
Abstract
The epidermal growth factor receptor (EGFR) is an oncogenic trans-membranous receptor, which is overexpressed in multiple human cancers. However, the role of EGFR in gastric cancer (GC) is still elusive. In this study, we aimed to investigate the expression and molecular mechanisms of EGFR in GC cells. Forty cases of GC and the corresponding adjacent non-cancerous tissues (ANCT) were collected, and the expression of EGFR was assessed using immunohistochemistry in biopsy samples. Furthermore, EGFR signaling was blocked by constructed recombinant small hairpin RNA lentiviral vector (Lv-shRAGE) used to transfect into human GC SGC-7901 cells. The expression of AKT, proliferating cell nuclear antigen (PCNA) and matrix metallopeptidase-9 (MMP-9) was detected by real-time PCR and western blotting assays. Cell proliferative activities and invasive capability were, respectively, determined by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) and Transwell assays. Cell apoptosis and cycle distribution were analyzed by flow cytometry. EGFR was found highly expressed in cancer tissues compared with the ANCT and correlated with lymph node metastases. Knockdown of EGFR reduced cell proliferation and invasion of GC with decreased expression of AKT, PCNA and MMP-9 and induced cell apoptosis and cycle arrest. Upregulation of EGFR expression is associated with lymph node metastases of GC, and blockade of EGFR signaling suppresses growth and invasion of GC cells through AKT pathway, suggesting that EGFR may represent a potential therapeutic target for this aggressive malignancy.
Similar content being viewed by others


Introduction
Gastric carcinogenesis is a multi-step process with genetic alterations, including mutation, activation of oncogenes and suppression of tumor-suppressor genes, and overexpression of growth factors. Genetic instability, DNA methylation and mutations are known to participate in the early development of gastric cancer (GC). Epigenetic changes become important areas in GC research.1, 2 Evidence has indicated that GC is considered one of the most deadly tumors worldwide. Even with the decline in its incidence, the mortality rate of this disease has remained high, mainly due to its late diagnosis and to the lack of precise prognostic markers.3, 4 Despite improvements in medical technology, such as the development of new diagnostic imaging methods, GC remains a silent disease that is frequently diagnosed in advanced stages, which is responsible for its elevated mortality.5 Additionally, the presence of metastasis in the lymph nodes is a frequent event in this type of neoplasia and is considered an important prognostic marker, because it may contribute to the high rates of recurrence and/or GC mortality.6 However, the mechanisms of GC cells’ migration, invasion and metastasis have not been fully understood.
Mutations that lead to EGFR overexpression (known as upregulation) or overactivity have been associated with a number of cancers, including lung cancer, anal cancers and glioblastoma multiforme.7, 8, 9, 10 These somatic mutations involving EGFR lead to its constant activation, which produces uncontrolled cell division.11 In glioblastoma, a more or less specific mutation of EGFR, called EGFR III, is often observed.12 Mutations, amplifications or misregulations of EGFR or family members are implicated in about 30% of all epithelial cancers.13, 14
However, few studies have indicated that, EGFR expression is strongly reduced at the mRNA and protein levels in non-small cell lung cancer (NSCLC), and overexpression of EGFR diminishes tumor growth, while downregulation of EGFR supports NSCLC and may be considered as a critical step in tissue reorganization and the formation of NSCLC.15 EGFR expression is negatively correlated with depth of invasion in esophageal squamous cell carcinoma, and tumors with positive EGFR expression exhibit better prognosis than those with negative EGFR expression.16, 17 Therefore, in order to expound the role of EGFR in GC, we investigated the expression of EGFR in human GC and the effects of EGFR knockdown on biological behaviors of GC cells.
Materials and methods
Materials
Human SGC-7901 GC cell line used in the experiments was from the Institute of Biochemistry and Cell Biology (Shanghai, China). Lv-shRAGE, negative control vector and virion-packaging elements were from Genechem (Shanghai, China). The primers of RAGE (receptor for advanced glycation end products) and AKT were synthesized by Applied Biosystems (Carlsbad, CA, USA). All antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA). They were cultured in RPMI 1640 media supplemented with 10% fetal bovine serum (FBS) and maintained at 37 °C in a humidified incubator with a 5% CO2 atmosphere.
