
Abstract
The Permian Basin is a unique ecosystem located in the southwest of the USA. An unanswered question is whether the bacteria in the Permian Basin adapted to the changing paleomarine environment and survived in the remnants of Permian groundwater. In our previous study, a novel bacterial strain, Permianibacter aggregans HW001T, was isolated from microalgae cultures incubated with Permian Basin waters, and was shown to originate from the Permian Ocean. In this study, strain HW001T was shown to be the representative strain of a novel family, classified as ‘Permianibacteraceae’. The results of molecular dating suggested that the strain HW001T diverged ~ 447 million years ago (mya), which is the early Permian period (~ 250 mya). Genome analysis was used to access its potential energy utilization and biosynthesis capacity. A large number of transporters, carbohydrate-active enzymes and protein-degradation related genes have been annotated in the genome of strain HW001T. In addition, a series of important metabolic pathways, such as peptidoglycan biosynthesis, osmotic stress response system and multifunctional quorum sensing were annotated, which may confer the ability to adapt to various unfavorable environmental conditions. Finally, the evolutionary history of strain HW001T was reconstructed and the horizontal transfer of genes was predicted, indicating that the adaptation of P. aggregans to a changing marine environment depends on the evolution of their metabolic capabilities, especially in signal transmission. In conclusion, the results of this study provide genomic information for revealing the adaptive mechanism of strain HW001T to the changing ancient oceans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Permian Basin is a large sedimentary basin in the southwestern USA. It extends from the southeastern Lubbock, Texas, to the south of Odessa and Midland, and westward to adjacent southern New Mexico (Bein and Dutton 1993). It represents a unique ecosystem, and is the remains of an ancient sea that existed during the Permian period (~ 250 million years ago) (Hong et al. 2013; Wright 2011). Permian water has relatively low ammonia concentration (0.19 ± 0.01 μmol/L), normal N/P ratio (20.95 ± 0.45) and low salinity (1.65 ± 0.10%) (Hong et al. 2013). In addition, modern Permian groundwater is an aerobic and relatively stable environment with high concentrations of bicarbonate, nitrite, nitrate, phosphate, and iron (Gruber 2008; Hong et al. 2013). In contrast to modern Permian groundwater, the Permian Ocean is believed to have experienced not only oceanic eutrophication, but also acidification, anoxia, and ecological perturbation during the Permian period, which eventually led to mass extinction (Sun et al. 2018). These marine environmental changes could affect their metabolism and the evolution of organisms living in the habitat. Gradually, the constant mutation rate of some bacteria helps them adapt to changing environmental conditions (Denamur and Matic 2006).
In general, Proteobacteria, especially Gammaproteobacteria, are abundant in Permian groundwater and are known for their extensive metabolic diversity (Mori et al. 2017). Bacterial strains cultured from the abundant Gammaproteobacteria in Permian groundwater may shed light on the mechanisms of adaption to the specific environment of ancient oceans. A bacterium designated P. aggregans strain HW001T was isolated from liquid cultures of the biofuel-producing microalga, Nannochloropsis oceanica IMET1, cultured in Permian groundwater (Wang et al. 2012). Using primers specific to the 16S rRNA gene of strain HW001T, the strain-specific amplicons were obtained only from original Permian groundwater, and not from other tested samples (Wang et al. 2012). This indicated that strain HW001T may be originated from Permian groundwater. Phylogenetic analysis indicated that P. aggregans strain HW001T is a member of a novel genus Permianibacter belonging to the family Pseudomonadaceae (Wang et al. 2014). In this study, we aimed to redefine the phylogenetic status of the strain P. aggregans HW001T, determine the timing of divergence, and investigate adaptive mechanisms that may play a role in its survival in the Permian Basin environment.
Results
Genomic characterization of strain HW001T
The genomic features of P. aggregans HW001T isolated from Permian groundwater are listed in Supplementary Table S1. Plasmids were not detected in the genome of P. aggregans strain HW001T. The genome size was 4,265,640 bp and the G + C content was 54.4%. Final annotation of P. aggregans strain HW001T produced 3816 predicted coding sequences (CDS). A total of 57 RNA sequences were detected, including 6 rRNA genes (5S, 16S and 23S) and 48 tRNA genes. The graphic circular plot of P. aggregans strain HW001T genome was colored by COG category (Fig. 1A). Among them, 452 genes encoding energy production and conversion were predicted. It was found that 29 GIs were identified across the chromosome of strain HW001T (Fig. 1B). Most of these GIs (68.34%) were dominated mainly by hypothetical proteins (Supplementary Table S2). Other proteins present were associated with DNA replication, transposition, fatty acid hydroxylation, group transfer, membrane transportation, metal resistance, and DNA-binding response regulators. Also, proteins involved in transcription regulation and various mobile elements were observed. The PHASTER server predicted only one incomplete prophage region in the genome of strain HW001T with a total size of 11.7 kb (Supplementary Fig. S1). The prophage region sequence was blasted to COG database, and it encodes key enzymes for some functions, such as various mobile elements.

Graphic circular plot of strain HW001T genome. A From outside to the center: Genes on forward strand (colored by COG categories), Genes on reverse strand (colored by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew. B Representation of genomic islands predicted by Islandviewer 4 in strain HW001T genome
Sequence identity differences
In our previous study, the 16S rRNA gene sequence of P. aggregans strain HW001T showed only 88.31% similarity with that of Pseudomonas protegens strain CHA0T, which was affiliated with Pseudomonadaceae (Wang et al. 2014). In this study, Blastn analysis showed that P. aggregans strain HW001T had a higher similarity (89.15%) with Cavicella subterranea strain W2.09-231T belonging to another family Moraxellaceae in the class of Gammaproteobacteria. This similarity value was higher than 86.5%, which is the threshold for classifying different families (Yarza et al. 2014). Genome-based analysis was conducted to investigate the phylogeny of P. aggregans strain HW001T. It was found that the ANI value (67.78%) between P. aggregans strain HW001T and Pseudomonas aeruginosa strain ATCC 10145T was the highest, whereas the sequence homology between P. aggregans strain HW001T and other strains was relatively low, generally around 66% (< 75%), indicating that the phylogeny of the P. aggregans strain HW001T needs to be redefined (Supplementary Table S3).
Phylogenetic analysis of the strain HW001T
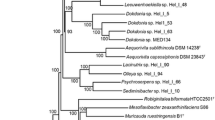
Phylogenetic analysis based on 16S rRNA gene sequence analysis using different tree algorithms [Fig. 2 (ML) and Supplementary Fig. S2 (NJ and ME)] showed that P. aggregans strain HW001T was distinct from other families in the class of Gammaproteobacteria and formed a single clade. This is different from our previous study, which showed that P. aggregans strain HW001T belongs to Pseudomonadaceae (Wang et al. 2014). For further verification, the genomic tree of P. aggregans strain HW001T and other Gammaproteobacteria strains was constructed based on single-copy genes (Fig. 3). Genomic recombination was performed using RDP4 and ClonalFrameML to mitigate the effects of homologous recombination (Supplementary Figs. S3, S4; Supplementary Table S4). The results of phylogenetic analysis of the whole genomes showed that the P. aggregans strain HW001T resides in a single clade, clearly different from Pseudomonadaceae and other known Gammaproteobacteria families.

