecular pathology predominantly amplicon-based NGS is applied. This is due to the fact that (i) the vast majority of sequence information generated by WGS and WES cannot be translated into clinical intervention, (ii) WGS and WES are not ready for FFPE based NGS, (iii) bioinformatics for WGS and WES data requires both, germline and tumor data, (iv) the coverage of WGS and WES appears to be too low especially in cases of low tumor cell content, (v) the turn-around times of WGS and WES are too long in a diagnostic setting and (vi) the costs (consumables, data analysis, bioinformatics and data handling) of WGS and WES are much too high. In contrast, targeted amplicon-based NGS strategies can overcome most of these disadvantages. Since the first reports that small amounts of FFPE-derived DNA are feasible for NGS-based analyses8 several protocols have been established that both adapt amplicon length as well as DNA extraction methods to the specific requirements of FFPE tissue and biopsy samples. The restriction of the gene panels employed for amplicon-based to targets with clinically actionable mutations enables relatively small amplicon numbers. In turn, the small number of amplicons provides very high coverages despite intensive multiplexing (parallel processing of a considerable number of samples), which thus is very cost effective. The resulting high coverage provides enough sensitivity even in situations of low tumor cell content or tumor cell heterogeneity and provides. the basis for thresholds to ignore artifacts introduced by formalin fixation. Finally, due to the restriction to known druggable genes, the bioinformatics analysis of amplicon-based NGS data is feasible with commercial software products and basic bioinformatic knowledge. Regarding the turn-around-time, amplicon-based NGS can be very much adapted to the needs and the sample numbers of the respective molecular diagnostic laboratory (Figure 2). This provides processing times per sample which are very similar to those accepted for conventional Sanger- or pyro-sequencing.

Integration of next-generation sequencing in diagnostic pathology exemplified by the Ion AmpliSeq technology. This and others procedure proved to be very valuable in molecular analyses of FFPE tumor tissue. This is of particular relevance in predictive molecular pathology as prerequisite of targeted therapy of cancer.
A question which was extensively discussed concerns the reliability of amplicon-based NGS for diagnostic purposes since Sanger sequencing or pyro-sequencing was regarded as gold standard since many years. Amplicon-based NGS—despite less complex as compared to WGS and WES—is a comprehensive approach because of the simultaneous generation of many amplicons which are subsequently individually sequenced in parallel by NGS. As a result, each amplicon is represented by a huge number (several hundreds up to thousands) of individual sequences which may differ at various nucleotide positions. Furthermore different methods exist to generate the multiplex amplicons (pull-down enrichment, primer extension and ligation or multiplex PCR) as well as to produce the sequences (mainly measurement of fluorescent dye incorporation and imaging (Illumina) or semiconductor sequencing (ThermoFisher). In addition to the NGS methods, there are further steps in the entire procedure, which might interfere with the sequencing results such as DNA extraction, the primer composition of the gene panels and the bioinformatics interpretation. To clarify the comparability of the data NGS data produced at different sites and applying various methods and techniques we initiated a multi-center study in the context of the German Consortium for Translational Cancer Research (DKTK). Based on molecular predefined tissue samples (Sanger sequencing) and cell line dilutions we produced Illumina MiSeq data and Life Technologies PGM data at 5 different pathology centers using exactly the same samples. Although the number of sequence reads per amplicon obtained for the regions of interest differed due to the local design of the sequencing approach, the identification of the predefined mutations was successfully achieved at all sites involved and by all NGS methods applied independent from the gene panels used (manuscript in preparation). However we recognized a significant impact of the DNA extraction method used which may cause significant differences in the allelic frequencies of the detected mutations or even complete drop-out of some cases especially in combination with the respective NGS technique used. In conclusion, it should be kept in mind that amplicon-based NGS is a complex approach which has to be carried out, controlled and supervised by trained technicians, scientists and pathologists in order to avoid the production of insufficient data and a misinterpretation of the results. Therefore the setup of quality assurance schemes taking these new technologies into account are required as soon as possible. Under these preconditions amplicon-based NGS is ready to step into the routine in molecular pathology. In line with this in late 2013, with the Illumina MiSeqDx the first next-generation high-throughput sequencer was approved by the FDA (Food and Drug Administration) thus paving the way for the application of numerous genome-based tests.9
Apart from its value in molecular pathology, the high sensitivity of amplicon-based NGS now enables non-invasive analyses such as early detection of relapse-determining mutations in blood or plasma samples from patients under treatment.10 This process, today commonly addressed as liquid biopsy, has led to an improved understanding of the development of therapy resistance and opened the path for a future monitoring of cancer patients.11
Perspectives on proteomics: the next level of molecular tumor profiling?
Pan-cancer mutational profiling efforts by the consortia TCGA (the cancer genome atlas) or ICGC (international cancer genome consortium) and many other groups have demonstrated a high complexity of the mutational landscapes in most cancers. The average squamous cell lung cancer, for instance, has been shown to harbor over 800 genetic aberration including exon mutations, copy number variations and rearrangements.6 Although only a minority of the observed mutations is believed to belong to the group of driver mutations that are causally linked with cancer pathology, the functional and therefore clinical implications of many less frequent mutations for individual patients are largely unknown. And even if a (rare) mutation is known to affect protein function, in principle, modulations of such effects through other mutations or epigenetic as well as posttranscriptional and posttranslational regulation may differ substantially among tumors. A solution would be to complement genomic with proteomic profiling of tumor tissue to help relate mutational profiles to protein changes as more direct indicators of tumor cell function. Proteomic tests have already been shown to provide prognostic information, on—as, for example, in the case of Veristrat (Biodesix)—the efficacy of EGFR inhibitory therapy on NSCLC.12, 13 However, while a step towards a better characterization of tumors, this mass spectrometry based test of patient serum relies only on eight abstract features from the obtained mass spectra that were correlated statistically with clinical outcome. To achieve a better understanding of the mechanisms underlying cancer pathology and to facilitate therapy decision making based on causality rather than statistical correlations more comprehensive and functional characterizations are needed, including protein expression and, as one of the major regulatory mechanisms of signaling pathways, protein phosphorylation measurements. Antibody-based protein array techniques or pathway profiling kits are already commercially available, shotgun or targeted mass spectrometry based proteomics are technically more complex. However, the advantage of mass-spec based approaches lies in providing a more global (simultaneous measurements of several thousand proteins) and unbiased view of the functional implications of mutational profiles and will likely play an important role in future molecular tumor profiling.
External quality assessment (EQA)/quality control (QC)
EQA is a systematic process for the assessment of diagnostic and predictive tests. A number of test samples are distributed to the participating centers for analysis. Within these trials participants may or may not certify for different diagnostic applications (eg estrogen/progesteron receptor, HER2, Ki67, EGRF, RAS, ALK). The goal is to achieve a high level of accuracy, standardization and reproducibility among different centers.14 The QuIP initiative (‘Qualitätssicherungs-Initiative Pathologie’) serves as an example of QC at the national and European level. Here, ALK-QCs might serve as an example: The first (2012) showed as success rate of 60.3% (32 of the 53 participating institutes were certified),15 in the second (2014) 92.5% (37/40) succeeded, demonstrating a successful learning curve.
A new challenging QC initiative started by a consortium of Institutes of Pathology in Germany is dealing with amplicon-based next-generation sequencing (NGS) for detection of pathogenic mutations in the genes for BRCA1/2. This became relevant since a new PARP inhibitor was approved for the treatment of high-grade serous ovarian and peritoneal carcinomas if a pathogenic BRCA1/2 mutation could be demonstrated in the tumor tissue of the patients. The method of choice for this purpose in amplicon-based NGS although other sequencing techniques are not excluded.
To open EQA to a broad community of pathologists in June 2014, a joint agreement was signed by the German Society of Pathology, the Association of German Pathologists and the European Society of Pathology. Each organization accepts the other’s quality assessment programs.