Drugs and reagents
Dulbecco’s Modified Eagle medium (DMEM) and FBS were from Thermo Fisher Scientific (Waltham, MA, USA); TRIzol Reagent and Lipofectamine 2000 were from Invitrogen (Carlsbad, CA, USA); M-MLV Reverse Transcriptase was from Promega (Madison, WI, USA); SYBR Green Master Mixture was from Takara (Otsu, Japan). ECL-PLUS/Kit was from GE Healthcare (Piscataway, NJ, USA). Clinical samples and tissue microarray was prepared for immunohistochemical test. Human GC tissues and the corresponding adjacent non-cancerous tissues (ANCT) were obtained from biopsy prior to chemotherapy in a total of 40 consecutive cases of GC admitted to our hospital from January 2008 to December 2012. The study was approved by Medical Ethics Committee of Xinjiang Medical University, and written informed consent was obtained from the patients or their parents before sample collection.Two pathologists reviewed all cases.
Tissue microarray
The advanced tissue arrayer (ATA-100, Chemicon International, Tamecula, CA, USA) was used to create holes in a recipient paraffin block and to acquire cylindrical core tissue biopsies with a diameter of 1 mm from the specific areas of the donor block. The tissue core biopsies were transferred to the recipient paraffin block at defined array positions. The tissue microarrays contained tissue samples from 40 formalin-fixed paraffin-embedded cancer specimens with known diagnosis and the corresponding ANCT from these patients. The block was incubated in an oven at 45 °C for 20 min to allow complete embedding of the grafted tissue cylinders in the paraffin of the recipient block and then stored at 4 °C until microtome sectioning.
Immunohistochemical (IHC) staining
Anti-EGFR antibody (Santa Cruz Biotechnology) was used for IHC detection of the expression of EGFR protein in tissue microarrays. Tissue microarray sections were processed for IHC analysis of EGFR protein as follows. IHC examinations were carried out on 3-mm-thick sections.For anti-EGFR IHC, unmasking was performed with 10 mM sodium citrate buffer, pH 6.0, at 90 °C for 30 min. For anti-EGFR IHC, antigen unmasking was not necessary. Sections were incubated in 0.03% hydrogen peroxide for 10 min at room temperature, to remove endogenous peroxidase activity, and then in blocking serum (0.04% bovine serum albumin, A2153, Sigma-Aldrich, and 0.5% normal goat serum X0907, Dako Corporation, in phosphate-buffered saline (PBS)) for 30 min at room temperature. Anti-EGFR antibody was used at a dilution of 1:200. The antibody was incubated overnight at 4 °C. Sections were then washed three times for 5 min in PBS. Non-specific staining was blocked with 0.5% casein and 5% normal serum for 30 min at room temperature. Finally, staining was developed with diaminobenzidine substrate, and sections were counterstained with hematoxylin. PBS replaced EGFR antibody in the negative controls.
Western blotting analysis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed as described for zymography with the modification that the gel was polymerized on Net-Fix for PAG (Serva, Heidelberg, Germany) in the absence of gelatin. Three fold for matrix metallopeptidase-9 (MMP-9) detection by the use of microsep ultrafiltration devices (Pall Filtron, Dreireich, Germany). Samples (40 ml) were mixed with loading buffer and separated either under reducing conditions in the presence of DL-DTT with prior boiling or under non-reducing conditions without boiling. After electrophoresis, proteins were transferred to polyvinyl difluoride membranes (Pall Filtron) using a semidry blotting apparatus (Pharmacia, Piscataway, NJ, USA) and probed with mouse monoclonal antibodies to EGFR (0.4 mg ml−1) or MMP-9 (0.2 mg ml−1), followed by incubation with peroxidase-labeled secondary antibodies (all antibodies were purchased from Amersham, Braunschweig, Germany). Detection was performed by the use of an chemiluminescence system (Amersham) according to the manufacturer’s instructions.
Reverse transcriptase-PCR
RNA isolation and reverse transcription were performed as previously described. Oligonucleotide primer sequences were as follows: β-actin (264 bp), forward: 5′-GAGACCTTCAACACCCCAGCC-3′; reverse:5′-AATGTCACGCACGATTTCCC-3′; EGFR (233 bp), forward: 5′-CTGGCGGAGCACAACGAACT-3′, reverse:5′-AGGATATCTCCATTGGGCTGAAAG-3′; and MMP-9 (201 bp), forward: 5′-TCCCCATCGCCATCCCC-3′, reverse: 5′-CACCATGGCCTCGGCTGG-3′. To all the above genes, amplification was performed under the same cycling conditions (1 min at 94 °C, 50 s at 57 °C, 1 min at 72 °C), except the number of cycles that were specified for each gene (31 EGFR and 32 for MMP-9).