Maximum-likelihood tree based on partial 16S rRNA gene sequences of the strain Permianibacter aggregans HW001T and other type strains of each family in the class Gammaproteobacteria

The RAxML maximum likelihood phylogenomic tree based on 102 single-copy gene families in the class Gammaproteobacteria. Five reference strains from Alpha- and Betaproteobacteria were used as the outgroup for genomic tree. The scale bar indicates 1.0 substitution per nucleotide position for genomic tree. G + C content and genome size are indicated in the two left columns
Molecular dating
The timing of divergence of P. aggregans strain HW001T was inferred from molecular phylogenetic chronogram and calibration of various fossils using penalized likelihood in r8s (r8s-PL) and Bayesian estimation with uncorrelated relaxed rates among lineages (BEAST). Molecular dating based on r8s software (Fig. 4) suggested that P. aggregans strain HW001T diverged around 447 (± 7) mya. The resulting dated phylogeny based on BEAST software (Supplementary Fig. S5 and Supplementary Table S5) was similar to r8s, and the P. aggregans strain HW001T diverged around 508 mya (95% credibility interval: 477.3590, 582.3631). These dating times are in the early Permian period (~ 250 million years ago) (Hong et al. 2013; Wright 2011). Cyanobacteria (2700 mya), akinetes (1991 mya), and Rhizobium (129 mya) were also closed to the reported period. The strain was thought to be encased in an underwater basin that contains the remnants of an ancient ocean that existed during the Permian period. In order to identify potential shifts in bacterial physiology by genome-wide evolution, we asked which genes associated with environmental adaptation were present/absent in the genome of P. aggregans strain HW001T.

A chronogram of Gammaproteobacteria performed using the r8s software. Nodes with fossil record corrections are indicated with an asterisk
Functional gene composition and comparison
According to the results of genome annotation and genome analysis, P. aggregans strain HW001T was enriched in genes encoding secondary metabolite biosynthesis, transport and catabolism, cell motility and defense mechanism, compared with other more recently diverged Gammaproteobacteria (Supplementary Fig. S6). Specific genes were annotated in the genome of P. aggregans strain HW001T, including nitrogen metabolism [pA (periplasmic nitrate reductase) and pB (cytochrome c-type protein)], amino acid metabolism [aspC (aspartate aminotransferase) and puuE (4-aminobutyrate aminotransferase)], putrescine transport system (potFGHI), butanol dehydrogenase (bdhAB, 3 copies), argHA (argininosuccite lyase), mogA (molybdopterin adenylyltransferase) and lapB (ATP-binding cassette) (Supplementary Table S6). The metabolic pathways of P. aggregans strain HW001T include oxidative phosphorylation, ABC transporters and associated proteins, nitrogen metabolism, sulfur metabolism, extracellular polymeric substance (EPS), and carbon fixation (Fig. 5).

Heterotrophic metabolic pathways of strain HW001 highlighting the major functions identified by genomic analysis
Carbohydrate-active enzymes
The genome of P. aggregans strain HW001T contains at least 57 genes encoding carbohydrate-active enzymes, including glycosyl transferases (GTs, 22 genes), glycoside hydrolases (GHs, 14 genes), carbohydrate-binding modules (CBMs, 9 genes), carbohydrate esterases (CEs, 6 genes) and auxiliary activities (AAs, 6 genes) (Supplementary Table S7). GTs and GHs belong to 8 and 11 known families, respectively, described in the CAZy database. Over half of the annotated P. aggregans strain HW001T GTs were associated with glycosyl transferase (GT families 2 and 4; 13 copies). Five genes (GH family 13) encode alpha amylase. In addition, genes encoding Beta-galactosidase, Beta-glucanase, exo-1,4-beta-glucosidase, peptidoglycan transglycosylase, and Beta-glucosaminidase were annotated. Furthermore, genes encoding catalase (AA family 2; 2 copies), oxidoreductase (AA family 3), and multimeric flavodoxin (AA family 6; 2 copies) were detected in the genome of P. aggregans strain HW001T.
Nitrogen and sulfur metabolism
It is well known that nitrate acts as an electron acceptor, affecting the biological activity of microbial cells (Ogilvie et al. 1997). In the presence of NarGHI, NapAB, NirBD, and NrfAH genes in P. aggregans strain HW001T, dissimilatory nitrate reduction (nitrate = > ammonia) was determined to be the main metabolic pathway for ammonia production. glnA ([EC:6.3.1.2], 4 copies) is the most common gene involved in nitrogen utilization, converting ammonia to glutamine and glutaminate. However, in the absence of the NosZ gene, P. aggregans strain HW001T could not catalyze the production of nitrogen from nitrate. Three nitrate/nitrite transporter genes encoding NRT were annotated. They may have the function of regulating nitrogen balance. Its assimilating sulfate reduction (sulfate = > H2S) pathway is a key mode of sulfur metabolism.
Extracellular polymeric substance (EPS)
In our previous study (Wang et al. 2012), it was determined that P. aggregans strain HW001T could aggregate microalgae, such as Nannochloropsis oceanica IMET1 and N. oceanica CT-1. Surface EPS is mainly composed of polysaccharides, proteins, nucleic acids, and lipids, which may exert an important role in the aggregation process (Xiao et al. 2018). Several complete glycan biosynthesis pathways were detected in the genome of P. aggregans strain HW001T, including peptidoglycan (DAP-type and Lys-type, Supplementary Fig. S7) and lipopolysaccharide biosynthesis (Supplementary Fig. S8). A complete phosphatidylethanolamine (PE) biosynthesis pathway [phosphatidic acid (PA) = > phosphatidylserine (PS) = > PE] was successfully annotated, including genes encoding for phosphatidate cytidylyltransferase (CDS1, HW001_00670 and HW001_02758), phosphatidylserine decarboxylase (psd, HW001_02719), and CDP-diacylglycerol–-serine O-phosphatidyltransferase (CHO1, HW001_02101). Additionally, a transmembrane protein (EpsH, HW001_02119) and a putative exopolysaccharide exporter (EPS-E, HW001_00554) were annotated based on KEGG database.
ABC transporters
At least 45 genes in the genome of P. aggregans strain HW001T encode a variety of ABC transporters, including a metal ion transfer systems (molybdate, sodium, iron (III), and iron complex), organic molecular transfer system (putrescine, phospholipid, phosphate, phosphonate, dipeptide, lipopolysaccharide, and lipoprotein), and other transport systems (heme, cell division, putative ABC, ABC-2 type) (Fig. 5). These identified transporters regulate intracellular homeostasis and provide sufficient materials for the survival of P. aggregans strain HW001T.