Molecular diagnostics of tumors (molecular classification)
Clinicians’ demand in tissue-based diagnostic analyses of solid and hematological tumors is becoming more and more challenging. This is in particular true if therapeutical options need companion diagnostics. While pathology reports on tumor type, WHO-code, pTNM status, dignity, i.e. benign, malignant, in situ carcinoma or borderline lesion; histogenesis and prognostic remain the skeleton of tumor classification the predictive relevance is limited. This means conventional histopathology has to be refined by molecular data often very relevant for selecting the optimal treatment. The most relevant examples are given below.
Breast cancer
In breast cancer, the classical morphology-based classification has been largely replaced by the molecular classification.16 Based on gene expression profiling as well as immunohistochemistry, at least five different subtypes can be distinguished: luminal-A, luminal-B/HER2 negative, luminal-B/HER2-positive, HER2-positive/non-luminal and triple negative. At the St Gallen conference 2011 the molecular subtypes were introduced as the many classification system for clinical decisions in breast cancer, this classification was slightly modified at the St Gallen 2013 meeting.17
Breast cancer subtypes can be determined by gene expression analysis. However, in clinical practice, the standard approach is the immunohistochemical investigation of estrogen receptor, progesterone receptor and HER2. The differential diagnosis of luminal-A versus luminal-B tumors can be a challenge in some situations. It has been suggested to used Ki67 as a marker for luminal-A versus B tumors, however, the cut-points for Ki67 are still under debate and international efforts for further standardization are still ongoing.
In the last years, several diagnostic assays have been developed to define a low risk group of luminal-A tumors that have an excellent prognosis with endocrine therapy alone. The OncotypeDX18 assay, the Endopredict19 assay as well as the PAM50/Prosigna20 test are based on mRNA analysis of gene expression in formalin-fixed paraffin-embedded tissue and can be used to defined low risk tumors that might not need a chemotherapy treatment. In addition, some of the tests (Endopredict21 and Prosigna22) have been used to identify which patients would benefit from extended endocrine therapy of more than 5 years.
In addition to mRNA-based gene expression profiling, mutation-based classification are under evaluation in breast cancer. The most commonly mutated genes are p53 and PIK3CA. For PIK3CA, it has recently described that tumors with PIK3CA mutations have a reduced response to neoadjuvant anti-HER2 therapy, further validation studies are ongoing.23
Ovarian cancer
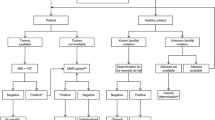
Sequence analysis of the breast and ovarian cancer susceptibility genes BRCA1 and BRCA2 genomic status is the first routinely performed molecular test for ovarian cancer. According to data provided during The Cancer Genome Atlas (TCGA) project high-grade serous carcinoma, which is the most frequent histological subtype of ovarian cancer (70%), reveals germline mutations within BRCA1 or BRCA2 in approximately 20% of primary tumors, additional 6% show somatic mutations.24 BRCA mutations lead to a homologous recombination deficiency (HRD), which is characterized by a reduced ability to repair DNA double strand breaks.25 The inability to perform DNA repair adequately is the cause of an increased cancer risk on the hand, but also the reason for the high sensitivity of BRCA mutated tumors to agents that induce DNA single strand breaks and thereby produce a synthetic lethality. Particularly, BRCA mutated ovarian cancer is highly sensitive to platinum-based chemotherapy, which seems to be the reason for the comparably favorable prognosis of this molecular tumor subtype.26 Poly(ADP-ribose) polymerase inhibitors (PARPi) are the most recently approved targeted therapeutics for ovarian cancer. In January 2015 the European Medicines Agency (EMA) has approved the PARP inhibitor olaparib (Astra Zeneca) for maintenance therapy of recurrent platinum-sensitive high-grade serous ovarian, tubal or peritoneal carcinoma revealing germline or somatic BRCA mutations. The approval was based on a phase II randomized clinical trial, which showed a significant progression-free survival (PFS) advantage in the olaparib vs the placebo arm (median PFS 8.4 months vs 4.8 months, P<0.001).27 The survival advantage was most pronounced in BRCA mutated carcinomas (median PFS 11.2 months vs 4.3 months P<0·0001), however, response was also seen in patients with BRCA wild-type status (median PFS 7.4 months vs 5.5 months, P=0·0075).28 This points to the possibility that apart from BRCA genomic status other molecular aberrations—most likely those causing HRD (also referred to as ‘BRCAness’)29 might be involved in the response to PARPi, however, BRCA mutations being the most frequent and best studied mechanism underlying HRD, BRCA sequence analysis is the companion diagnostic test for olaparib to date. Because of the occurrence of somatic mutations, analysis of tumor tissue in patients who have not yet been tested positive for a hereditary BRCA mutation (both patients with wild-type and unknown germline status) is the most sensitive method to detect all patients suitable for an olaparib maintenance therapy (Figure 3). The reported rates of somatic mutations vary, depending on the sample size and selection criteria of the cohort investigated, however is likely to be higher in patients with platinum-sensitive high-grade serous cancer than in the total population of ovarian cancer patients (10–14%).30 Both genes are of considerable size (BRCA1: 24 exons, 5.592 bases; BRCA2: 27 exons, 10.257 bases) and although some mutational hotspots occur, most mutations are widely distributed along the exome rendering the sequence analysis challenging (http://cancer.sanger.ac.uk/cosmic/gene/overview?ln=BRCA1; http://cancer.sanger.ac.uk/cosmic/gene/overview?ln=BRCA2). Next-generation sequencing (NGS) techniques, which are increasingly used in molecular pathology, are therefore the methodological option for BRCA analysis in a routine diagnostic setting. Apart from the need to sufficiently cover the BRCA1/2 exome during sequence analysis, the interpretation of detected variants requires a high bioinformatics expertise, as multiple polymorphisms exist in each given individual, necessitating a functional and clinical annotation of each variant. Several publicly available databases are in use for clinical annotation (eg the Breast Cancer Information Core (BIC) of the NIH), and are until now based on the estimation of familial breast and ovarian cancer risk. Due to the very recent approval of PARPi for ovarian cancer treatment, no robust data according to the predictive capacity of particular sequence variants for response to PARPi exist to date, however, those data are expected to accumulate with the clinical use of PARPi within the next years.

Algorithm for predictive BRCA testing in tumor tissue. Patients with recurrent, high-grade serous ovarian, tubal, or primary peritoneal carcinoma may be considered for an olaparib maintenance therapy. For patients with unknown BRCA status or patients who have previously been tested negative for a BRCA germline mutation BRCA status should be determined in tumor tissue, which enables the detection of germline and somatic mutations (green). Patients in whom a tumoral BRCA mutation is detected are eligible for therapy. Patients who have previously been tested positive for a germline BRCA mutation are eligible for therapy and do not need further testing (red).
Colorectal cancer
Antibody mediated blockade of the epidermal growth factor receptor (EGFR) is a therapeutic option in the treatment of advanced colorectal cancer. In several clinical trials it was shown, however, those only patients with cancers bearing no mutation in the KRAS gene benefit from EGFR-targeting antibodies like cetuximab and panitumumab.31, 32 KRAS is an intracellular downstream component of the EGFR signaling cascade. Mutations of the gene result in a constitutional activation of the EGFR pathway and thus might completely abolish the effects of an upstream inhibition of EGFR.
In 2008, therefore, exclusion of KRAS hot spot mutations in exon 2 have become mandatory for the application of EGFR-targeting antibodies in the first-line therapy of advanced colon cancer. The list of genetic alterations to be excluded for EGFR antibody therapy has been extended to KRAS exons 3,4 and NRAS exons 2–4 in 2013 after non-responsiveness has also been found for these alterations.
Other genetic changes like BRAF and PI3K mutations have also been described to associate with lacking EGFR antibody response33 but the data generated in a different study34 is too controversial to include these markers in routine predictive molecular testing and therapy decision making to date. It is foreseeable, however, that the number of markers predicting therapy response in CRC will rise with novel data generated from larger CRC cohorts and with the introduction of novel therapy strategies which may include antibody targeting of BRAF in the near future.