Quantification of protein expression
The expression of EGFR was semiquantitatively estimated as the total immunostaining scores, which were calculated as the product of a proportion score and an intensity score. The proportion and intensity of the staining was evaluated independently by two observers. The proportion score reflected the fraction of positive staining cells (score 0, <5%; score 1, 5–10%; score 2, 10–50%; score 3, 50–75%; score 4, >75%), and the intensity score represented the staining intensity (score 0, no staining; score 1, weak positive; score 2, moderate positive; score 3, strong positive). Finally, a total expression score was given ranging from 0 to 12. Based on the analysis in advance, EGFR was regarded as negative expression in GC tissues if the score <2 and positive expression if the score ⩾2.
Cell culture and infection
SGC-7901 GC cells were cultured in DMEM medium supplemented with 10% heat-inactivated FBS, 100 U ml−1 of penicillin and 100 μg ml−1 of streptomycin. They were all placed in a humidified atmosphere containing 5% CO2 at 37 °C. On the day of transduction, SGC-7901 cells were replated at 5 × 104 cells per well in 24-well plates containing serum-free growth medium with polybrene (5 mg ml−1). When reached 50% confluence, cells were transfected with recombinant Lv-shRAGE or control virus at the optimal MOI (multiplicity of infection) of 50 and cultured at 37 °C and 5% CO2 for 4 h. Then the supernatant was discarded, and the serum containing growth medium was added. At 4 days of posttransduction, transduction efficiency was measured by the frequency of green fluorescent protein-positive cells. Positive stable transfectants were selected and expanded for further study. The clone that LvshEGFR was transfected was named as the Lv-shEGFR group and in which the negative control vectors were transfected was named as the NC group.
Cell proliferation assay
Cell proliferation was analyzed with the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay. Briefly, the cells transfected with Lvsh EGFR were incubated in 96-well plates at a density of 1 × 105 cells per well with DMEM medium supplemented with 10% FBS. Cells were treated with 20 μl MTT dye at 0, 24, 48 and 72 h and then incubated with 150 μl of dimethyl sulfoxide for 5 min. The color reaction was measured at 570 nm with enzyme immunoassay analyzer (Bio-Rad Laboratories, Hercules, CA, USA). The proliferation activity was calculated for each clone.
Transwell invasion assay
Transwell filters were coated with matrigel (3.9 mg μl−1, 60–80 μl) on the upper surface of a polycarbonic membrane (diameter 6.5 mm, pore size 8 mm). After incubating at 37 °C for 30 min, the matrigel solidified and served as the extracellular matrix for analysis of tumor cell invasion. Harvested cells (1 × 105) in 100 μl of serum-free DMEM were added into the upper compartment of the chamber. A total of 200 μl conditioned medium derived from NIH3T3 cells was used as a source of chemoattractant and was placed in the bottom compartment of the chamber. After 24 h incubation at 37 °C with 5% CO2, the medium was removed from the upper chamber. The non-invaded cells on the upper side of the chamber were scraped off with a cotton swab. The cells that had migrated from the matrigel into the pores of the inserted filter were fixed with 100% methanol, stained with hematoxylin, mounted and dried at 80 °C for 30 min. The number of cells invading through the matrigel was counted in three randomly selected visual fields from the central and peripheral portion of the filter using an inverted microscope (Piscataway, NJ, USA) ( × 200 magnification). Each assay was repeated three times.
Cell apoptosis analysis
To detect cell apoptosis, SGC-7901 cells were trypsinized, washed with cold PBS and resuspended in binding buffer according to the instruction of the apoptosis kit. Fluorescein isothiocyanate -AnnexinV and propidium iodide (PI) were added to the fixed cells for 20 min in darkness at room temperature. Then, Annexin V-binding buffer (Becton Dickinson, Mountain View, CA, USA) was added to the mixture before the fluorescence was measured on a FAC sort flow cytometer. The cell apoptosis was analyzed using the Cell Quest software (Becton Dickinson, Franklin Lakes, NJ, USA). Three separate experiments were performed for each clone.
Cell cycle analysis
To detect cell cycle variation, SGC-7901 cells were trypsinized, washed with PBS and fixed with 80% cold ethanol overnight at −20 °C. After PBS washing, the fixed cells were stained with PI in the presence of RNase A for 30 min at room temperature in darkness. Each sample was filtered through a 50-μm nylon filter to obtain single-cell suspensions. The samples were then analyzed on FAC sort flow cytometer. ModFit3.0 software (Verity Software House, Topsham, ME, USA) was used for cell cycle analysis. Three separate experiments were performed for each clone.