Two-component system
Various two-component systems, including PhoQ-PhoP (magnesium transport), CusS-CusR (copper tolerance), EnvZ-OmpR (osmotic stress response), RstB-RstA BaeS-BaeR (envelope stress response), DesK-DesR (membrane lipid fluidity regulation), BarA-UvrY (central carbon metabolism), AlgZ-AlgR (alginate production), PhoR-PhoB (phosphate starvation response), GlnL-GlnG (nitrogen regulation), PilS-PilR (type 4 fimbriae synthesis), and FlrB-FlrC (polar flagellar synthesis) were annotated in the genome of P. aggregans strain HW001T.
Quorum sensing
In general, quorum sensing can regulate various metabolic pathways, which may be related to the transition of bacterial lifestyles between free-living (low substrate utilization, low population density) and particle associated (high substrate utilization, high population density) (Gram et al. 2002). In this study, 25 genes encoding quorum sensing were found in the genome of P. aggregans strain HW001T. Specifically, the gene (HW001_01847) encoding a putative adhesion enzyme was annotated as a fibronectin type III domain protein or BapA (Supplementary Fig. S9). The protein was considered to be essential for biofilm formation and host colonization of Salmonella enterica serovar Enteritidis (Latasa et al. 2005). The analysis of codon usage in the genome of P. aggregans strain HW001T revealed that the codon adaptation index (CAI) of the gene was 0.694, whereas the average CAI was 0.714, indicating that P. aggregans strain HW001T likely obtained this gene in the past (Supplementary Table S8). However, the results based on the HGTector2 showed the gene was not transferred from other species. Only a gene encoding ferric uptake regulator (Fur), which controls the expression of enzymes that protect against reactive oxygen species (ROS) damage, was predicted to transfer from Gammaproteobacteria (Troxell and Hassan 2013).
Analysis of gene gain and loss
To study the evolution of P. aggregans strain HW001T, we clustered all protein sequences from 54 strains into different protein families (see Materials and methods), and estimated the gain and loss of protein families using phylogenetic gain–loss-duplication model implemented in Dollo parsimony (Supplementary Tables S9, S10). A total of 19 gene families were predicted to be gained (Supplementary Table S9). Gains may come from horizontal gene transfer, mainly involving genes encoding methyltransferase, formyl transferase, and transcriptional regulator. Of the total 155 gained genes at nodes 17 and 19, further analysis showed that 5 (~ 4.63%) and 9 (~ 19.15%) of them were identified as cellular process and signaling based on the eggNOG database, respectively (Fig. 6).

Evolutionary histories of strain HW001T using COUNT with Dollo parsimony. A The Bayesian tree is generated from Figtree. The numbers of gain and loss events were marked at leaves and nodes on the phylogenetic tree. The blue words represent the total number of gene families at different nodes. “ + ”s represent gain events and “ − ”s represent loss events. The lightly yellow star represents the major gene gain/loss event. B The pie chart shows the numbers of gained genes classified by COG database
Of the 419 deletion gene families in strain HW001T, 351 were annotated by the COG and KEGG databases. Among all deletion gene families, genes were mainly related to information storage and processing (~ 10.02% of the total deletion gene families), cellular process and signaling (~ 21.96%), and metabolism (~ 20.05%). Many of them are involved in cell wall/membrane/envelope biogenesis (COG M category, 23 families), amino acid transport and metabolism (COG E category, 24 families), transcription (COG K category, 23 families), signal transduction mechanism (COG T category, 13 families), energy production and conversion (COG C category, 14 families), inorganic ion transport and metabolism (COG P category, 27 families; Supplementary Table S10).
HGT for strain HW001T
The results of HGT gene analysis indicated that more than 6% of strain HW001T genes may have been horizontally transferred from other bacteria (Supplementary Table S11). Among all putative HGTs, genes related to signaling and cellular processes (15.98% of the total putative HGTs), genetic information processing (5.33%), carbohydrate metabolism (4.92%), amino acid metabolism (3.28%), and environmental information processing (2.46%), were the top five most abundant functional classifications. Among candidate donors, most genes were acquired from the phylum Proteobacteria (171), with Gammaproteobacteria transferring the largest number (93). Many genes (68), including 10 genes encoding amino acid transport and metabolism, were acquired from some unclassified bacteria.
Global distribution
The results of the global distribution survey of P. aggregans strain HW001T based on IMNGS analysis showed that 476 of the environmental samples (0.11%) had 16S rRNA genes with > 97% similarity to P. aggregans strain HW001T (Supplementary Table S12). Among them, SRR2041107, SRR2041108, SRR2041114, SRR2041168, and SRR2041112 with high abundance target 16S rRNA gene of 0.0338%, 0.0113%, 0.0063%, 0.0063%, and 0.0059%, respectively, were all collected from the aquatic samples of coral pond water in Davis, California, USA. Also, targeted 16S rRNA genes were found in samples collected from the beach sand metagenomes, maize rhizosphere saline soil, Suaeda salsa rhizosphere soil, and lettuce rhizosphere arable soil of Pensacola, Florida, USA.
Further investigation of the physico-chemical parameters of the Permian groundwater showed that compared with the other three habitat types (open ocean, coral reef, and marine-derived lake sites) in the global ocean sampling data, the living environment of P. aggregans strain HW001T was similar to that of the east coast of North America (Fig. 7; Supplementary Fig. S10). It is worth noting that the concentration of silicate in Permian Basin water was higher than that in the GOS data. These results suggested that the P. aggregans strain HW001T or its closely related groups may be mainly distributed in the adjacent regions of the USA, and tend to propagate in the Permian aquatic environments as well as some rhizosphere soils.

Comparison of physiological and biochemical characteristics between the sampling sites of strain HW001T and other GOS data
Discussion
Divergence time
Molecular dating, a standard method for inferring timetrees, is a fundamental step in drawing biological conclusions from nucleotides or amino acid sequence data (Ho and Duchêne 2014; Mello 2018). It may reveal the diversification of major taxa and their association with Earth’s history (Misof et al. 2014). The divergence time of P. aggregans strain HW001T in this study was inferred from molecular phylogenetic chronogram and various fossils calibrated using r8s-PL and BEAST. The results indicated that strain HW001T should have diverged around 447 mya, the Ordovician Period (480 mya-440 mya years ago), which was earlier than the Permian period (~ 250 million years ago) (Hong et al. 2013; Wright 2011). In our previous study, strain HW001T was proved to be derived from the Permian ground water, which was used for cultivating the biofuel-producing microalgae, Nannochloropsis oceanica IMET1 (Wang et al. 2012). Therefore, we believed that HW001T is a bacterium that existed in the Permian Ocean in the early Permian Period, which adapted to the changing marine environments at that time, and survived in the groundwater until the present.