Another application of molecular pathology in CRC refers to a principal genetic mechanism causing the disease. In approximately 10–15% of CRCs microsatellite instability (MSI-H), a consequence of DNA mismatch repair deficiency, is found. MSI-H is a hallmark alteration of HNPCC/Lynch syndrome-associated tumors, but is also found in sporadic colon cancers.35, 36, 37 MSI-H has a significant impact on tumor biology. This is reflected by a more favorable prognosis of this molecular CRC subtype and a lacking response of MSI-H CRCs to 5′fluorouracil monotherapy.38
To date, knowledge of the MSI status is required to address two routine applications/requirements:
1. Following the revised Bethesda guidelines,39 colon cancer is tested for MSI in order to evaluate the possibility of Lynch syndrome. In combination with immunohistochemical analysis of the mismatch repair proteins (MLH1, MSH2, PMS2, MSH6) and EPCAM40 the molecularly determined MSI status guides further genetic counseling.
2. Moreover, the MSI status is determined in poorly differentiated cancers according to conventional histologic grading. Only in the absence of MSI-H, poor histologic/high-grade differentiation (G3–4) is considered a prognostic marker while MSI-H cancers are considered low grade, irrespective of the conventional histologic appearance.
Non-small cell lung cancer (NSCLC)
To date the detection of EGFR- and ALK-alterations is the standard of molecular testing in NSCLC but should be further completed by testing for other markers as ROS1, MET or even KRAS under certain conditions.1, 41, 42, 43
EGFR and ALK mainly occur in advanced-staged adenocarcinomas (ADC), whereas the frequency in squamous cell carcinomas (SCC) is very low.44 However, under certain conditions (eg young patient, non-smoker) testing should be considered in the latter, as well.44 KRAS- and EGFR-testing is performed by PCR (more and more NGS), ALK-, ROS1- and MET-testing mainly by Fluorescence in situ hybridization (FISH).41, 45, 46 Recent studies demonstrated, that a ‘carefully validated’ ALK-immunohistochemistry (IHC) is eligible for multi-center routine testing15, 46, 47 at least as a screening tool and in samples that may be unsuitable for the FISH approach (eg very small amount of tumor cells, high amount of osseous tissue).48
KRAS-alterations (chromosome 12) are quite frequent (~30%). To date, “only” blockage of downstream kinases in the RAS-RAF-MEK-ERK pathway is under investigation, e.g. with the MEK1/MEK2-inbibitor selumetinib in combination with docetaxel.49, 50 However, to date a targeted KRAS-therapy is not available.49, 50 As mutual exclusiveness is discussed for KRAS, EGFR, ALK and ROS1, some authors suggest upfront KRAS testing, with further broad testing in a KRAS wild-type situation only.48, 49, 50 In our opinion this testing approach costs tissue and time (as results should be reported within 5–10 days43). Furthermore, comutations (even if very rare) might occur and single patients could get lost for a promising therapy.
For patients with EGFR (~15%, chromosome 7) activating mutations (exon 19 deletion and exon 21 L858R point mutation) an EGFR-TKI (eg erlotinib, gefitinib or afatinib) is standard first-line therapy if a currative tumor resection is not possible.48, 49, 50, 51 Most EGFR exon 20 insertion mutants are resistant to EGFR inhibitors, except the A763_Y764insFQEA, which is sensitive to erlotinib and gefitinib.51, 52 Secondary resistance to EGFR inhibitors may occur due to an additional EGFR-mutation (T790M) or MET-amplification,49, 51, 53 therefore testing for the latter (see below) becomes more and more standard and combination of EGFR-TKI and MET-inhibitors are discussed.49, 50, 51
For patients with ALK-inversions/translocations (~3%, chromosome 2) the ALK-TKI crizotinib reached approval in 2011 (FDA) and 2012 (EMA).43, 44, 45, 46 Meanwhile “second generation” ALK inhibitors have been invented, facing the question of brain metastasis and ALK-resistance.48, 49, 50 Acquired ALK-resistance occurs due to secondary ALK-mutations, ALK-amplification, as well as EGFR-, cKit- or KRAS-mutations.54, 55 Therefore, rebiopsy in these patients is highly recommended as this will have further impact on the consecutive therapy management.49, 50
However, not only acquired secondary mutations but especially tumor heterogeneity is an highly important issue discussed in mechanisms of therapy resistance. Patients with a heterogeneous EGFR profile showed less response to gefitinib, as well as an earlier tumor progress and a shorter median survival.56 These kind of aspects (eg major clones and subclones) should be further kept in mind when comparing results of biopsy and rebiopsy. Additionally, an evolving field is the so called ‘liquid biopsy’ encompassing the detection of circulating tumor cells (CTC), circulating tumor DNA (ctDNA), circulating RNA or microRNAs. To date, further robust validation and large independent prospectively designed comparative trials need to be done.57
Interestingly the above mentioned drug crizotinib (originally designed as MET-inhibitor) is an ALK/ROS1/MET-inhibitor that also showed good response data in patients with ROS1-translocation and MET-amplification.58, 59 Recently the FDA granted breakthrough therapy designation for crizotinib in ROS1-positive NSCLC. ROS1-testing (chromosome 6, translocation, ~0.5–2%, mainly ADC) is performed by means of FISH.41, 49, 60 IHC and PCR are possible, however, (so far) not multi-center standardized.50, 61
Testing for MET (chromosome 7, mutation/amplification/overexpression ~10%) is not only an option in the context of EGFR-resistance as described above,49, 62 as therapy response was shown with ALK-TKIs and MET-inhibitors in patients with MET-amplification and alterations at MET exon 14.59, 63, 64 However, a clinical phase III trial (MET-inhibitor) was stopped recently and its role will need further evaluation.65
MET alterations are more frequent in ADC than in SCC, at the moment diagnosis is mainly made by FISH (different scoring systems!) and IHC (be aware of discordant IHC/FISH results!).60
To summarize, at the moment EGFR, ALK and ROS1 are in the focus of ADC-NSCLC testing and new drugs targeting even more than one alteration are under investigation (for an overview please see and read Table 1, page 441 in ref. 66).
Further alterations41, 50 that should be considered (encouraging studies) in case of negativity of the above described markers are:
-
RET (chromosome 10; inversion/translocation; 1–2%, FISH).
-
HER2 (chromosome 17; Exon 20 insertion, deletion, 1–2%, PCR, but not protein expression as known in breast and gastric cancer).
-
BRAF (chromosome 7, activating point mutation V600E, fusion protein, 1–3%, PCR).
Thus parallel testing for EGFR, ALK and ROS1 encompassing an ALK-IHC seems a pragmatic manner. Figure 4 combines the markers discussed here in the context of a local (reflex) testing approach.

NSCLC molecular testing algorithm at the Charité University Hospital (ADC: adenocarcinoma, LCC: large cell carcinoma, NOS: not other specified).
So far the data for SCC are not as encouraging as for ADC: The main two targets to test for are FGFR1 (chromosome 8, 20%, amplification, only 1% in ADC, detection by FISH) and DDR2 (chromosome 1, point mutation, 2%, PCR).50
The implementation of NGS in the daily routine might bring us different mutations in one tumor (not only concerning the above mentionned targets) in the context of tumor heterogeneity. This will not only broaden the spectrum of targeted therapy but will also help to better understand the questions of drug resistance.
Malignant melanoma
The incidence of malignant melanoma has increased rapidly in the past few years, partly due to changes in diagnostic criteria and improvement of screening methods, but also due to behavioral changes such as increased exposure to UVR.67 A first breakthrough in the therapy of malignant melanoma was achieved by the approval of several targeted therapies for patients with BRAF mutated metastatic melanoma, Vemurafenib and later Dabrafenib.68, 69 About 50% of metastatic melanoma harbor a mutation in the oncogene BRAF, with V600E being the most common one (75% of V600 mutations), followed by V600K (20%) and V600R.70
The first results of the BRIM3 study revealed a prolonged overall survival of 13.9 months (vemurafenib) compared to 9.6 months (dacarbicine) in BRAF mut patients.68 One of the biggest challenges in the therapy of BRAF mut patients with these targeted therapies is still the primary and secondary resistance which most patients develop over time. The success of the therapy is often restricted by the activation of the MAPK signaling pathway in BRAF WT cells and the resulting activation of proliferation followed by a fast relapse.71, 72
Thus, the most important prerequisites for an optimal therapy of the patients, now and in future, are reliable and reproducible methods for BRAF testing and the development of new treatment strategies, particular by combining different approaches, to increase therapy success and to achieve long-term responses.