Statistical analysis
SPSS 20.0 (Becton Dickinson, Mountain View, CA, USA) was used for the statistical analysis. Kruskal–Wallis H test and Chi-square test were used to analyze the expression rate in all the groups. One-way analysis of variance was used to analyze the differences between groups. The least significant difference method of multiple comparisons was used when the probability for analysis of variance was statistically significant. Statistical significance was set at P<0.05.
Results
Expression of EGFR protein in human GC
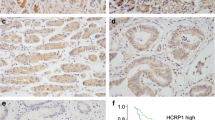
The expression of RAGE protein was evaluated using IHC staining. The positive expression of RAGE protein was detected in the cytoplasm of GC and ANCT cells (Figure 1). The positive rates of EGFR expression were examined in 70.0% (28/40) of the GC tissues and 45.0% (18/40) in a small fraction of ANCT. There was a significant difference between them (P=0.039).

The expression of epidermal growth factor receptor (EGFR) protein in human gastric cancer ( × 200). The expression of EGFR protein was evaluated using immunohistochemical staining. The positive expression of EGFR protein was detected in the cytoplasm of gastric cancer cells and was increased in gastric cancer tissues compared with the adjacent non-cancerous tissues (ANCT). Scale bars=75 μm.
Correlation of EGFR expression with clinicopathological characteristics of gastric adenocarcinoma patients
The correlation of EGFR expression with various clinicopathological characteristics was analyzed. As shown in Table 1, EGFR expression did not associate with the age, gender and tumor size of the gastric adenocarcinoma patients (each P>0.05). Upregulation of EGFR expression did not associate with T stage and N stage (P=0.620; P=0.593). Moreover, positive expression of EGFR was correlated with lymph nodes metastases of the tumors (P=0.026).
Effect of EGFR knockdown on AKT expression
In pilot studies, the infection efficiency of Lv-shEGFR vector (MOI=50) was >70.0% as observed by fluorescent microscopy in SGC-7901 cells (Figure 2). Then the effect of EGFR knockdown on AKT expression was identified by real-time PCR and western blotting assays in GC cells. Real-time PCR showed lower mRNA expression levels of EGFR and AKT in the Lv-shRAGE group than in the NC group (each **P<0.01; Figure 3a). Consistent with this result, the protein expression levels of EGFR and AKT, indicated by western blotting assay were remarkably downregulated in the Lv-shEGFR group in comparison with the NC group (each **P<0.01; Figure 3b).

The infection efficiency of Lv-shEGFR vector in gastric cancer cells. In pilot studies, the infection efficiency of Lv-shEGFR vector (multiplicity of infection=50) was >70.0% as observed by fluorescent microscopy in Lv-shEGFR-transfected SGC-7901 cells. Scale bars=75 μm.

Effect of epidermal growth factor receptor (EGFR) knockdown on AKT expression. The effect of EGFR knockdown on AKT expression was identified by real-time PCR (a) and western blotting assays (b) in gastric cancer cells, which indicated the lower expression levels of EGFR and AKT in the Lv-shEGFR group than those in the negative control (NC) group in gastric cancer SGC-7901 cells (each *P<0.01).
Effect of EGFR knockdown on cell proliferation
Deregulated cell proliferation is a hallmark of cancer. In order to examine the effect of EGFR knockdown on cell growth, we investigated the proliferative activities of GC cells by MTT assay. Knockdown of EGFR could significantly diminish the proliferative activities of SGC-7901 cells in a time-dependent manner (**P<0.01) (Figure 4a). In addition, we also detected the expression of proliferating cell nuclear antigen (PCNA) by real-time PCR and western blotting assays to determine whether knockdown of EGFR suppressed the endogenous PCNA expression. The results showed that the amount of PCNA was significantly decreased in the Lv-shEGFR group compared with the NC group (**P<0.01) (Figures 4b and c), suggesting that knockdown of EGFR might inhibit GC cell proliferation through downregulation of the PCNA expression.

Effect of epidermal growth factor receptor (EGFR) knockdown on cell proliferation. (a) The proliferative activities of gastric cancer SGC-7901 cells were assessed by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay, indicating that knockdown of EGFR significantly diminished the proliferative activities of SGC-7901 cells in a time-dependent manner (*P<0.01). The amount of PCNA, indicated by real-time (b) and western blotting assays (c), was significantly decreased in the Lv-shEGFR group compared with that in the negative control (NC) group (*P<0.01).