Metabolic capabilities of the strain HW001T
Chemoheterotrophic metabolic capabilities
Previous studies have shown that P. aggregans strain HW001T utilized organic carbon sources (e.g., cellobiose, maltose, starch, glucose, gluconate, sucrose and adipic acid) rather than inorganic bicarbonate for growth (Wang et al. 2014). In this study, we successfully annotated many branched chain amino acid transporters, peptide transporters and amino acid efflux proteins in the genome of P. aggregans strain HW001T, further demonstrating that proteins are important carbon sources for P. aggregans strain HW001T. In the P. aggregans genome, numerous ABC-type transporter systems of putrescine are present, similar to the finding in the marine bacterium Silicibacter pomeroyi (Moran et al. 2004). In contrast, P. aggregans does not have a spermidine transport system, but can synthesize and utilize spermidine and putrescine through the polyamine biosynthesis pathway (arginine = > agmatine = > putrescine = > spermidine). One reason why P. aggregans tends to metabolize organic carbon is that this promotes dissimilatory nitrate reduction processes, which are useful for cellular respiration and electron transport (Yin et al. 2002). Due to the presence of various CBM described families, such as CBM5, CBM50, CBM12, and CBM48, strain HW001T is believed to be able to adhere to chitin, glycogen, and cell walls. Also, genes encoding for 4-oxalocrotonate tautomerase were detected in the genome of strain HW001T, indicating that it could generate intermediates of the TCA cycle via the conversion of aromatic compounds. Additionally, the detection of 5-phospho-alpha-D-ribose 1-diphosphate PRPP biosynthesis and the pentose phosphate pathway in this strain suggests the potential for the production of some nucleotide and amino acid precursors (Luo et al. 2021). Moreover, HGT, as an important evolutionary process in prokaryotes, largely affecting the diversity of gene repertoires (Zhaxybayeva et al. 2009). For example, the genes presented encoding chitinase [EC:3.2.1.14] and beta-glucosidase [EC:3.2.1.21] (bglX and bglB) were apparently the products of HGT events. On the one hand, strain HW001T has multiple organic matter transport systems that can be used to utilize organic matter. One the other hand, gene families related to inorganic substrate transport, such as nitrate/nitrite transport (nrtA) and sulfate/thiosulfate transport (cysR), have been lost over a long evolutionary history, reflecting the shifts of bacterial physiology during the early Permian Period. In short, a large series of transporters, abundance of genes encoding carbohydrate-active enzymes, and many genes encoding degradative and synthetic proteins suggested that HW001T possess chemoheterotrophic metabolic capabilities to target a wide range of organic carbon-containing compounds.
Multifunctional strategies for other environmental stresses
In our previous studies, we observed EPS on the cell surface of P. aggregans (Wang et al. 2012, 2014). EPS not only contributes nutrients to the environment, but also protects bacteria from toxic substances. At the same time, EPS may be used as energy storage to cope with external environmental pressure (Xiao and Zheng 2016). P. aggregans cells are encapsulated by multi-layer peptidoglycans (DAP-type and Lys-type). These layers on the cell wall may improve mechanical strength, help bacteria to regulate the external osmotic pressures, and survive in harsh environments. Conversely, the degree of cross-linking of peptidoglycan is associated with the structural integrity of cells (Höltje 1998).
A unique physiological marker of biofilm-producing bacteria is the high level of intracellular content of cyclic di-GMP (c-di-GMP), which is a secondary messenger that plays an essential role in determining the lifestyle of a wide range of bacteria (Chew and Yang 2016). Intracellular c-di-GMP is synthesized by several diguanylate cyclases (DGCs), and degraded by phosphodiesterases (PDEs). Multiple DGCs and PDEs offer P. aggregans strain HW001T great flexibility to regulate its intracellular c-di-GMP content enabling adaptation to different environmental conditions (Chew and Yang 2016). Histidine kinases (HK) are sensory proteins of two-component systems that control the response of many bacteria to different stimuli, i.e., mainly changes in environmental parameters (Fernández et al. 2019). Many of the genes encoding the LytTR family are predicted to be transferred from Oceanospirillales and other members of Proteobacteria. In addition, the gain of transposase may help to catalyze the donor cleavage and strand transfer reactions of HW001T through HGT from Gammaproteobacteria. There was also a putative HGT gene pspA encoding for phage shock protein A involved in responses to various stresses (ethanol, heat, and osmotic shock) (Brissette et al. 1990; Kleerebezem et al. 1996). Furthermore, the two-component system KdpD/KdpE is well known for its regulatory role in potassium (K+) transport antimicrobial stress, osmotic stress and oxidative stress (Freeman et al. 2013). A series of these gene families were lost, including genes encoding KdpA, KdpB, KdpC, KdpD, and KdpE, indicating that strain HW001T had this two-component system in the past. Also, gains/losses of other signal transduction proteins, flagellar biosynthesis proteins, quorum sensing systems, and two-component system regulatory proteins ensured the survival of P. aggregans strain HW001T cells in changing environmental conditions. Among all putative HGTs, genes related to signaling and cellular processes (15.98% of the total putative HGTs) and genetic information processing (5.33%) were the top two enriched functional classifications. In this regard, the adaptation of P. aggregans to a changing marine environment depends on the evolution of their metabolic capabilities, especially in signal transmission.
Genes encoding the ATP-dependent DNA helicases PIF1 and DEAD box helicase were predicted also with the results showing that they might have transferred from other Proteobacteria. These genes contribute largely to DNA repair (Rand et al. 2003). Several putative HGT genes encoding for multidrug resistance proteins imported from unclassified Bacteria may play a significate role in coping with stress. In addition, genes encoding superoxide dismutase (SOD2), which is essential for the detoxification of reactive oxygen species (ROS), was detected. ROS are generated during aerobic respiration and ammonia oxidation (Kim et al. 2016). To protect cells from oxidative stress and repair oxidatively damaged cytoplasmic proteins, thioredoxin reductase encoded by the gene trxB in HW001T could be helpful (Cheng et al. 2017). Additionally, inferred HGT genes essential for protection from ROS were predicted to be transferred from Gammaproteobacteria, such as speE. speE encoding spermidine synthase which could also protect DNA in thermal biotopes (Cheng et al. 2009). These genes would enable HW001T to adapt to low-oxygen conditions.
Ecological implications
According to IMNGS analysis and comparison of physico-chemical parameters with global ocean sampling (GOS) data, P. aggregans strain HW001T was mainly distributed in the regions adjacent to the USA, and tends to propagate in the Permian aquatic environments as well as some coastal soils or sediment environments. P. aggregans was abundant under eutrophic conditions, especially in coral pond water. This suggests that coral ponds contain substances that meet the metabolic needs of P. aggregans. These organisms could be applied as potential cleaning agents to degrade and remove organic carbon in eutrophic environments, thereby reducing water eutrophication. In addition to aquatic environments, P. aggregans may be a member of the rhizosphere community in some coastal soils. Relics of marine bacterial communities can be preserved in sediments for many years (Langenheder et al. 2016). Therefore, it is necessary to study the bacterial community in sediments of the Permian Basin to help understand the adaptive metabolism of P. aggregans.