BRAF testing
Today, a multitude of different methods for BRAF testing is available, among them different sequencing technologies and real time PCR assays. The so called “next-generation sequencing” (NGS) became popular in the past few years, allowing for testing of multiple markers in one run, and will also become of interest for malignant melanoma due to the increasing requirements for molecular testing.
Furthermore, BRAF testing can also be done by immunohistochemistry.73 However, the currently available antibody only allows for the testing of BRAF V600E mutations and thus should only be used as a screening method, combined with a second technology for a subsequent analysis of all negative cases for mutations other than V600E.
NRAS and KIT
Besides BRAF mutations malignant melanomas harbor several other oncogenic alterations, like mutations in the GTPase NRAS and the tyrosine kinase KIT. In about 20% of all malignant melanoma an activating mutation in NRAS can be found which is mainly associated with a worse prognosis.74 Since NRAS cannot be targeted directly, testing for NRAS alterations in malignant melanoma initially became of interest as a potential predictive factor for downstream applications e.g. for MEK inhibition.
KIT mutations in malignant melanoma are rare. About 15% of malignant melanoma harbor KIT mutations,75 mainly in exon 11 and exon 13. KIT inhibitors like Sunitinib, Dasatinib and Imatinib, although primarily not developed for the treatment of malignant melanoma, turned out to be interesting therapy options for patients with KIT positive melanoma.
Combinatorial therapies
Due to the limited long-term effect of BRAF inhibitors, efforts are being made to combine different strategies to improve therapy outcome. One promising approach is the complete inhibition of the MAPK pathway by combining BRAF and MEK inhibitors. The first results of a Phase I/II study for the combination of dabrafenib and trametinib showed an increase of median PFS from 5.8 with dabrafenib alone to 9.4 months with the dabrafenib plus trametinib and showed even less toxicity compared to the treatment with one of the agents alone.76 Furthermore, the use of ERK inhibitors is being tested for patients with MAPK pathway dependent resistance to RAF or MEK inhibitors.77
Recently, the newly-discovered approach of treatment with targeted therapies combined with novel immunotherapies, e.g. with the anti-CTLA4 antibody ipilimumab or anti-PD-1 and –PD-L1 antibodies nivolumab and lambrolizumab showed great promise for a prolonged therapeutic efficacy.78
Cancer of unknown primary (CUP)
In about 5% of tumors the anatomical site of origin cannot be determined despite extensive examinations.79, 80 These cancers are termed “cancer of unknown primary”, CUP. Whether CUP represents a group of cancers with common biologic features, irrespective of the site of the primary tumor or if each CUP retains the signature of the tissue it developed from and which can be used for site-directed therapies, is still a matter of discussion.
Until recently most CUP patients were treated with empiric broad-spectrum chemotherapeutic regimes, resulting in median survival times of only 9 months. Only a subset of patients received a relatively-site-specific therapy, based on the presence of certain clinical or pathological features which prolonged the survival times.81
To determine the tissue of origin in CUP, clinical and pathological data as well as results from advanced imaging technologies are integrated and in most instances are sufficient. In cases where this procedure is not successful molecular assays can be of help in order to provide the patient with a site-specific therapy. At present three multi-marker profiling assays for the determination of the tissue of origin in CUP are commercially available, all of which are suitable for formalin-fixed tissue and only require little tumor tissue. The CancerType ID Assay (Biotheranostics, CA, USA) analyses the expression levels of 87 candidate and 5 housekeeping genes by qPCR.82 The gene expression signature is then compared against a proprietary database consisting of 30 tumor types (54 subtypes). The Tissue of Origin Test (Response Genetics) is a microarray-based messenger RNA profiling test.83 The expression levels of 2000 genes are measured and a similarity score in comparison to a database of 15 tumor classes (58 types/subtypes) is constructed. The Cancer Origin Test (Rosetta Genomics) profiles the expression of 64 micro RNAs by microarray and enables the identifications of 42 tumor types.84 The accuracy of the assays has been evaluated in retrospective studies where the primary tumor was found later on and demonstrated good concordance.85 Also, when comparing the gene expression assays with immunohistochemistry the performance was similar, with an advantage of the gene expression assays in tumors with poor differentiation.86, 87 Based on the available studies and a meta-analysis all assays seem to perform equally well. Therefore the decision if and what molecular test to use should be made for each case dependent on the clarity of the immunohistological results, availability of tissue, the tumor subtypes available for comparison in the respective test databases or access to a certain test. Furthermore it should be kept in mind that a clear clinical benefit has not been shown (and is difficult to demonstrate) for the molecular tests, due to a lack of suitable prospective studies.
Chances for an improved treatment of CUP patients also lie in the use of next-generation sequencing, either as whole genome, exome or panel sequencing. Using these approaches common underlying molecular changes can be identified and, if druggable gene mutations are present, used for the application of targeted therapies. The use of the tumor-specific mutation pattern as the primary criteria for the choice of therapy, irrespective of the tumor origin, has been demonstrated in small scale studies88 and will be used in so called basket studies, like e.g. the NCI MATCH trial starting in the middle of 2015.
Lymphoid malignancies
The main effort in research on lymphoid malignancies during the last 30 years has been directed towards a precise, reproducible and world wide accepted classification that has ultimately led to the identification of nearly 110 different variants. This approach has had however only minimal influence on treatment that still heavily relies on the empiric administration of combination chemotherapy. The only major therapeutic change has been the incorporation of the anti-CD20 monoclonal antibody rituximab that can be considered as the first biomarker-based treatment for B-cell malignancies. The demonstration of CD20 expression on the neoplastic cells is a prerequisite for Rituximab application but it cannot predict response to treatment. The comprehensive assessment of genetics by means of whole-exome and whole-genome sequencing has revealed that genomic alterations are enriched within and across lymphoma variants. In addition, these studies have revealed that each individual lymphoma harbors a unique combination of genomic alterations. Therefore the major task of future lymphoma research will be the determination of key driver mutations and of possible oncogenic combinations of mutations that can serve as predictive biomarkers beyond the current classification. Some currently used predictive biomarkers are listed on Table 1 and discussed below.
Diffuse large B-cell lymphoma represents the most common subtype of Non-Hodgkin lymphoma world wide accounting for 30–40% of all newly diagnosed cases. It represents an aggressive neoplasm that is fatal without treatment. Current immunochemotherapy employing rituximab and a combination of cyclophoshamide, doxorubicin, vincristine and prednisolon (R-CHOP) can cure the majority of DLBCL patients, but most patients who fail R-CHOP will ultimately die from their disease. Molecular studies have elucidated a complex biologic heterogeneity of DLCBL. In particular, determination of the cell of origin can deliver important predictive information with DLBCLs of the activated B-cell (ABC) molecular subtype exhibiting an inferior outcome after R-CHOP treatment than DLBCLs of the germinal center cell (GCB) subtype (3-year progression-free survival 40% Vs 75%).89, 90 Unfortunately, there is currently not a standardized commercially available test that allows the precise determination of the molecular DLBCL subtype. Gene expression profiling represents the gold standard but it is not routinely available, as it requires fresh-frozen tissue specimens. In order to cope with paraffin-embedded tissue specimens, several immunohistochemistry-based algorithms have been established that are in fact an imperfect substitution for gene expression profiling and also suffer from the poor reproducibility of immunehistochemistry.91
Besides determination of the molecular cell of origin, other established biologic predictive factors that can influence therapy of DLBCL are MYC and BCL2. Rearrangements of MYC oncogene can be identified in approximately 10% of patients with DLBCL. The resulting increased expression of MYC protein is associated with increased proliferation of neoplastic cells and several studies could demonstrate that MYC rearrangement is associated with a poorer outcome in R-CHOP-treated DLBCL patients.92, 93 In addition, recent studies have shown that the impact of MYC is heavily influenced by BCL2. So called “double hit” lymphomas harboring MYC and BCL2 translocation represent a small group (approximately 5% of DLBCL cases) that are usually refractory to treatment with a median survival of approximately 8 months.94 In addition, overexpression of MYC protein due to an up-regulation by other mechanisms than gene translocation can be detected by immunohistochemistry in up to 30% of DLBL patients. Interestingly a negative prognostic impact of such MYC protein overexpression is observed only in patients who simultaneously overexpress BCL2 protein. Such “dual expressers” account for approximately 25% of DLBCL patients who have a significantly poorer outcome than patients who express only one or neither protein.95, 96, 97 It is thus conceivable that all DLBCL cases should be routinely assessed for translocations and protein overexpression of MYC and BCL2 in order to identify patients that could be subjected to alternative therapies.