Effect of EGFR knockdown on cell invasion
To determine the effect of EGFR knockdown on cell invasion, Transwell assay was carried out. The invasive potential was determined on the basis of the ability of cells to invade a matrix barrier containing laminin and type IV collagen, the major components of the basement membrane. Representative micrographs of the Transwell filters can be seen in Figure 5a. The invasive potential of GC cells was significantly weakened in the Lv-shEGFR group than in the NC group (**P<0.01; Figure 5b). In addition, the endogenous expression of MMP-9, indicated by real-time PCR and western blotting assays, was significantly decreased in the Lv-shEGFR group compared with that in the NC group (**P<0.01; Figures 5c and d), indicating that knockdown of EGFR might inhibit the invasive potential of GC cells through downregulation of MMP-9 expression.

Effect of epidermal growth factor receptor (EGFR) knockdown on cell invasion. (a) The effect of EGFR knockdown on cell invasion was investigated by Transwell assay. The invasive potential was determined on the basis of the ability of cells to invade a matrix barrier containing laminin and type IV collagen, the major components of the basement membrane. (b) The invasive potential of gastric cancer cells was significantly weakened in the Lv-shEGFR group compared with that in negative control (NC) group in SGC-7901 cells (**P<0.01). The amount of MMP-9, indicated by real-time (c) and western blotting assays (d), was significantly decreased in the Lv-shEGFR group compared with that in NC group (**P<0.01). Scale bars=panel (a), 75 μm.
Effect of EGFR knockdown on the migration of SGC-7901 cells
To investigate the roles of EGFR on the migration of SGC-7901 cells, EGFR was silenced by shRNA in SGC-7901 cells, and the migration of these cells was examined by Transwell migration assay. The results showed that following EGFR knockdown the migration ability of SGC-7901 cells decreased by 61% (Figure 6), suggesting that EGFR is vital for SGC-7901 cells to exert the capacities of migration and hence might have roles in lung cancer metastasis.

Effect of epidermal growth factor receptor (EGFR) knockdown on cell migration (a) Transwell migration assay for negative control (NC) or Lv-shEGFR-transfected SGC-7901 cells. (b) Histogram represented the relative migration ability calculated from 4 to 5 fields under a light microscope. *P<0.05.
Effect of EGFR knockdown on cell apoptosis and cycle distribution
To determine the effects of EGFR knockdown on apoptosis and cycle distribution in GC cells (SGC-7901), flow cytometric analysis was performed. The apoptotic indices of GC cells in the Lv-shEGFR group were markedly higher than in the NC group (**P<0.01; Figure 7a). Cell cycle kinetics showed that the G0/G1 phase fraction was increased, whereas the S phase fraction was decreased, and cell cycle was arrested in the G0/G1 phase in the Lv-shEGFR group compared with that in the NC group (Figure 7b).

Effect of epidermal growth factor receptor (EGFR) knockdown on cell apoptosis and cycle distribution. (a) The apoptotic indices of gastric cancer SGC-7901 cells were markedly higher in the Lv-shEGFR group than those in the negative control (NC) group (**P<0.01). (b) Cell cycle kinetics showed that the G0/G1 phase fraction was increased, whereas the S phase fraction was decreased, and cell cycle was arrested in the G0/G1 phase in the Lv-shEGFR group compared with the NC group in SGC-7901 cells.
Discussion
GC, one of the most common malignancies worldwide, is the second most frequent cause of cancer death.14, 15 The high mortalityof GC is a consequence of late-stage of diagnosis; the 5-year survival rate for advanced stages is extremely poor and around 5–15%.16, 17 Although diagnosis and treatment of GC have improved, the survival rate has not increased substantially in the couple of years. Therefore an improved understanding of the molecular pathways involved in the progression of GC will be helpful in improving prevention, diagnosis and therapy of this disease.18, 19, 20, 21, 22
The present study indicated that EGFR was highly expressed in GC, but the correlation of EGFR expression with the clinical characteristics of patients with GC was not further analyzed due to insufficient clinical data. However, Ye et al.23 have reported that EGFR expression is closely associated with the invasion and metastasis with patients with GC, which provides us an experimental basis for the functional study of EGFR in GC. Interestingly, Xie et al.24 have identified the novel function of EGFR in regulating oval cell activation and tumor development in inflammation-associated liver carcinogenesis. TheHMGB1/EGFR inflammatory pathway promotes tumor growth by regulating mitochondrial bioenergetics, suggesting that EGFR may represent an important target for the treatment of cancer.25, 26
Up to now, the function of EGFR in GC is unclear, but some studies revealed the role of EGFR in other cancers, in which blockade of EGFR decreased growth and metastases of both implanted tumors and tumors developing spontaneously in susceptible mice.27, 28 Targeting EGFR decreases proliferation in breast cancer cells, induces cell apoptosis and inhibits prostate cancer growth. Loss of EGFR function also inhibits the angiogenesis and progression of colorectal cancer but prolongs the survival in pancreatic cancer.29 In order to confirm the function of EGFR in GC, the present study indicated that knockdown of EGFR expression suppressed the growth and invasion and induced cell apoptosis and cycle arrest in GC cells. Further work is needed to target EGFR for possible early intervention and prophylaxis in patients at risk for developing cancer. Our previous studies have found that ethyl pyruvate can inhibit growth and metastasis of GC cells via regulation of the HMGB1-EGFR pathway, suggesting that inhibition of EGFR by ethyl pyruvate may have a critical role in the treatment of GC in conjunction with other therapeutic agents.