Taxonomy
The chemotaxonomic and metabolic characteristics of P. aggregans strain HW001T differed considerably from other strains in Gammaproteobacteria (Supplementary Table S13). For example, GC content, enzymatic activities and carbohydrate utilization rate were different from those of other families. Based on genotypic, phylogenetic, chemotaxonomic and phenotypic assays, P. aggregans strain HW001T may be classified to a novel family, named Permianibacteraceae fam. nov. in the order of Psedudomadales.
Description of Permianibacteraceae fam. nov.
Permianibacteraceae (Per.mi.a.ni.bac.ter.ace'ae. N.L. masc. n. Permianibacter, the type genus of the family; N.L. suff. -aceae ending to denote the name of a family; N.L. fem. pl. n. Permianibacteraceae the family of Permianibacter).
This genus is classified to a new family because of its large physiological differences and phylogenetic distance from other members of Gammaproteobacteria. The description is identical to that of the genus Permianibacter, in which the cells are aerobic, oxidase-positive but catalase and urease-negative Gram-stain-negative rods ca. 1.6–2.7 µm long and 0.4 µm wide, without endospores. The G + C content of genomic DNA was ~ 55.4 mol%. The major fatty acids were iso-C15: 0, summed feature 9 (iso-C17: 1 ω9c), and C16: 0. Q-8 is the main respiratory quinone. Polar lipid profile consists of phosphatidylethanolamine, an unidentified aminophospholipid, and some other unidentified lipids.
Conclusion
In this study, genomic analysis of P. aggregans strain HW001T provided the first glimpse of the genome landscape of the novel family Permianibacteraceae. With the genome expansion of P. aggregans HW001T, its evolved metabolic and physiological characteristics can ensure its survival in a changing marine environment. The results of this study indicated that HW001T has a chemoheterotrophic metabolism targeting organic carbon, which promotes electron transfer to improve its resistances to oxygen stress. In addition, the integration of various EPSs, two-component systems, and its own quorum sensing may help ensure cell survival under eutrophication, acidification and other environmental pressures. P. aggregans strain HW001T is mainly distributed in regions adjacent to the USA, and has a tendency to reproduce in plant rhizosphere soils. In the future, more in-depth experimental verification will be carried out on P. aggregans HW001T, especially its silicate metabolism ability and diverse quorum sensing systems.
Materials and methods
Genomic DNA extraction and sequencing
P. aggregans strain HW001T (= CICC 10856T = KCTC 32485T) was cultured in marine 2216E broth (BD Biosciences) at 30 °C and 150 rpm for 3 days. Aliquots (2 ml) of liquid cultures were centrifuged (6000 rpm, 5 min) and cell pellets were collected for genomic DNA extraction using the Ultra-Clean microbial DNA isolation kit (MoBio Laboratories, USA). DNA concentration and purity were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). The genomic DNA (50 μl) with the final concentration of 300 ng/μl was sent to Beijing Genomics Institute (BGI, Shenzhen, China) for sequencing using Illumina (HiSeq 4000, USA) and Pacbio RSII sequencing platform. The sequencing results were assembled into different scales of contigs using SOAPdenovo (V1.05) and RS_HGAP Assembly3.
Genome annotation and analysis
Protein-encoding gene prediction of P. aggregans strain HW001T was conducted using the Prokaryotic Genome Annotation System pipeline (Version 1.11) (Seemann 2014). The encoded predicted proteins were classified based on Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Swiss-Prot, Non-redundant protein sequences (NR), and Clusters of Orthologous Groups (COG) databases (e value < 0.00001). Carbohydrate-active enzymes were identified with the assistance of MetaCyc (Caspi et al. 2014) and CAZy database (Drula et al. 2022). The genomic similarity between P. aggregans strain HW001T and other Gammaproteobacteria isolates was investigated by calculating the average nucleic acid identities (ANIs) using EZBioCloud online service (https://www.ezbiocloud.net/tools/ani) (Yoon et al. 2017). Prediction and annotation of functional proteins of other Gammaproteobacteria isolates were also based on GO, KEGG, and COG databases. The number and size of genomic islands (GIs) were determined with IslandViewer 4 server, an integrated interface of four different GI prediction methods: IslandPick, IslandPath-DIMOB, SIGI-HMM, and Islander (Bertelli et al. 2017). Prophages of strain HW001T were predicted using the online API in PHASTER (Arndt et al. 2016).
Genome recombination
It is clearly stated that recombination may obscure phylogenetic signals and may result in exaggerated branch lengths and increased evolutionary distances between strains (Knight et al. 2015). In order to mitigate its effects, recombinant genomic regions should be excluded from any phylogenetic reconstruction. Multiple alignment of the genome sequences for strain HW001T and the closed strains was performed by using Mauve (v20150226) multiple alignment software (Darling et al. 2004). RDP4 software was used to detect possible recombination breakpoints and potential recombination strains (Martin et al. 2017). In addition, in order to determine reliable recombination events, nine different methods (RDP, MaxChi, Chimaera, SiScan, GENECONV, BootScan, Phylpro, LARD, and 3Seq) were embedded in the RDP program with the corrected P value cutoff of 106.
To estimate mutation and recombination rates, the RAxML tree was first constructed based on the results of the RDP program (members in the tree were Acinetobacter gerneri DSM 14,967, Alcanivorax jadensis T9, Alteromonas macleodii ATCC 27,126, Diplorickettsia massiliensis 20B, Kangiella sediminilitoris KCTC 23,892, Rickettsiella grylli TrM1, and Permianibacter aggregans HW001). For further details, please refer to the Phylogenetic analysis section. The MAFFT alignment was calculated using their genomic sequences. Both RAxML predefined tree and MAFFT alignment were applied as the input to ClonalFrameML v1.12 (Didelot and Wilson 2015). ClonalFrameML was used to detect gene clusters at loci with elevated base substitution densities, identify multiple recombination events and generate a final corrected tree. Default priors R/θ = 10−1, 1/δ = 10−3, ν = 10−1 and mean branch length of 10−4 were used. The reliability of each node was supported by 100 pseudo-bootstrap replicates, as suggested by Didelot et al. (Didelot and Wilson 2015). The R package “ape” v3.348 and the R package “PopGenome” v2.1.649 were used to compute the mean patristic branch length and transition/transversion ratio, respectively. The priors obtained from this mode were used as the initialization values to rerun ClonalFrameML under the “per-branch model” mode with a branch dispersion value of 0.1 (Oliveira et al. 2017).
