
Abstract
The American mink (Mustela vison) is known as a successful non-native species in Europe, impacting native species’ population sizes and habitats. This study investigates the genetic structure and diversity of American mink populations in France over two decades (1997–2016). The analysis involves feral and farmed mink sampled from various regions, using ten autosomal microsatellite loci for genotyping. The objective is to identify the putative existence of genetic lineages, especially between feral and farmed individuals, and to assess changes in genetic structure over time. Results reveal high genetic diversity and inbreeding within populations, with evidence of genetic structure influenced by both farm releases and feral colonization. The study highlights the reflection of the genetic structure in farm populations in the feral populations within the first period (1997–2007), and a decline of a lineage over time in the second period (2007–2016) with the emergence of a new genetic cluster, potentially influenced by factors such as selection, phenotypic changes, and interactions with pathogens. Overall, this research contributes to the understanding of the dynamics of American mink populations in France and their genetic variability, emphasizing the importance of ongoing monitoring and management efforts to mitigate the impact of this invasive species, especially on endangered or/and endemic species such as European mink (Mustela lutreola) and Iberian desman (Galemys pyrenaicus).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Invasive alien species have been widely recognized as one of the major threats of biodiversity due to anthropogenic changes at both global and local scales (Lockwood et al. 2007; Genovesi 2009). They can directly impact the habitat and ecology of native species they interact with as they affect native species’ population sizes and habitat ranges (Zalewski et al. 2010). An example of such a successful invader is the American mink (Mustela vison or Neovison vison) in parts of Europe, which was introduced from North America for fur farming in the early twentieth century. Following accidental escapes, as well as intentional releases, this mink is present in 28 European countries (Bonesi and Palazon 2007; Reid et al. 2016). This species was also introduced and established in parts of South America and Asia (Shimatani et al. 2010; Mora et al. 2018). In France it is considered as an invasive species (Savouré-Soubelet et al. 2024). The generalist and opportunistic aspects of this mustelid’s diet impacted native populations of both aquatic and terrestrial species, reducing prey species of seabirds in Brittany, especially the Roseate Tern (Sterna dougallii) loosing between 25 and 32% of its French population each year (Jacob and Capoulade 2010). Lorvelec et al. (2024) also observed eleven-time fewer marine birds on Brittany breeding grounds of Tomé Island from 2004 to 2019, with drastic decrease of the European herring gull (Larus argentatus), largely attributed to the American mink’s invasion confirmed in 2012. Great proportion of predation of the endangered Iberian desman (Galemys pyrenaicus) endemic to the French Pyrenees and the Iberian Peninsula was also described in Spain (Romero 2015). While the feeding plasticity of this non-native species can be considered to limit competition with other carnivores (Hammershoj et al. 2004; Carlsson et al. 2010), added predation pressure on native prey species is not negligible (Krawczyk et al. 2013; Mezzetto et al. 2021).
The American mink can however be in direct and indirect competition for resources with other carnivorous mammals such as the critically endangered European mink (Mustela lutreola), with evidence of direct aggression from the invader towards the native species observed in other parts of Europe (Melero et al. 2008; Sidorovich et al. 2010; Podra et al. 2013). Current distribution of both species in France advocate without confirmation for European Mink displacement from the American mink (DREAL Nouvelle Aquitaine et al. 2021). The American mink can also play a role in disease transmission among native species, especially in carnivores, as they can carry for example the Aleutian Disease Virus (ADV) and could potentially be the source of the ADV in Europe through fur farms (Zaleska-Wawro et al. 2021; Vahedi et al. 2023). Greater seroprevalence of ADV in American mink has been observed compared to other species in France, and circulation in American mink populations can potentially increase transmission in native species (Fournier-Chambrillon et al. 2004). They can also carry other parasites in common with other mustelids, thus potentially affecting native populations (Torres et al. 2008). With 47 native species impacted in Europe by the American mink, this species features prominently in the list compiled by Genovesi et al. (2012).