AKT is overexpressed in many tumor cell lines and in some human tumors, including GC, and has a critical role in tumor growth and metastasis.30, 31, 32 PCNA is a nuclear protein that is expressed in proliferating cells, may be required for maintaining cell proliferation and used as a marker for cancer cell proliferation.33 MMP-9, expressed on the tumor cell surface, is an important molecule involved in tumor metastasis, activates pro-MMP to exacerbate the malignancy and is considered a powerful indicator of distant metastasis of cancer.34, 35, 36 AKT signaling can promote tumor growth and invasion through regulation of the expression of PCNA and MMP-9.37 Moreover, EGFR leads to the stimulation of the AKT pathway in carcinoma cells, thus enhancing their metastatic capacity. In the present study, our finding showed that knockdown of EGFR downregulated the expression of AKT, PCNA and MMP-9 and suppressed the bioactivities of GC cells, suggesting that EGFR may be involved in the development and progression of GC via AKT-mediated regulation of the PCNA and MMP-9 expression.38, 39, 40
In conclusion, our findings indicate that upregulation of EGFR expression is associated with lymph node metastases of GC, and blockade of EGFR signaling suppresses growth and invasion of GC cells through AKT pathway, suggesting that EGFR may represent a potential therapeutic target for this aggressive malignancy.
References
Luo HQ, Han L, Jiang Y . Meta-analysis of six randomized control trials of chemotherapy plus anti-HER monoclonal antibody for advanced gastric and gastroesophageal cancer. Asian Pac J Cancer Prev 2014; 15 (13): 5343–5348.
Lyu X, Huang J, Liu J, Wang W, Su Y, Zhang W et al. Detection and significance of epidermal growth factor receptor mutation in esophageal, esophagogastric junction and gastric cancers. Zhonghua Zhong Liu Za Zhi 2014; 36 (5): 346–350 Chinese.
Li Y, Tan BB, Zhao Q, Fan LQ, Wang D, Liu Y . ZNF139 promotes tumor metastasis by increasing migration and invasion in human gastric cancer cells. Neoplasma 2014; 61 (3): 291–298.
Liu G, Jiang C, Li D, Wang R, Wang W . MiRNA-34a inhibits EGFR-signaling-dependent MMP7 activation in gastric cancer. Tumour Biol 2014: 36–45.
Smyth EC, Sclafani F, Cunningham D . Emerging molecular targets in oncology: clinical potential of MET/hepatocyte growth-factor inhibitors. Onco Targets Ther 2014; 7: 1001–1014.
Rolfo C, Bronte G, Sortino G, Papadimitriou K, Passiglia F, Fiorentino E et al. The role of targeted therapy for gastrointestinal tumors. Expert Rev Gastroenterol Hepatol 2014; 7: 1–11.
Guan SS, Chang J, Cheng CC, Luo TY, Ho AS, Wang CC et al. Afatinib and its encapsulated polymeric micelles inhibits HER2-overexpressed colorectal tumor cell growth in vitro and in vivo. Oncotarget 2014; 5 (13): 4868–4880.
Luo BH, Xiong F, Wang JP, Li JH, Zhong M, Liu QL et al. Epidermal growth factor-like domain-containing protein 7 (EGFL7) enhances EGF receptor-AKT signaling, epithelial-mesenchymal transition, and metastasis of gastric cancer cells. PLoS One 2014; 9: 332–343.