Phylogenetic analysis
For phylogenetic analysis of P. aggregans strain HW001T, phylogenetic trees based on 16S rRNA gene sequence were constructed using maximum-likelihood (ML), neighbor-joining (NJ), and minimum-evolution (ME) algorithms in MEGA software package (version 7) (Kumar et al. 2016). To study the whole genome evolution of the life history of strain HW001T, the phylogenetic relationship between P. aggregans strain HW001T and 115 other Gammaproteobacteria strains was analyzed. The genomes of Sinorhizobium meliloti 1021 and Rhodospirillum rubrum ATCC 11,170 affiliated with Alphaproteobacteria, and genomes of Ralstonia solanacearum GMI1000 and Chromobacterium violaceum ATCC 12,472, and Neisseria meningitidis MC58 affiliated with Betaproteobacteria were used as outgroups. First, orthologous gene families were identified using the GET_HO-MOLOGUES package (Contreras-Moreira and Vinuesa 2013), which carries out the algorithm of the OrthoMCL software (Li et al. 2003). Then a comprehensive data cluster of 83,234 orthologous gene families covering 116 strains was compiled. From these orthologous gene families, 20,903 single-copy gene families were obtained. Through screening, 102 single-copy shared gene families were selected for downstream phylogenomic analysis. These members in each gene family were aligned at the amino acid sequence level using MAFFT software (Katoh and Standley 2014) and deleted columns with gaps. Considering that different genomic regions may evolve independently, PartitionFinder software was used to perform data partition models (Lanfear et al. 2012). Then 31 partitions were obtained, which were identified as the best partition scheme based on the Bayesian information criterion (BIC). These 31 partitions were used to predict the LG amino acid substitution matrix (Le and Gascuel 2008) as the best model. In addition, the gamma distribution of rate variations (Yang 1996) and the predicted invariable site model was appropriate to all partitions. The resulting alignment was constructed using the RAxML version 8 (Stamatakis 2014) with LG substitution matrix and gamma model. Then, prediction of genome recombination was performed using ClonalFrameML (as shown in the previous section (Genome recombination)) to mitigate the effects of homologous recombination. Finally, a phylogenetic tree was mid-point rooted and performed using FigTree v1.4.4 (Rambaut 2018).
Molecular dating
Two widely used dating methods, penalized likelihood implemented in the r8s software (r8s-PL) and Bayesian estimation with uncorrelated relaxed rates among lineages (BEAST), were used to process the dating analysis of strain HW001T. To estimate divergence dates in r8s (Luo et al. 2013; Sanderson 2003), a genome-wide molecular phylogenetic tree was constructed using RAxML version 8 software with a data partition model identified by PartitionFinder to calculate divergence time of P. aggregans strain HW001T, as shown in the previous section (Phylogenetic analysis) (Lanfear et al. 2012). A total of 89 genomes were sampled, including several that evolved from ancestral branches with the fossil records. A cyanobacterium, Gloeobacter violaceus PCC 7421, was used as the outgroup of the phylogenetic tree. This tree was calibrated by imposing constraints on several ancestral nodes, including cyanobacteria occurring around 2700–3500 mya (David and Alm 2011; Falcón et al. 2010), and akinetes that deviated from cyanobacteria at > 1,500 mya (David and Alm 2011). Dating analyses were conducted on the basis of 100 bootstrapped trees with the identical topology. The chronogram of Gammaproteobacteria was displayed by using FigTree v1.4.4 software and modified by Adobe Illustrator CS6.
Another phylogeny was time-calibrated using the Bayesian algorithm in BEAST v2.6.4 (Bouckaert et al. 2014). For all analyses in BEAST, the same fixed topology (ML) based on the results of r8s-PL was used. Strict clock method with a Birth Death speciation process was used for all analyses. The range of origin dates of Rhizobium (in nodules) diverged from Agrobacterium (not in nodules) was calibrated with about 100–120 mya (Ochman and Wilson 1987). A normal prior was set with a mean value at the midpoint between the minimum and maximum values (node = 110.0). For all nodes, default prior was used, the mean and stdev were set to 1.0, and offset values were used. The assessment of chain convergence was done using Tracer 1.7.1 (Rambaut et al. 2018). A maximum clade credibility tree with mean heights was constructed with TreeAnnotator 2.2.1 (Burnin percentage: 50; Posterior probability limit: 0.0).
Gene gain/loss prediction through ancestral reconstruction of genome content
For further comparative analysis, only genomes with completeness > 80% and contamination < 5% were considered. The phylogenetic tree and clusters of homologous protein were reconstructed for the remaining 54 genomes affiliated with Gammaproteobacteria and Betaproteobacteria (the outgroup in the reference tree). For further details, please refer to the Phylogenetic analysis section. An “all-against-all” protein sequence similarity search was conducted with DIAMOND v. 0.9.18 (Buchfink et al. 2015) (“more sensitive” mode with a maximum e value cutoff of 10–5 and retaining up to 2500 hits). OrthoFinder v. 2.3.3 was applied to reconstruct orthologous gene families with default parameters (Emms and Kelly 2015). This yielded a total of 12,572 protein families. In order to predict gene gain and loss in P. aggregans strain HW001T, a maximum likelihood (ML) birth-and-death model was initially selected in the Count software (Csűös 2010; Nakjang et al. 2013). A gain–loss-duplication model without any restrictions on lineage-specific rates was used to maximize the likelihood of phyletic pattern (vector of observed family sizes at terminal taxa). The gain–loss-duplication model was computed with the ML model parameters implemented in Count with Dollo parsimony. This approach is very strict, prohibiting multiple gain of genes (Hua et al. 2018). Using this method may reconstruct gene gain and loss events at both potential ancestors and observed species (Hua et al. 2018). In addition, the rates of gain, loss, and duplication were conducted based on four discrete gamma distributions. Functional annotation of gains and losses was performed using InterProScan v. 5.39–77.0 (Jones et al. 2014) and eggNOG 5.0 (Huerta-Cepas et al. 2019), including mapping InterPro entries to GO annotations.
Horizontal gene transfer predictions
To further examine the role of HGT in the adaptation of strain HW001T, the automated pipeline HGTector2 (Zhu et al. 2014) was used to infer putative HGT genes. In this process, DIAMOND combined with the homologs of predicted genes retrieved from the NCBI-nr database was applied to search protein sequence similarity. The parameter settings mainly include sequence identity ≥ 30%, E value ≤ 1e−20, and coverage of query sequence ≥ 50%.