In France, the American mink was introduced in the 1920s for fur farming; and first observations of feral animals were recorded since 1950s (Léger et al. 2018). A long-term monitoring study from 2000 to 2015 recorded evidence of the expansion of the American mink over France with three main established populations: (1) the historical region of Brittany, Normandy and Pays de la Loire dating from 1960s—renamed as Brittany in the following study—(2) the South West region including south of Nouvelle-Aquitaine and West of Occitanie, renamed as South West which emerged in the 1980s, and (3) the South East of Occitanie starting from the 2000s—renamed South East—(Léger et al. 2018). Moreover, local data or related to the presence of a mink farm, without a source of dispersion, are observed particularly in the region subsequently named Central West, between Brittany and South West. Concerns were raised on the overall expansion of the American mink throughout Western France, questioning when the established populations would merge into one, which would allow genetic flow and potentially increase the species invasiveness.
The use of molecular tools such as genotyping allows for a better understanding of the dynamics leading to the range expansion of the American mink in Europe. Different studies have been conducted on the population genetics of the American mink throughout Europe, observing greater genetic diversity within feral populations compared to captive conspecifics (Bifolchi et al. 2010; Garcia et al. 2017; Korablev et al. 2018; Mora et al. 2018). This greater diversity is also observed in regions where the American mink is native (Kidd et al. 2009; Bowman et al. 2017). Insights from the population genetic structure can inform management practices by tracking the dispersal of the population and limiting their expansion by regulating targeted populations (Sakai et al. 2001; LeRoux and Wieczorek 2009).
The present study aimed to investigate the genetic structure and diversity of American mink over time for multiple regions in France. We monitored several populations over two decades, which included feral mink and individuals held in fur farms, through genotyping of 10 autosomal microsatellite loci. Those neutral nuclear genomic markers vary from 1 to 20 nucleotides long tandem repeats that are unstable with elevated mutation rates. Having flanking regions highly conserved is also evolutionary relevant for studying population structure and diversity (Selkoe and Toonen 2006). The objective was to determine if different genetic clusters could be identified within our sample set, especially between feral and farmed individuals from respective regions (Central West and South West of France for captive individuals) and if the genetic structure changed between 1997–2007 and 2008–2016 periods. We hypothesized that genetic clusters would reflect the historical emergence of the three main established populations observed by Léger et al. (2018) and predicted that those population have recently met and started to homogenize.
Methods
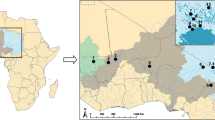
Samples were collected on free-ranging feral American mink trapped and culled by duly licensed trappers (“French American mink network”) in the context of invasive population management. Samples from farmed individuals were obtained thanks to the collaboration of the breeders concerned, during the pelting season following Fournier-Chambrillon et al. (2010). All samples were preserved in 90° alcohol before analysis. A total of 355 samples were collected from American mink over a twenty-year period between 1997 and 2016 and divided by periods as followed: ‘old’ between 1997 and 2007 and ‘recent’ ranging from 2008 to 2016, as well as if individuals were either free-ranging (feral) or held within fur farms (farm, Table 1). For subsequent analyses, individuals were also grouped according to their geographic origin, sorted into four main regions: Brittany, Central West, South West and South East (Fig. 1). Ten groups were finally defined according to individual environment, period and geographical origin (Table 1).

Distribution of samples, colors indicate hypothetical geographical clusters used to define the groups
Genomic DNA was isolated using the DNeasy Blood and Tissue Kit (QIAGEN) from tissue samples. Negative controls and aerosol resistant pipettes were used. Multilocus genotypes were obtained by PCR amplification of 10 autosomal microsatellites (Fleming et al. 1999; Cabria et al. 2007). The forward primer of each locus was 5’-end labeled with a fluorescent dye (Table A). The following three multiplex sets were designed: mix 1 (MLUT25, MLUT27, Mvis099), mix 2 (MLUT04 Mvis022) and mix 3 (MER009, Mvis075, Mvis072, MER41, MER022). PCRs were carried out in 10 μl volumes containing 1 μl of primer mix (containing each 2 μM primer), 5 μl of Multiplex PCR Master Mix (QIAGEN) and 1 μl of DNA. All amplifications were performed as follows: 95 °C for 15 min followed by 40 cycles (94 °C for 30 s, annealing at 57 °C for 90 s, extension at 72 °C for 60 s) and a final extension step at 60 °C for 30 min. PCR products were genotyped on an Applied Biosystems 3130XL Genetic Analyzer using 2 μl of amplified DNA, 10 μl of Hi-Di formamide and 0.15 μl of GeneScan-500 (LIZ) size standard (Applied Biosystems). Length variation determination (alleles and genotypes) was performed using GENEMAPPER 4.0 (Applied Biosystems). MICRO-CHECKER 2.2.3 (Van Oosterhout et al. 2004) was used to detect stutter errors and to estimate the proportion of null alleles at each locus for each cluster defined by the preliminary STRUCTURE analysis (see below). Genotypes were then corrected accordingly. To construct consensus multilocus genotypes, we followed the rules described hereafter (adapted from Bonesi et al. 2013): an allele was only accepted if observed at least twice. We thus accepted heterozygous genotypes that were observed twice. A homozygote was accepted after three positive PCRs gave the same single allele; otherwise, we coded the locus as being potentially heterozygous with only one allele identified and one allele missing.