Hairy cell leukemia (HCL) is an uncommon malignancy of mature B cells with a characteristic morphology and immunophenotype primarily affecting adult males. HCL is characterized by the involvement of the red pulp of the spleen, sinusoids of the liver, bone marrow, and peripheral blood ad extremely rarely by a lymphadenopathy.98 Apart from the classical HCL (cHCL) that shows expression of CD20, CD22, CD25, Annexin A1, DBA.44, tartrate-resistant acid phosphatase (TRAP), CD103, CD123 and T-bet two other subsets exist: the HCL variant (HCLv) with a different immunophenotype than cHCL with a lack of CD25, annexin A1, and/or TRAP expression,99 and the HCL expressing the immunoglobulin VH4-34 rearrangement.100 Identification of these variants is of importance as most patients with cHCL show an excellent prognosis with the treatment with cladribine or pentostatin with or without an anti-CD20 monoclonal antibody, while HCLv and VH4-34 HCL have a relatively poor response to this treatment and are associated with a shorter overall survival. The discovery of BRAF V600E mutation in the tumor cells of patients with hairy cell leukemia (HCL)101 revealed that this represents not only a diagnostic but also a prognostic biomarker as it can differentiate between cHCL constantly harboring this mutation, while HCLv and VH4-34 HCL are BRAF mutation negative.102 In addition, detection of BRAF mutation can also serve as a predictive biomarker in cHCL: although the majority of these patients show an excellent response to the mentioned therapy with purine analogs, up to 40% may have a relapse. As the treatment of the relapse can be difficult by decreased responsiveness to chemotherapy, progressive cytopenia and opportunistic infections other therapeutic options are discussed in the literature. Few case reports have convincingly shown that vemurafenib (an oral inhibitor of BRAF serine threonine kinase) is a promising option for patients with cHCL.103
Waldenström macroglobulinemia (WM) is an incurable B-cell neoplasm that belongs to the entity of lymphoplasmocytic lymphoma (LPL) and is defined as LPL with bone marrow involvement and an IgM monoclonal gammopathy of any concentration (WHO).
Using whole-genome sequencing two activating somatic mutations could be discovered in WM namely one affecting the Toll-like receptor and the other the CXCR4 receptor signaling.104 Most of the patients with WM have a mutation L265P in MYD88 gene, while 1/3 of the patients show a mutation in the C terminus of CXCR4. The MYD88 mutation triggers the interleukin-1 receptor associated kinase (IRAK) and the Bruton's tyrosine kinase (BTK) resulting in activation of the malignant cell growth, while 2 classes of CXCR4 have been described: a nonsense (ns) and a frameshift (fs) mutation. A recent study could delineate the prognostic value of these mutations: WM patients with MYD88 L265P and CXCR4 ns mutations were overrepresented in the group of aggressive cases presenting with higher bone marrow disease involvement, higher IgM levels and hyperviscosity syndrome, while patients with MYD88 and CXCR4 wild-type presented with the lowest bone marrow involvement. Patients with MYD88 wild-type showed significantly higher mortality than patients with MYD88 L265P. Therefore determination of MYD88 and CXCR4 mutation status can be also of predictive value in the future as targeted therapies are going to be developed for WM patients.
Mesenchymal tumors
Within the heterogeneous group of human soft tissue tumors, GISTs are the most common mesenchymal tumors in the gastrointestinal tract. Discovery of KIT receptor tyrosine kinase expression and KIT mutations opened up novel therapeutic options. Approximately 85% of GISTs harbor a KIT mutation or a rare PDFGR- (platelet-derived growth factor) receptor alpha mutation.105 Oncogenic KIT mutations occur within different domains and the location is relevant for sensitivity to targeted inhibitors, and in some cases for prognosis.106 Most KIT mutations are found in the juxta-membrane domain encoded by exon 11, PDGFR alpha mutations are often D842V substitutions within the PDGFR alpha tyrosine kinase 2 domain encoded by exon 18.106 Less than 1% of GIST cases feature BRAF V600E mutations,107 and around 10% of adult GIST and most GISTs in children show none of the above mentioned mutations.108 Some studies analyzed the different mutation types of GISTs that occur in patients with special syndromes, e.g. neurofibromatosis type 1 comparing to sporadic GISTs.109
Nowadays Imatinib-based therapy is recommended as a first-line treatment of patients with non-resectable and/or metastatic GIST as well as for adjuvant therapy in high risk patients.98, 110, 111, 112 Tumors with a KIT mutation in exon 11 are most sensitive to Imatinib, while GISTs with a PDGFRA mutation in exon 18 (D842V) seem to be resistant to this therapy.113, 114 GISTs with a KIT mutation in exon 9 need a higher dose of Imatinib (>400 mg/day) to gain a longer progression-free survival.115 Furthermore, some patients develop a secondary resistance under Imatinib therapy. Sunitinib malate as a multi-targeted tyrosine kinase inhibitor has been approved for second-line therapy in these patients with disease progression under Imatinib therapy.116, 117
Within the last two years genetic landscapes determined via whole-genome or exome sequencing of other childhood mesenchymal tumors including Ewing Sarcoma (ES) and Rhabdomyosarcoma (RMS;118) have been published. Shern et al. unraveled an extensive genetic alteration map for Rhabdomysoarcoma. Their data indicate that in addition to PAX3/7 fusions also other genetic alterations including well-known oncogenes such as NRAS, KRAS and FGFR can occur in a subgroup of RMS patients, albeit with low incidence. The observation that the RTK/RAS/PIK3CA axis is affected in a large proportion of patients should be considered in more detail and included in novel clinical trials. Current efforts treating the PAX-fusion target IGF1R in RMS patients with the anti-IFG1R antibody either in combination with chemotherapy or other signaling pathway inhibitors such as temsirolimus have not proven satisfying results and lack a stringent biomarker support.119, 120 Genomic landscapes of Ewing sarcoma were published by several groups via whole-genome/exome or targeted sequencing,121, 122 see also review by Sand et al.123). In general, relatively low overall numbers of genetic alterations such as CNVs, indels and other structural variants not being EWSR1-ETS were discovered. Among the most frequent alterations were mutations within the STAG2, CDKN2A and TP53 genes. Interesting, mutational inactivation or loss of STAG2 alone or together with TP53 alterations seems to be a prognostic marker as it is frequently associated with disease dissemination and poor outcome of the patients.121 More recently, Agelopoulos et al.124 confirmed these results but in addition discovered higher numbers of mutations in relapsed tumors and copy number gain of the FGFR1 gene in roughly one-third of the patients. Preclinical investigations using an FGFR1 inhibitor showed promising results and might lead to improved clinical trials and biomarker development for Ewing sarcoma in the near future.
In addition to RMS and ES, also osteosarcomas have been analyzed in detail via exome sequencing.125 This analysis revealed an interesting pattern of ‘kataegis’ (localized hypermutations) in half of the samples tested, structural alterations and copy number alterations, yet rather few targetable mutations. The most frequently altered genes comprise the TP53, RB1, ATRX and DLG2 genes. While no structural alterations where determined via sequencing in the EGFR family, clinical trials are already under way testing the efficacy of both kinase inhibitors and antibodies against members of the EGFR family in osteosarcomas and other mesenchymal cancers such as synovial sarcomas (see review by Wang et al.126) These approaches are based on observations that sarcomas can overexpress EGFR and other members of this RTK family, however the results rather dampen the expectations and await further detailed investigation.