Statistical analysis
The codon adaptation index (CAI) value of genome gain of P. aggregans strain HW001T was determined using the CAI calculator (http://genomes.urv.cat/CAIcal/) (Puigbò et al. 2008). Physico-chemical parameters were obtained from the CAMERA database and used to compare with in situ environmental parameters in the present-day environments of P. aggregans strain HW001T (Toulza et al. 2012). The 54 Global Ocean Sampling (GOS) sites consist of four habitat types, including 24 coastal, 22 open ocean, 4 coral reefs, and 4 marine-derived lake (Antarctic) sites. According to the Pearson method, the coefficient relationship between sampling sites was calculated by “psych” in R (Revelle 2013; Team 2013). The interaction between P value (< 0.01) and ρ value (|ρ|> 0.7) was applied to construct networks using Cytoscape software (Kohl et al. 2011). Based on the 16S rRNA gene sequence of P. aggregans strain HW001T, the Integrated Microbial Next Generation Sequencing (IMNGS, https://www.imngs.org) server was applied to investigate the global distribution of P. aggregans strain HW001T. Threshold was set at 97% sequence similarity and minimum size was set to 200 bp (Lagkouvardos et al. 2016).
Data availability
The genome sequence has been submitted to the GenBank database under BioProject PRJNA526813 and accession number CP037953.
References
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21
Bein A, Dutton AR (1993) Origin, distribution, and movement of brine in the Permian Basin (USA): a model for displacement of connate brine. Geol Soc Am Bull 105:695–707
Bertelli C, Laird MR, Williams KP, Group SFURC, Lau BY, Hoad G, Winsor GL, Brinkman FS (2017) IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res 45:W30–W35
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537
Brissette JL, Russel M, Weiner L, Model P (1990) Phage shock protein, a stress protein of Escherichia coli. Proc Natl Acad Sci USA 87:862–866
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60
Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A (2014) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 42:D459–D471
Cheng L, Zou Y, Ding S, Zhang J, Yu X, Cao J, Lu G (2009) Polyamine accumulation in transgenic tomato enhances the tolerance to high temperature stress. J Integr Plant Biol 51:489–499
Cheng C, Dong Z, Han X, Wang H, Jiang L, Sun J, Yang Y, Ma T, Shao C, Wang X, Chen Z, Fang W, Freitag NE, Huang H, Song H (2017) Thioredoxin A is essential for motility and contributes to host infection of Listeria monocytogenes via redox interactions. Front Cell Infect Microbiol 7:287
Chew SC, Yang L (2016) Biofilms. In: Caballero B, Finglas P, Toldrá F (eds) Encyclopedia of food and health. Elsevier, Oxford, pp 407–415
Contreras-Moreira B, Vinuesa P (2013) GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol 79:7696–7701
Csűös M (2010) Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26:1910–1912
Darling ACE, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403
David LA, Alm EJ (2011) Rapid evolutionary innovation during an Archaean genetic expansion. Nature 469:93
Denamur E, Matic I (2006) Evolution of mutation rates in bacteria. Mol Microbiol 60:820–827
Didelot X, Wilson D (2015) ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol 11:e1004041
Drula E, Garron ML, Dogan S, Lombard V, Henrissat B, Terrapon N (2022) The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res 50:D571–D577
Emms DM, Kelly S (2015) Orthofinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16:157
Falcón LI, Magallón S, Castillo A (2010) Dating the cyanobacterial ancestor of the chloroplast. ISME J 4:777–783
Fernández P, Porrini L, Albanesi D, Abriata LA, Dal Peraro M, De Mendoza D, Mansilla MC (2019) Transmembrane prolines mediate signal sensing and decoding in Bacillus subtilis DesK histidine kinase. mBio 10:e02564-19
Freeman ZN, Dorus S, Waterfield NR (2013) The KdpD/KdpE two-component system: integrating K+ homeostasis and virulence. PLoS Pathog 9:e1003201
Gram L, Grossart H-P, Schlingloff A, Kiørboe T (2002) Possible quorum sensing in marine snow bacteria: production of acylated homoserine lactones by Roseobacter strains isolated from marine snow. Appl Environ Microbiol 68:4111–4116
Gruber N (2008) The marine nitrogen cycle: overview and challenges. Nitrogen Mar Environ 2:1–50
Ho SYW, Duchêne S (2014) Molecular-clock methods for estimating evolutionary rates and timescales. Mol Ecol 2324:5947–5965
Höltje J-V (1998) Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev 62:181–203
Hong Y, Youshao W, Chen F (2013) Archaea dominate ammonia oxidizers in the Permian water ecosystem of midland basin. Microbes Environ 28:396–399
Hua ZS, Qu YN, Zhu Q, Zhou EM, Qi YL, Yin YR, Rao YZ, Tian Y, Li YX, Liu L, Castelle CJ, Hedlund BP, Shu WS, Knight R, Li WJ (2018) Genomic inference of the metabolism and evolution of the archaeal phylum Aigarchaeota. Nat Commun 9:1–11
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, von Mering C, Bork P (2019) eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47:D309–D314
Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong S-Y, Lopez R, Hunter S (2014) InterProScan 5: Genome-scale protein function classification. Bioinformatics 30:1236–1240
Katoh K, Standley DM (2014) MAFFT: iterative refinement and additional methods. Methods Mol Biol 1079:131–146
Kim JG, Park SJ, Sinninghe Damsté JS, Schouten S, Rijpstra WIC, Jung WY, Kim SJ, Gwak JH, Hong H, Si OJ, Lee SH, Madsen EL, Rhee SK (2016) Hydrogen peroxide detoxification is a key mechanism for growth of ammonia-oxidizing archaea. Proc Natl Acad Sci USA 113:7888–7893
Kleerebezem M, Crielaard W, Tommassen J (1996) Involvement of stress protein PspA (phage shock protein A) of Escherichia coli in maintenance of the protonmotive force under stress conditions. EMBO J 15:162–171
Knight DR, Elliott B, Chang BJ, Perkins TT, Riley TV (2015) Diversity and evolution in the genome of Clostridium difficile. Clinical Microbiol Rev 28:721–741
Kohl M, Wiese S, Warscheid B (2011) Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol 696:291–303
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lagkouvardos I, Joseph D, Kapfhammer M, Giritli S, Horn M, Haller D, Clavel T (2016) IMNGS: a comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci Rep 6:33721
Lanfear R, Calcott B, Ho SY, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Langenheder S, Comte J, Zha Y, Samad MS, Sinclair L, Eiler A, Lindstrom ES (2016) Remnants of marine bacterial communities can be retrieved from deep sediments in lakes of marine origin. Environ Microbiol Rep 8:479–485
Latasa C, Roux A, Toledo-Arana A, Ghigo J-M, Gamazo C, Penadés JR, Lasa I (2005) BapA, a large secreted protein required for biofilm formation and host colonization of Salmonella enterica serovar Enteritidis. Mol Microbiol 58:1322–1339
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320
Li L, Stoeckert CJ, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189