F-statistics (pairwise FST, DJost and FIS), allelic richness (Ar), the Hardy–Weinberg Equilibrium (HWE), FIS calculations and the expected (He) and observed (Ho) heterozygosity were calculated for each defined group using diveRsity (Keenan et al. 2013) in R version 3.5.2 (R Development Core Team 2008). Group differentiation was assessed by analysis of molecular variance (AMOVA) using the poppr package. It was calculated as FST for all groups and between pairs of groups (Peakall and Smouse 2012). One model per period was conducted to evaluate variation between sampling localities and between mink environments, animals being free-ranging or held in farms. The significance of the estimative was obtained by using 9,999 permutations and a confidence interval of 95%, by 10,000 resampling. In addition, Principal coordinate analyses (PCoA) were carried out using GenAlex (Peakall and Smouse 2012) for the identification of genetic diversity patterns within the full dataset, and separately for each decade (old: 1997–2007 and recent: 2008–2016) based on Euclidean distances generated between samples.
The genetic structure of the American mink groups was inferred using Bayesian clustering analysis with STRUCTURE 2.3 software (Pritchard et al. 2000) on the entire dataset, and divided by decades (old: 1997–2007 and recent: 2008–2016). We ran 10 iterations for each K value from 1 to 10 using the admixture model. A total of 106 MCMC repetitions were performed after a burn-in period of 20%. The results of the 10 iterations for each K value were summarized and averaged using the CLUMPP method (Jakobsson and Rosenberg 2007) implemented in CLUMPAK (Kopelman et al. 2015). The optimal number of clusters was investigated using the inlikelihood of the data (Pritchard et al. 2000) as well as the ΔK method (Evanno et al. 2005), implemented in STRUCTURE HARVESTER (Earl and von Holdt 2012). We determined an admixture pattern for each individual when attribution to one cluster q-value was below 90%. Effective population sizes calculation were conducted using BOTTLENECK 1.2 (Cornuet and Luikart 1996) according to periods for feral mink, as well as between clusters identified using STRUCTURE.
Results
All ten microsatellite loci were polymorphic in the 355 mink sampled. The mean allelic richness was 3.44, the mean number of alleles per locus was 6.7, and the mean observed heterozygosity was 0.56 with overall similar values between populations. However, slightly lower values are found within the recent period compared to the old period (Table 2). We observe a greater number of loci per group that deviated from Hardy–Weinberg Equilibrium in the recent period (Table 2). More specifically, in the South East and Central West ferals, as well as farm individuals from the South West, potentially indicating population substructures but is more likely uneven sampling due to low sample size. Additionally, FIS values above 0.15 are found overall for both periods apart from the Central west recent farm and feral (Table B).
The three models of AMOVA indicated most genetic variance within individuals (mean = 90.8%, mean FIT = 0.091, p < 0.001) and then among individuals between geographical regions (mean = 7.6%, mean FIS = 0.077, p < 0.001). Variances between periods and between environments (feral-farm) for both periods (old and recent) were 0.23%, 1.04% and 3.4%, respectively while not reaching statistical significance (Table 3).
Principal component analysis (PCoA) detected low genetic differentiation further explained by both axis (X-axis: 10.36%, Y-axis: 7.49%; Fig. 2A). However, PCoA did detect a genetic structure, isolating a set of feral individuals sampled in Brittany and the South West region on the X-axis (recent period, Fig. 2A, C). Despite showing continuous spatial variation, an orthogonal gradient through X-axis is observed when only considering the old period, exhibiting genetic differentiation between mink sampled in the South West compared to Central West conspecifics (Fig. 2B).