Vaiopoulos AG, Kostakis ID, Gkioka E, Athanasoula KCh, Pikoulis E, Papalambros A et al. Detection of circulating tumor cells in colorectal and gastric cancer using a multiplex PCR assay. Anticancer Res 2014; 34 (6): 3083–3092.
Choi MR, An CH, Chung YJ, Choi YJ, Yoo NJ, Lee SH . Mutational and expressional analysis of ERBB3 gene in common solid cancers. APMIS 2014; 23: 113–124.
Musiani D, Konda JD, Pavan S, Torchiaro E, Sassi F, Noghero A et al. Heat-shock protein 27 (HSP27, HSPB1) is up-regulated by MET kinase inhibitors and confers resistance to MET-targeted therapy. FASEB J 2014; 23: 456–468.
Xu L, Qu X, Li H, Li C, Liu J, Zheng H et al. Src/caveolin-1-regulated EGFR activation antagonizes TRAIL-induced apoptosis ingastric cancer cells. Oncol Rep 2014; 32 (1): 318–324.
Aprile G, Giampieri R, Bonotto M, Bittoni A, Ongaro E, Cardellino GG et al. The challenge of targeted therapies for gastric cancer patients: the beginning of a long journey. Expert Opin Investig Drugs 2014; 23 (7): 925–942.
Kurokawa Y, Matsuura N, Kawabata R, Nishikawa K, Ebisui C, Yokoyama Y et al. Prognostic impact of major receptor tyrosine kinase expression in gastric cancer. Ann Surg Oncol 2014; 11: 432–441.
Nielsen TO, Friis-Hansen L, Poulsen SS, Federspiel B, Sorensen BS . Expression of the EGF family in gastric cancer: downregulation of HER4 and its activating ligand NRG4. PLoS One 2014; 9 (4): e94606.
Wei J, Zhao ZX, Li Y, Zhou ZQ, You TG . Cortactin expression confers a more malignant phenotype to gastric cancer SGC-7901 cells. World J Gastroenterol 2014; 20 (12): 3287–3300.
Zhang EB, Kong R, Yin DD, You LH, Sun M, Han L et al. Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget 2014; 5 (8): 2276–2292.
Yan M, Parker BA, Schwab R, Kurzrock R . HER2 aberrations in cancer: implications for therapy. Cancer Treat Rev 2014; 40 (6): 770–780.
Naruke A, Azuma M, Takeuchi A, Ishido K, Katada C, Sasaki T et al. Comparison of site-specific gene expression levels in primary tumors and synchronous lymph node metastases in advanced gastric cancer. Gastric Cancer 2014; 9: 116–124.
Jiang L, Chen Y, Sang J, Li Y, Lan T, Wang Y et al. Type II cGMP-dependent protein kinase inhibits activation of key members of the RTK family in gastric cancer cells. Biomed Rep 2013; 1 (3): 399–404.
Nagatsuma AK, Aizawa M, Kuwata T, Doi T, Ohtsu A, Fujii H et al. Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer 2014; 14: 783–797.
Zhang J, Cao J, Li J, Zhang Y, Chen Z, Peng W et al. A phase I study of AST1306, a novel irreversible EGFR and HER2 kinase inhibitor, in patients with advanced solid tumors. J Hematol Oncol 2014; 7: 22.
Ye B, Jiang LL, Xu HT, Zhou DW, Li ZS . Expression of PI3K/AKT pathway in gastric cancer and its blockade suppresses tumor growth and metastasis. Int J Immunopathol Pharmacol 2012; 25: 627–636.
Xie J, Chen M, Zhou J, Mo MS, Zhu LH, Liu YP et al. miR-7 inhibits the invasion and metastasis of gastric cancer cells by suppressing epidermal growth factor receptor expression. Oncol Rep 2014; 31 (4): 1715–1722.
Jiang L, Chen Y, Li Y, Lan T, Wu M, Wang Y et al. Type II cGMP-dependent protein kinase inhibits ligand-induced activation of EGFR ingastric cancer cells. Mol Med Rep 2014; 9 (4): 1405–1409.
Chaturvedi R, Asim M, Piazuelo MB, Yan F, Barry DP, Sierra JC et al. Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastricepithelial cells with DNA damage. Gastroenterology 2014 146 7: 1739.e14–1751.e14.
Gala K, Chandarlapaty S . Molecular pathways: HER3 targeted therapy. Clin Cancer Res 2014; 20 (6): 1410–1416.
Agaimy A, Rau TT, Hartmann A, Stoehr R . SMARCB1 (INI1)-negative rhabdoid carcinomas of the gastrointestinal tract: clinicopathologic and molecular study of a highly aggressive variant with literature review. Am J Surg Pathol 2014; 38 (7): 910–920.