Luo H, Csürös M, Hughes AL, Moran MA (2013) Evolution of divergent life history strategies in marine Alphaproteobacteria. mBio 4:e00373-13
Luo ZH, Narsing Rao MP, Chen H, Hua ZS, Li Q, Hedlund BP, Dong ZY, Liu BB, Guo SX, Shu WS, Li WJ (2021) Genomic insights of “Candidatus Nitrosocaldaceae” based on nine new metagenome-assembled genomes, including “Candidatus Nitrosothermus” gen nov. and two new species of “Candidatus Nitrosocaldus.” Front Microbiol 11:3412
Martin DP, Murrell B, Khoosal A, Muhire B (2017) Detecting and analyzing genetic recombination using RDP4. In: Keith JM (ed) Methods in molecular biology. Humana, Totowa, pp 433–460
Mello B (2018) Estimating timetrees with MEGA and the TimeTree resource. Mol Biol Evol 35:2334–2342
Misof B, Liu S, Meusemann K, Peters RS, Donath A, Mayer C, Frandsen PB, Ware J, Flouri T, Beutel RG, Niehuis O, Petersen M, Izquierdo-Carrasco F, Wappler T, Rust J, Aberer AJ, Aspöck U, Aspöck H, Bartel D, Blanke A et al (2014) Phylogenomics resolves the timing and pattern of insect evolution. Science 346:763–767
Moran MA, Buchan A, González JM, Heidelberg JF, Whitman WB, Kiene RP, Henriksen JR, King GM, Belas R, Fuqua C (2004) Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 432:910
Mori JF, Scott JJ, Hager KW, Moyer CL, Kusel K, Emerson D (2017) Physiological and ecological implications of an iron- or hydrogen-oxidizing member of the Zetaproteobacteria, Ghiorsea bivora, gen. nov., sp. nov. ISME J 11:2624–2636
Nakjang S, Williams TA, Heinz E, Watson AK, Foster PG, Sendra KM, Heaps SE, Hirt RP, Martin Embley T (2013) Reduction and expansion in microsporidian genome evolution: new insights from comparative genomics. Genome Biol Evol 5:2285–2303
Ochman H, Wilson AC (1987) Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J Mol Evol 26:74–86
Ogilvie BG, Rutter M, Nedwell DB (1997) Selection by temperature of nitrate-reducing bacteria from estuarine sediments: species composition and competition for nitrate. FEMS Microbiol Ecol 23:11–22
Oliveira PH, Touchon M, Cury J, Rocha EP (2017) The chromosomal organization of horizontal gene transfer in bacteria. Nat Commun 8:1–11
Puigbò P, Bravo IG, Garcia-Vallve S (2008) CAIcal: a combined set of tools to assess codon usage adaptation. Biol Direct 3:38
Rambaut A (2018) Figtree, a graphical viewer of phylogenetic trees, version 1.4. 4. Institute of evolutionary biology, University of Edinburgh
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904
Rand L, Hinds J, Springer B, Sander P, Buxton RS, Davis EO (2003) The majority of inducible DNA repair genes in Mycobacterium tuberculosis are induced independently of RecA. Mol Microbiol 50:1031–1042
Revelle WR (2013) psych: procedures for personality and psychological research. Available http://CRAN.R-project.org/package=psych Version = 1.3.10. Accessed 20 Dec 2013
Sanderson MJ (2003) r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19:301–302
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Sun H, Xiao Y, Gao Y, Zhang G, Casey JF, Shen Y (2018) Rapid enhancement of chemical weathering recorded by extremely light seawater lithium isotopes at the Permian-Triassic boundary. Proc Natl Acad Sci USA 115:3782–3787
Team RC (2013) R: A language and environment for statistical computing. Available http://www.R-project.org. Accessed 2 Feb 2013
Toulza E, Tagliabue A, Blain S, Piganeau G (2012) Analysis of the Global Ocean Sampling (GOS) project for trends in iron uptake by surface ocean microbes. PLoS ONE 7:e30931
Troxell B, Hassan HM (2013) Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front Cell Infect Microbiol 3:59
Wang H, Laughinghouse HD, Anderson MA, Chen F, Willliams E, Place AR, Zmora O, Zohar Y, Zheng T, Hill RT (2012) Novel bacterial isolate from Permian groundwater, capable of aggregating potential biofuel-producing microalga Nannochloropsis oceanica IMET1. Appl Environ Microbiol 78:1445–1453
Wang H, Zheng T, Hill RT, Hu X (2014) Permianibacter aggregans gen. nov., sp. nov., a bacterium of the family Pseudomonadaceae capable of aggregating potential biofuel-producing microalgae. Int J Syst Evol Microbiol 64:3503–3507
Wright WR (2011) Pennsylvanian paleodepositional evolution of the greater Permian Basin, Texas and New Mexico: depositional systems and hydrocarbon reservoir analysis. AAPG Bull 95:1525–1555
Xiao R, Zheng Y (2016) Overview of microalgal extracellular polymeric substances (EPS) and their applications. Biotechnol Adv 34:1225–1244
Xiao R, Yang X, Li M, Li X, Wei Y, Cao M, Ragauskas A, Thies M, Ding J, Zheng Y (2018) Investigation of composition, structure and bioactivity of extracellular polymeric substances from original and stress-induced strains of Thraustochytrium striatum. Carbohyd Polym 195:515–524
Yang Z (1996) Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol Evol 11:367–372
Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer K-H, Whitman WB, Euzéby J, Amann R, Rosselló-Móra R (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645
Yin S, Chen D, Chen L, Edis R (2002) Dissimilatory nitrate reduction to ammonium and responsible microorganisms in two Chinese and Australian paddy soils. Soil Biol Biochemi 34:1131–1137
Yoon S-H, Ha S-M, Lim J, Kwon S, Chun J (2017) A large-scale evaluation of algorithms to calculate average nucleotide identity. Ant v Leeuwenhoek 110:1281–1286
Zhaxybayeva O, Swithers KS, Lapierre P, Fournier GP, Bickhart DM, DeBoy RT, Nelsond KE, Nesbøe CL, Doolittlea WF, Gogarten JP, Noll KM (2009) On the chimeric nature, thermophilic origin, and phylogenetic placement of the Thermotogales. Proc Natl Acad Sci USA 106:5865–5870
Zhu Q, Kosoy M, Dittmar K (2014) HGTector: an automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genomics 15:1–18
Acknowledgements
Funding for this study was provided by the Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2019ZD0606), National Natural Science Foundation of China (92051118), Guangdong Science and Technology Department (2019A1515011139), and 2020 Li Ka Shing Foundation (LKSF) Cross-Disciplinary Research Grant (2020LKSFG07A).
Author information
Authors and Affiliations
Contributions
HW and RTH conceived and designed the experiments; SZ performed the experiments and drafted the manuscript; HW, RTH, and SZ revised the manuscript; HW supervised the project. The final manuscript was approved by all of the authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Animal and human rights statement
This article does not contain any studies performed with human and animals.
Additional information
Edited by Jiamei Li.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, S., Hill, R.T. & Wang, H. Genomic characterization and molecular dating of the novel bacterium Permianibacter aggregans HW001T, which originated from Permian ground water. Mar Life Sci Technol 5, 12–27 (2023). https://doi.org/10.1007/s42995-023-00164-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42995-023-00164-3