PCoA generated from Euclidian genetic distances between individuals for A the full dataset, B the old period and C the recent period
The Bayesian clustering analysis showed that the optimal number of clusters by the Evanno method was K = 3 for the overall dataset (K1, K2 and K3), as well as K = 2 for both old and recent periods (KA old and KB old, KA recent and KB recent; Table C, Fig. 3; Table 4). One of the genetic clusters (K2, purple in Fig. 3) is overall present in all populations and is shared in high frequency with all samples collected in the recent period, especially in farmed individuals. In addition, the second most frequent cluster (K1, orange in Fig. 3) is present in higher frequency in samples collected in the old period represented by 43–14% of mink attributed to this cluster by populations (Fig. 3, q-value > 0.9). However, this cluster counts zero strict attribution in recent populations apart from South West ferals (6%), showing a decline in this potential lineage over time in both farmed and feral individuals. Finally, the third cluster (K3, turquoise in Fig. 3) is mostly present in recent feral samples, especially in Brittany and South West, being marginally represented in other regional populations.

Bayesian clustering attribution of samples according to their hypothetical geographical populations, environment and period. On maps are represented with pie charts sample attribution to each cluster with q > 0.90. Chart size is proportional to sample size for each group. Horizontal bar plots represent the K = 3 clusters attribution for the overall data set for both decades
When considering both periods separately, Bayesian clustering analysis showed that the optimal number of clusters by the Evanno method was two clusters for the old period (KA and KB, Table C). Although admixture individuals were found in the four groups, a dominance of mink attributed to KA (Table 4) is found in farms of both regions while mink strictly attributed to KB are ferals from the South West. However, when considering the Bayesian clustering analysis of the recent period, all population are dominated by individuals attributed to KB (Table 4) and few admixture patterns. Nevertheless, a great proportion of samples was attributed to KA in the Brittany and South West region for feral individuals. This result seems to converge with results obtained on the full dataset regarding the third cluster (K3, turquoise), showing great resemblance of feral populations from Brittany and the South West in the recent period.
Finally, effective population sizes were calculated for feral populations, and were found greater in the recent period compared to the old period, as well as according to the clusters identified through Bayesian approach (Table 5).
Discussion
The present study demonstrates the relatively high genetic diversity of feral and farm mink populations over a 20-year period in France. Compared to Bifolchi et al. (0.62; 2010) that focused on Brittany, we observe similar overall mean heterozygosity levels (0.58; Table 2), which are also relatively similar to those obtained in other non-native countries such as Patagonia (0.38; Mora et al. 2018), Japan (0.48; Shimatani et al. 2010), China (0.50–0.63; Zhang et al. 2021), Scotland (0.57; Fraser et al. 2013), Poland (0.59; Zalewski et al. 2010), Spain (0.61; Lecis et al. 2008) and Russia (0.69; Korablev et al. 2018). However, comparisons between studies must be interpreted with caution due to the use of multiple markers that are not all common between studies.
Relatively high inbreeding is observed overall within the present dataset (Table 2), as well as deviation from Hardy–Weinberg Equilibrium, showing a deficit of heterozygosity within our dataset. These results potentially show a Wahlund effect that is anticipated when distinct gene pools are combined (Hartl and Clark 1997) and structures in subpopulations at lower geographical scale is confirmed when looking for genetic partitions using Bayesian methods. Indeed, multiple, and independent introductions from different source populations with distinct genetic compositions occurred for the introduction of the American mink (Bifolchi et al. 2010), and the resulting population structure can include subpopulations with different allele frequencies. This leads to a Wahlund effect when these subpopulations interbreed.
We also observe high allelic richness, despite lower mean values for feral groups from the recent period (2008–2016). Those values can be explained by the low sample size of some populations (Table 1), which are therefore not entirely representative of all mink in the areas sampled. No distinct geographical patterns are found in this dataset when it comes to genetic diversity, advocating for the success of the invasion of France for the American mink, as in other countries in Europe and in the world (Bonesi and Palazon 2007). Indeed, high genetic diversity and gene flow can make a species competitive for its establishment in a new environment and its expansion. High genetic variability in a population can foster phenotypic plasticity that can mitigate with new encounters with unknown pathogens and niche breadth. Moreover, genetic potential can be augmented by the number of founders and multiple introduction events when it comes to invasive species (Dlugosch and Parker 2008). In comparison, previous studies on the native and critically endangered European mink showed lower allelic diversity and heterozygosity levels in France compared to Eastern Europe, with significant results for a recent bottleneck event (for n = 106, Ho = 0.379, Michaux et al. 2005 and n = 73, Ar = 2.6, Ho = 0.389 ± 0.182, Cabria et al. 2015).