Central nervous system tumors
Depending on certain cytological and histological criteria the World Health Organization (WHO) assigns four grades for CNS tumors (WHO grade I to IV). While these grades are mainly based on histopathological criteria characterizing malignancy, they also include and thus predict the expected clinical course of the patient.127
Based on the rapid progress in the molecular understanding of the pathogenesis of brain tumors, in particular of glial tumors, new molecular signatures have been identified, which serve as reliable diagnostic, prognostic and predictive molecular markers (Figure 5). Based on several clinical studies three molecular markers—namely, 1p/19q co-deletion, MGMT promoter methylation and mutations in the IDH1/2 genes—just to mention the to date most prominent ones, were routinely used to improve and manage patients’ outcome.

(a) malignant glial tumor with numerous vascular proliferations (Glioblastoma WHO grade IV), (b) the tumor cells show a nuclear loss of the Atrx Protein according to a mutation in the transcription factor ATRX, which is a molecular feature of astroglial differentiation. Endothelial cells and reactive astrocytes are positive. (c) the gemistocytic tumor cells strongly react with antibodies against the mutation specific marker IDH1 indicating an IDH1(R132H) mutation. IDH mutant malignant glioma show longer overall survival than those with IDH wild-type neoplasms. (d) IDH1(R132H) mutation was confirmed by pyro-sequencing.
Combined loss of 1p/19q in glial tumors
It has been shown that about 80% of oligodendroglioma (grade II), 60% of anaplastic oligodendroglioma (grade III) and 50% of mixed glioma are characterized by combined allelic deletions on chromosome 1p and 19q.85 Two clinical studies demonstrated that patients with anaplastic glioma benefit from a combined radio- plus PCV chemotherapy if the tumor harbor a combined 1p/19q loss.128, 129 MGMT promoter methylation: The O6-methylguanine DNA methyltransferase (MGMT) is a DNA repair enzyme that removes the alkylation of the O6 position of guanine, which protects DNA alkylation and finally apoptosis. Methylation of the MGMT promoter results in gene silencing due to reduced proficiency to repair DNA damage induced by alkylating chemotherapeutic agents like temozolomide. MGMT promoter methylation occurs in about 40% of primary glioblastoma (WHO grade IV) and is associated with an increased survival after radiotherapy and chemotherapy with temozolomide.130, 131 Recently, two independent multicentric studies demonstrated the predictive relevance of MGMT status in older glioblastoma patients (>65 years,) which benefit from a temozolomide therapy alone if MGMT is methylated.
Mutations of IDH1 and IDH2
IDH (Isocitrate-Dehydrogenase)1 and its mitochondrial isoform IDH2 encode for proteins which catalyze isocitrate to α-ketoglutarate and play important roles in the cellular control of oxidative processes. IDH1/2 mutations functionally results in global changes of the tumor-epigenome. The presence of somatic IDH1/2 point mutations in the vast majority of low-grade glioma and secondary glioblastoma is helpful in the differentiation to primary glioblastoma, pilocytic astrocytoma and other primary brain tumors, which are not characterized by these mutations. The presence of IDH1/2 mutations in anaplastic glioma and glioblastoma has also a prognostic relevance as patients with IDH mutant tumors show longer overall survival than those with IDH wild-type neoplasms.
The continuous identification of novel molecular signatures, apart from the above mentioned ones, forces state-of-the-art neuropathology to broaden its diagnostic molecular spectrum by persistently implementing such molecular markers within the standard repertoire of diagnostic procedures used to characterize brain tumors.
In current meetings concerning the revision of the fourth edition of the WHO classification of central nervous system tumors it has been discussed if for some entities (in particular oligodendroglial and some pediatric nervous system tumors), molecular information are required to provide an ‘integrated’ diagnosis and only an descriptive histological diagnosis is acceptable if no molecular diagnostic testing is available. For instance in case of mixed glioma (oligoastrocytoma), independent of the histological phenotype the IDH1/2, ATRX and 1p/19q status will define the overall diagnosis.132
Conclusions
In addition to what is written in the previous paper1 we can state that the development of targeted therapy is dramatic, the number of new drugs in clinical studies is enormous and the resulting challenges for predictive pathology/companion diagnostic are indeed substantial. Since currently the vast majority of the assays are tissue-based the responsibility of accurate performance lies in the hand of pathologists. The scientific societies have to be alert to cover this chance. Education of doctors and technicians, quality control of technical procedures and the intellectual interpretation of the results are crucial to provide reliable results. Clearly this will play an increasing role in the future structure of tissue-based diagnostics.
The multilayer analyses of malignant tumors produce an increasing number of data creating a high complexity of information for more or less each tumor. To manage this situation interdisciplinary becomes an absolute important prerequisite in cancer treatment. This is true in daily work and in scientific projects. A consequence of this situation is the necessity to build up comprehensive cancer centers which can provide the broad spectrum of knowledge and experience.
In addition an up-dated design of clinical studies is needed to bring the possible advantages of the new multiplex approach to patients. Here all medical disciplines, the regulatory authorities and pharma industry have to stay and think together how the progress can be translated fast and safe into the clinic. This may become a severe challenge in particular for rare tumor types.
References
Dietel M, Johrens K, Laffert M, Hummel M, Blaker H, Muller BM et al. Predictive molecular pathology and its role in targeted cancer therapy: a review focussing on clinical relevance. Cancer Gene Ther 2013; 20: 211–221.
Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013; 368: 2385–2394.
Willyard C . 'Basket studies' will hold intricate data for cancer drug approvals. Nat Med 2013; 19: 655.
Sleijfer S, Bogaerts J, Siu LL . Designing transformative clinical trials in the cancer genome era. J Clin Oncol 2013; 31: 1834–1841.
Duan N, Kravitz RL, Schmid CH . Single-patient (n-of-1) trials: a pragmatic clinical decision methodology for patient-centered comparative effectiveness research. J Clin Epidemiol 2013; 66: S21–S28.
Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489: 519–525.
Klauschen F, Andreeff M, Keilholz U, Dietel M, Stenzinger A . The combinatorial complexity of cancer precision medicine. Oncoscience 2014; 1: 504–509.
Kerick M, Isau M, Timmermann B, Sultmann H, Herwig R, Krobitsch S et al. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genomics 2011; 4: 68.
Collins FS . Hamburg MA. First FDA authorization for next-generation sequencer. N Engl J Med 2013; 369: 2369–2371.
Jamshidi F, Nielsen TO, Huntsman DG . Cancer genomics: why rare is valuable. J Mol Med 2015; 93: 369–381.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014; 6: 224ra24.
Taguchi F, Solomon B, Gregorc V, Roder H, Gray R, Kasahara K et al. Mass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: a multi cohort cross-institutional study. J Natl Cancer Institute 2007; 99: 838–846.
Kuiper JL, Lind JS, Groen HJ, Roder J, Grigorieva J, Roder H et al. VeriStrat((R)) has prognostic value in advanced stage NSCLC patients treated with erlotinib and sorafenib. Br J Cancer 2012; 107: 1820–1825.
van Krieken JH, Normanno N, Blackhall F, Boone E, Botti G, Carneiro F et al. Guideline on the requirements of external quality assessment programs in molecular pathology. Virchows Arch 2013; 462: 27–37.
von Laffert M, Warth A, Penzel R, Schirmacher P, Kerr KM, Elmberger G et al. Multicenter immunohistochemical ALK-testing of non-small-cell lung cancer shows high concordance after harmonization of techniques and interpretation criteria. J Thorac Oncol 2014; 9: 1685–1692.
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA et al. Molecular portraits of human breast tumours. Nature 2000; 406: 747–752.
Goldhirsch A, Winer EP, Coates AS, Gelber RD, Piccart-Gebhart M, Thurlimann B et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol 2013; 24: 2206–2223.
Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004; 351: 2817–2826.
Filipits M, Rudas M, Jakesz R, Dubsky P, Fitzal F, Singer CF et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res 2011; 17: 6012–6020.
Gnant M, Filipits M, Greil R, Stoeger H, Rudas M, Bago-Horvath Z et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: using the PAM50 Risk of Recurrence score in 1478 postmenopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann Oncol 2014; 25: 339–345.
Dubsky P, Brase JC, Jakesz R, Rudas M, Singer CF, Greil R et al. The EndoPredict score provides prognostic information on late distant metastases in ER+/HER2- breast cancer patients. Br J Cancer 2013; 109: 2959–2964.
Filipits M, Nielsen TO, Rudas M, Greil R, Stoger H, Jakesz R et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res 2014; 20: 1298–1305.
Loibl S, von Minckwitz G, Schneeweiss A, Paepke S, Lehmann A, Rezai M et al. PIK3CA mutations are associated with lower rates of pathologic complete response to anti-human epidermal growth factor receptor 2 (her2) therapy in primary HER2-overexpressing breast cancer. J Clin Oncol 2014; 32: 3212–3220.
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474: 609–615.
Roy R, Chun J, Powell SN . BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 2012; 12: 68–78.
Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol 2012; 30: 2654–2663.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 2012; 366: 1382–1392.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 2014; 15: 852–861.
Rigakos G, Razis E . BRCAness: finding the Achilles heel in ovarian cancer. Oncologist 2012; 17: 956–962.
Dann RB, DeLoia JA, Timms KM, Zorn KK, Potter J, Flake DD 2nd et al. BRCA1/2 mutations and expression: response to platinum chemotherapy in patients with advanced stage epithelial ovarian cancer. Gynecol Oncol 2012; 125: 677–682.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26: 1626–1634.
Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359: 1757–1765.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010; 11: 753–762.
Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer 2012; 48: 1466–1475.
Thibodeau SN, Bren G, Schaid D . Microsatellite instability in cancer of the proximal colon. Science 1993; 260: 816–819.
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M . Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993; 363: 558–561.
Kim H, Jen J, Vogelstein B, Hamilton SR . Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994; 145: 148–156.
Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 2010; 28: 3219–3226.
Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Institute 2004; 96: 261–268.
Kloor M, Voigt AY, Schackert HK, Schirmacher P, von Knebel Doeberitz M, Blaker H . Analysis of EPCAM protein expression in diagnostics of Lynch syndrome. J Clin Oncol 2011; 29: 223–227.
McCoach CE, Doebele RC . The minority report: targeting the rare oncogenes in NSCLC. Curr Treat Options Oncol 2014; 15: 644–657.
Thunnissen E, Bubendorf L, Dietel M, Elmberger G, Kerr K, Lopez-Rios F et al. EML4-ALK testing in non-small cell carcinomas of the lung: a review with recommendations. Virchows Arch 2012; 461: 245–257.
Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. Arch Pathol Lab Med 2013; 137: 828–860.
Reck M, Popat S, Reinmuth N, De Ruysscher D, Kerr KM, Peters S . Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014; 25: iii27–iii39.
Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 2011; 12: 1004–1012.
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363: 1693–1703.
Blackhall FH, Peters S, Bubendorf L, Dafni U, Kerr KM, Hager H et al. Prevalence and clinical outcomes for patients with ALK-positive resected stage I to III adenocarcinoma: results from the European Thoracic Oncology Platform Lungscape Project. J Clin Oncol 2014; 32: 2780–2787.
Sampsonas F, Ryan D, McPhillips D, Breen DP . Molecular testing and personalized treatment of lung cancer. Curr Mol Pharmacol 2014; 7: 22–32.
Stinchcombe TE . Novel agents in development for advanced non-small cell lung cancer. Ther Adv Med Oncol 2014; 6: 240–253.
Warth A, Stenzinger A, Weichert W . Novel morphological and molecular aspects of lung cancer. Der Pathologe 2013; 34: 419–428.
Powell CABE, Bubendorf L, Dacic S, Dziadziusko R, Geisinger K et al. Molecular testing for treatment selection in lung cancer. In: Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG (eds), WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. 4th edn. 2015; Lyon, pp 22–25.
Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 2013; 5: 216ra177.
Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther 2013; 12: 220–229.
Tibaldi C . Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small-cell lung cancer. Pharmacogenomics 2014; 15: 133–135.
Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 2012; 18: 1472–1482.
Taniguchi K, Okami J, Kodama K, Higashiyama M, Kato K . Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci 2008; 99: 929–935.
Ilie M, Hofman V, Long E, Bordone O, Selva E, Washetine K et al. Current challenges for detection of circulating tumor cells and cell-free circulating nucleic acids, and their characterization in non-small cell lung carcinoma patients. What is the best blood substrate for personalized medicine? Ann Transl Med 2014; 2: 107.
Bos M, Gardizi M, Schildhaus HU, Heukamp LC, Geist T, Kaminsky B et al. Complete metabolic response in a patient with repeatedly relapsed non-small cell lung cancer harboring ROS1 gene rearrangement after treatment with crizotinib. Lung Cancer 2013; 81: 142–143.
Ou SH, Kwak EL, Siwak-Tapp C, Dy J, Bergethon K, Clark JW et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol 2011; 6: 942–946.
Jurmeister P, Lenze D, Berg E, Mende S, Schaper F, Kellner U et al. Parallel screening for ALK, MET and ROS1 alterations in non-small cell lung cancer with implications for daily routine testing. Lung Cancer 2015; 87: 122–129.
Yoshida A, Tsuta K, Wakai S, Arai Y, Asamura H, Shibata T et al. Immunohistochemical detection of ROS1 is useful for identifying ROS1 rearrangements in lung cancers. Mod Pathol 2014; 27: 711–720.
Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA 2007; 104: 20932–20937.
Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH Jr., Blumenschein GR Jr. et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 2013; 31: 4105–4114.
Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 2015; 5: 850–859.
Hirsch FR, Bunn PA Jr. . Herbst RS. "Companion diagnostics": has their time come and gone? Clin Cancer Res 2014; 20: 4422–4424.
Vijayvergia N, Mehra R . Clinical challenges in targeting anaplastic lymphoma kinase in advanced non-small cell lung cancer. Cancer Chem Pharmacol 2014; 74: 437–446.
Erickson C, Driscoll MS . Melanoma epidemic: facts and controversies. Clin Dermatol 2010; 28: 281–286.
McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014; 15: 323–332.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380: 358–365.
Tronnier M, Mitteldorf C . Treating advanced melanoma: current insights and opportunities. Cancer Management Res 2014; 6: 349–356.
Livingstone E, Zimmer L, Vaubel J, Schadendorf D . BRAF, MEK and KIT inhibitors for melanoma: adverse events and their management. Chinese Clin Oncol 2014; 3: 29.
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N . RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010; 464: 427–430.
Dinges HC, Capper D, Ritz O, Bruderlein S, Marienfeld R, von Deimling A et al. Validation of a manual protocol for BRAF V600E mutation-specific immunohistochemistry. Appl Immunohistochem Mol Morphol 2015; 23: 382–388.
Jakob JA, Bassett RL Jr., Ng CS, Curry JL, Joseph RW, Alvarado GC et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012; 118: 4014–4023.
Torres-Cabala CA, Wang WL, Trent J, Yang D, Chen S, Galbincea J et al. Correlation between KIT expression and KIT mutation in melanoma: a study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod Pathol 2009; 22: 1446–1456.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012; 367: 1694–1703.
Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov 2013; 3: 742–750.
Hu-Lieskovan S, Robert L, Homet Moreno B, Ribas A . Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challenges. J Clin Oncol 2014; 32: 2248–2254.
Pentheroudakis G, Golfinopoulos V, Pavlidis N . Switching benchmarks in cancer of unknown primary: from autopsy to microarray. Eur J Cancer 2007; 43: 2026–2036.
Pavlidis N, Fizazi K . Carcinoma of unknown primary (CUP). Crit Rev Oncol 2009; 69: 271–278.