Tajiri R, Ooi A, Fujimura T, Dobashi Y, Oyama T, Nakamura R et al. Intratumoral heterogeneous amplification of ERBB2 and subclonal genetic diversity in gastric cancers revealed by multiple ligation-dependent probe amplification and fluorescence in situ hybridization. Hum Pathol 2014; 45 (4): 725–734.
Shi M, Shi H, Ji J, Cai Q, Chen X, Yu Y et al. Cetuximab inhibits gastric cancer growth in vivo, independent of KRAS status. Curr Cancer Drug Targets 2014; 16: 763–784.
Terragni R, Casadei Gardini A, Sabattini S, Bettini G, Amadori D, Talamonti C et al. EGFR, HER-2 and KRAS in canine gastric epithelial tumors: a potential human model? PLoS One 2014; 9 (1): e85388.
Zhang J, Zhan Z, Wu J, Zhang C, Yang Y, Tong S et al. Relationship between EGF, TGFA, and EGFR gene polymorphisms and traditional Chinese medicine ZHENG in gastric cancer. Evid Based Complement Alternat Med 2013; 2013: 731071.
Zhang L, Zhao G, Hou Y, Zhang J, Hu J, Zhang K . The experimental study on the treatment of cytokine-induced killer cells combined with EGFR monoclonal antibody against gastric cancer. Cancer Biother Radiopharm 2014; 29 (3): 99–107.
Hong L, Han Y, Brain L . The role of epidermal growth factor receptor in prognosis and treatment of gastric cancer. Expert Rev Gastroenterol Hepatol 2014; 8 (1): 111–117.
Feng D, Ma Y, Liu J, Xu L, Zhang Y, Qu J et al. Cbl-b enhances sensitivity to 5-fluorouracil via EGFR- and mitochondria-mediated pathways in gastric cancer cells. Int J Mol Sci 2013; 14 (12): 24399–24411.
Zhang EB, Han L, Yin DD, Kong R, De W, Chen J . c-Myc-induced, long, noncoding H19 affects cell proliferation and predicts a poor prognosis in patients with gastric cancer. Med Oncol 2014; 31 (5): 914.
Aydin K, Okutur SK, Bozkurt M, Turkmen I, Namal E, Pilanci K et al. Effect of epidermal growth factor receptor status on the outcomes of patients with metastatic gastric cancer: a pilot study. Oncol Lett 2014; 7 (1): 255–259.
Ito A, Ito Y, Matsushima S, Tsuchida D, Ogasawara M, Hasegawa J et al. New whole-body multimodality imaging of gastric cancer peritoneal metastasis combining fluorescence imaging with ICG-labeled antibody and MRI in mice. Gastric Cancer 2014; 17 (3): 497–507.
Pang Q, Ge J, Shao Y, Sun W, Song H, Xia T et al. Increased expression of long intergenic non-coding RNA LINC00152 in gastric cancer and its clinical significance. Tumour Biol 2014; 35 (6): 5441–5447.
Wang Y, Feng X, Jia R, Liu G, Zhang M, Fan D et al. Microarray expression profile analysis of long non-coding RNAs of advanced stage human gastric cardia adenocarcinoma. Mol Genet Genomics 2014; 289 (3): 291–302.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhen, Y., Guanghui, L. & Xiefu, Z. Knockdown of EGFR inhibits growth and invasion of gastric cancer cells. Cancer Gene Ther 21, 491–497 (2014). https://doi.org/10.1038/cgt.2014.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cgt.2014.55
- Springer Nature America, Inc.
This article is cited by
-
Endosomal Arl4A attenuates EGFR degradation by binding to the ESCRT-II component VPS36
Nature Communications (2023)
-
A phase I study evaluating combined nimotuzumab and neoadjuvant chemoradiotherapy followed by surgery in locally advanced esophageal cancer
Cancer Chemotherapy and Pharmacology (2019)
-
Increased EGFR expression induced by a novel oncogene, CUG2, confers resistance to doxorubicin through Stat1-HDAC4 signaling
Cellular Oncology (2017)
-
CD24 associates with EGFR and supports EGF/EGFR signaling via RhoA in gastric cancer cells
Journal of Translational Medicine (2016)
-
The combinational therapy of trastuzumab and cetuximab inhibits tumor growth in a patient-derived tumor xenograft model of gastric cancer
Clinical and Translational Oncology (2016)