In the case of the American mink in Europe, most studies show that a distinct genetic structure is observed in feral mink compared to farm lineages. This is reflective of mink conditions of introduction, namely the proximity and number of sites for introduction, as well as the lineages released. This is observed in Russia (Korablev et al. 2018), Poland (Zalewski et al. 2011), Spain (Lecis et al. 2008) and Brittany for France (Bifolchi et al. 2010) and is illustrated by the free-ranging population originating from farm escapees of multiple lineages. The genetic structure of populations in the old period (1996–2007) in France within our study had similar patterns of cluster attribution, reflecting the strong influence of multiple introduction events from farms on feral population structures. The presence of a dominant cluster and admixture individuals in the old period (Table 4) already advocates for great dispersion among populations, with common, highly diverse genetic pools between farms. The genomic admixture that occurred during the mixing of different populations might have increased the fitness of individuals and accelerated the invasiveness of this species during this period.
Different genetic population structures have been observed in the recent period (2008–2016). First, we found a striking progressive reduction of the orange cluster (Fig. 3) between old and recent periods in both feral and farm mink. Second is the establishment of the turquoise cluster in the recent period (Fig. 3), which was only marginally present in admixture individuals in the old period, but which is now highly represented in Brittany and the South West compared to other populations. This cluster’s distribution in the South East and Central West remains to be confirmed due to our reduced sample size for feral mink. The distribution of those clusters is unlikely to be linked to farm escapees, compared to the genetic structure of the old period. This is mainly because since the beginning of national action plans for the European mink in 1999, extensive awareness-raising and training campaigns have been conducted among mink farmers to limit new introduction levels (DREAL Nouvelle Aquitaine et al. 2021). The absence of the turquoise cluster in farm populations thus reflects the lack of similarity between farm and feral populations, advocating for other means of genetic variation.
One explanation for the recent genetic structure of feral populations could reflect a relatively new natural colonization by a lineage of escapees coming from Brittany and the South West, forming two colonization fronts. As discussed by Zalewski et al. (2011), the long-distance dispersal ability of American mink males can shape and homogenize the genetic structure of populations founded by individuals of various origins (Melero et al. 2018). However, the lack of established population between Brittany and the South West region does not support this hypothesis. While long-distance male dispersal and multiple introductions in the past from the same lineage could explain the ubiquitous presence of the purple cluster (Fig. 3), our sample size in some populations cannot validate the same distribution for the turquoise cluster. Moreover, the very low abundance of this cluster in the old period for the South West raises the question whether this lineage was already present in Brittany during the old period, or/and if farm escapees from this lineage in the South West were missed in our sampling. Regardless of its geographical origin, there must have been drivers that fostered the maintenance and expansion of this lineage over time in feral populations and the reduction of the prevalence of the orange cluster. If this variation in genetic structure is explained by genetic drift, selection, or both, it remains to be tested through other markers, both neutral and selective. However, we can speculate that there is potential for lineage selection based on phenotype variation in other regions where the American mink is invasive.
When considering the depletion of the orange lineage, we can consider that it might have been less competitive than other lineages in both captivity and in the wild. In that sense, French breeding stock for fur farming originates from Danish lineages, and it is possible that one lineage was less favored by breeders during livestock renewal for various reasons (docility, fur density, fur color, etc. Fournier-Chambrillon, personal comment). A recent study also demonstrated that morphological changes arise after domestication once the American mink becomes feral (Pohle et al. 2023). According to Pohle et al. (2023), the brain size of well-established feral mink in Poland reached similarity to their wild North American ancestors compared to farmed individuals, and brain volume can potentially impact a wide range of changes in species behavior through cognitive capacity, hence advocating for phenotypic plasticity between feral lineages. In the same way, forelimb length and body shape were observed to be different between feral and farm American mink in Poland (Mucha et al. 2021), where the genetic background has a significant influence on the size of the offspring, despite the ecological aspect. Those examples illustrate the great and rapid variation in phenotype for this species during the invasive process, fostered by a high fecundity selected by breeders, which could explain the depletion/establishment of different lineages.