Greco FA, Pavlidis N . Treatment for patients with unknown primary carcinoma and unfavorable prognostic factors. Semin Oncol 2009; 36: 65–74.
Ma XJ, Patel R, Wang X, Salunga R, Murage J, Desai R et al. Molecular classification of human cancers using a 92-gene real-time quantitative polymerase chain reaction assay. Arch Pathol Lab Med 2006; 130: 465–473.
Pillai R, Deeter R, Rigl CT, Nystrom JS, Miller MH, Buturovic L et al. Validation and reproducibility of a microarray-based gene expression test for tumor identification in formalin-fixed, paraffin-embedded specimens. J Mol Diagn 2011; 13: 48–56.
Meiri E, Mueller WC, Rosenwald S, Zepeniuk M, Klinke E, Edmonston TB et al. A second-generation microRNA-based assay for diagnosing tumor tissue origin. Oncologist 2012; 17: 801–812.
Greco FA, Lennington WJ, Spigel DR, Hainsworth JD . Molecular profiling diagnosis in unknown primary cancer: accuracy and ability to complement standard pathology. J Natl Cancer Institute 2013; 105: 782–790.
Weiss LM, Chu P, Schroeder BE, Singh V, Zhang Y, Erlander MG et al. Blinded comparator study of immunohistochemical analysis versus a 92-gene cancer classifier in the diagnosis of the primary site in metastatic tumors. J Mol Diagn 2013; 15: 263–269.
Handorf CR, Kulkarni A, Grenert JP, Weiss LM, Rogers WM, Kim OS et al. A multicenter study directly comparing the diagnostic accuracy of gene expression profiling and immunohistochemistry for primary site identification in metastatic tumors. Am J Surg Pathol 2013; 37: 1067–1075.
Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res 2012; 18: 6373–6383.
Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002; 346: 1937–1947.
Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008; 359: 2313–2323.
Meyer PN, Fu K, Greiner TC, Smith LM, Delabie J, Gascoyne RD et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J Clin Oncol 2011; 29: 200–207.
Savage KJ, Johnson NA, Ben-Neriah S, Connors JM, Sehn LH, Farinha P et al. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood 2009; 114: 3533–3537.
Barrans S, Crouch S, Smith A, Turner K, Owen R, Patmore R et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010; 28: 3360–3365.
Friedberg JW . Double-hit diffuse large B-cell lymphoma. J Clin Oncol 2012; 30: 3439–3443.
Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012; 30: 3452–3459.
Green TM, Young KH, Visco C, Xu-Monette ZY, Orazi A, Go RS et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012; 30: 3460–3467.
Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013; 121: 4021–4031 quiz 4250.
Goodman GR, Bethel KJ, Saven A . Hairy cell leukemia: an update. Curr Opin Hematol 2003; 10: 258–266.
Matutes E, Wotherspoon A, Catovsky D . The variant form of hairy-cell leukaemia. Best Pract Res Clin Haematol 2003; 16: 41–56.
Arons E, Kreitman RJ . Molecular variant of hairy cell leukemia with poor prognosis. Leuk Lymphoma 2011; 52: 99–102.
Tiacci E, Schiavoni G, Forconi F, Santi A, Trentin L, Ambrosetti A et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the BRAF-V600E mutation. Blood 2012; 119: 192–195.
Xi L, Arons E, Navarro W, Calvo KR, Stetler-Stevenson M, Raffeld M et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012; 119: 3330–3332.
Jain P, Polliack A, Ravandi F . Novel therapeutic options for relapsed hairy cell leukemia. Leuk Lymphoma 2015; 4: 1–9.
Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR . Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood 2014; 123: 2791–2796.
Corless CL, Heinrich MC . Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol 2008; 3: 557–586.
Miettinen M, Lasota J . Gastrointestinal stromal tumors. Gastroenterol Clin North Am 2013; 42: 399–415.
Agaram NP, Wong GC, Guo T, Maki RG, Singer S, Dematteo RP et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008; 47: 853–859.
Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, Ladanyi M . Advances in sarcoma genomics and new therapeutic targets. Nature Rev Cancer 2011; 11: 541–557.
Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 2006; 15: 1015–1023.
Blay JY, Rutkowski P . Adherence to imatinib therapy in patients with gastrointestinal stromal tumors. Cancer Treat Rev 2014; 40: 242–247.
Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 2009; 373: 1097–1104.
DeMatteo RP, Ballman KV, Antonescu CR, Corless C, Kolesnikova V, von Mehren M et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor: ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg 2013; 258: 422–429.
Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005; 23: 5357–5364.
Lee JH, Kim Y, Choi JW, Kim YS . Correlation of imatinib resistance with the mutational status of KIT and PDGFRA genes in gastrointestinal stromal tumors: a meta-analysis. J Gastrointest Liv Dis 2013; 22: 413–418.
Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006; 42: 1093–1103.
Heng DY, Kollmannsberger C . Sunitinib. Recent Results Cancer Res 2010; 184: 71–82.
Patel S . Managing progressive disease in patients with GIST: factors to consider besides acquired secondary tyrosine kinase inhibitor resistance. Cancer Treat Rev 2012; 38: 467–472.
Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 2014; 4: 216–231.
Wagner LM, Fouladi M, Ahmed A, Krailo MD, Weigel B, DuBois SG et al. Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 2015; 62: 440–444.
Weiss A, Gill J, Goldberg J, Lagmay J, Spraker-Perlman H, Venkatramani R et al. Advances in therapy for pediatric sarcomas. Current Oncol Rep 2014; 16: 395.
Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 2014; 4: 1326–1341.
Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 2014; 4: 1342–1353.
Sand LG, Szuhai K, Hogendoorn PC . Sequencing overview of Ewing Sarcoma: a journey across genomic, epigenomic and transcriptomic landscapes. Int J Mol Sci 2015; 16: 16176–16215.
Agelopoulos K, Richter GH, Schmidt E, Dirksen U, von Heyking K, Moser B et al. Deep sequencing in conjunction with expression and functional analyses reveals activation of FGFR1 in Ewing sarcoma. Clin Cancer Res. 15 July 2015. pii: clincanres.2744.2014 (e-pub ahead of print).
Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 2014; 7: 104–112.
Wang H, Yang Q, Fu Z, Zuo D, Hua Y, Cai Z . ErbB receptors as prognostic and therapeutic drug targets in bone and soft tissue sarcomas. Cancer Invest 2014; 32: 533–542.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007; 114: 97–109.
Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol 2013; 31: 337–343.
van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 2013; 31: 344–350.
Malmstrom A, Gronberg BH, Marosi C, Stupp R, Frappaz D, Schultz H et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol 2012; 13: 916–926.
Wick W, Platten M, Meisner C, Felsberg J, Tabatabai G, Simon M et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol 2012; 13: 707–715.
Louis DN, Perry A, Burger P, Ellison DW, Reifenberger G, von Deimling A et al. International Society Of Neuropathology—Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 2014; 24: 429–435.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Dietel, M., Jöhrens, K., Laffert, M. et al. A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: a review focussing on clinical relevance. Cancer Gene Ther 22, 417–430 (2015). https://doi.org/10.1038/cgt.2015.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cgt.2015.39
- Springer Nature America, Inc.
This article is cited by
-
The paediatric cancer clinical research landscape in Spain: a 13-year multicentre experience of the new agents group of the Spanish Society of Paediatric Haematology and Oncology (SEHOP)
Clinical and Translational Oncology (2021)
-
Optimal specimen type for accurate diagnosis of infectious peripheral pulmonary lesions by mNGS
BMC Pulmonary Medicine (2020)
-
Comprehensive routine diagnostic screening to identify predictive mutations, gene amplifications, and microsatellite instability in FFPE tumor material
BMC Cancer (2020)
-
The Effects of Different mTOR Inhibitors in EGFR Inhibitor Resistant Colon Carcinoma Cells
Pathology & Oncology Research (2019)
-
A Guide to Total Quality Management System (TQMS) in Molecular Diagnostics from Experiences in Seeking Accreditation and Implementation
SN Comprehensive Clinical Medicine (2019)