Another aspect to consider is the relationship between the American mink and its pathogens, which could explain the changing genetic structure of the mink in France. A recent study in Poland investigated the link between reproductive output and the Aleutian Disease Virus (ADV) prevalence in feral American mink (Zalewski et al. 2023). Feral females infected with ADV delivered significantly smaller litters than uninfected females, reducing their litter size by 8%. Moreover, offspring survival within litters of infected females was lower than that within those of uninfected females. This link between infection and reproductive output can seriously impact feral mink populations. However, investigation within the French feral population is needed to better understand if the turquoise cluster carries such phenotypic changes compared to the orange cluster, despite known circulation of ADV in France (Fournier-Chambrillon et al. 2004).
Overall, this study allows for an overview of the genetic variation over time in multiple feral and farm populations of American mink in France. While no distinct geographical patterns are found, hence not reflecting the historical emergence of the three main established populations observed by Léger et al. (2018), we do observe the ubiquitous presence of a lineage, advocating for a unified population. Since all mink farms are now closed in France since 2021, the risk of new introductions is very limited. Now that this species is established in the French landscape, other factors such as genetic drift or selection will be crucial for its genetic variability and dominance over native species. Moreover, measuring the variability in mink phenotype according to the clusters identity could shed more light on the evolutionary history of the species within France. Further conservation actions testing for potential impacts of the species on the native species present within the American mink’s range also needs to be conducted across the territory. The fact that we observed greater effective population sizes in the recent period compared to 1997–2006 for feral populations advocates for a boosted dynamic within populations. Those results could have direct implication when it comes to the mitigation of the expansion of the American mink in France. American mink trapping is carried out almost everywhere on an individual basis by local associations of approved trappers. However intense, organized and coordinated control is carried out only in the priority intervention zones defined by the national action plan for European mink, essentially on the colonization fronts that directly threaten European mink relict nuclei in the Charente basin and in Spain: South front for the Brittany population, as well as North and South fronts for the South West population (DREAL Nouvelle Aquitaine et al. 2021). There is thus a lack of national coordination to measure and limit impacts of the American mink in all of France. Our study shows no critical geographical point of admixture, and no management units from the genetic aspect, limiting localized actions and therefore advocating for a large scale coordinated action. However, Vada et al. (2023) observed a decrease in American mink feral population size in 2017–2021 compared to 2012–2016 (-21%) in France, showing the impact of governmental measures to mitigate this species establishment (European Life program, National Action Plans from 1999 to 2030). Genotyping mink from 2017 onward could bring new insight on the potential consequences of population control on the genetic population diversity and structure. If comparing even more recent data with results from this study could show greater levels of heterozygosity deficiency, the reduction of genetic diversity, and an uneven distribution of genetic clusters, it would be possible to confirm the efficiency of American mink control actions.
Data availability
Supporting information has been made available online. Microsatellite data and information on samples can be found here: https://doi.org/10.6084/m9.figshare.25224758.
References
Bifolchi A, Picard D, Lemaire C, Cormier JP, Pagano A (2010) Evidence of admixture between differentiated genetic pools at a regional scale in an invasive carnivore. Conserv Genet 11(1):1–9. https://doi.org/10.1007/s10592-008-9780-1
Bonesi L, Hale M, Macdonald DW (2013) Lessons from the use of non-invasive genetic sampling as a way to estimate Eurasian otter population size and sex ratio. Acta Theriol 58(2):157–168. https://doi.org/10.1007/s13364-012-0118-5
Bonesi L, Palazon S (2007) The American mink in Europe: status, impacts, and control. Biol Conserv 134(4):470–483. https://doi.org/10.1016/j.biocon.2006.09.006
Bowman J, Beauclerc K, Hossain Farid A, Fenton HC, Klütsch CF, Schulte-Hostedde AI (2017) Hybridization of domestic mink with wild American mink (Neovison vison) in eastern Canada. Can J Zool 95(6):443–451
Cabria MT, González EG, Gómez-Moliner BJ, Zardoya R (2007) Microsatellite markers for the endangered European mink (Mustela lutreola) and closely related mustelids. Mol Ecol Notes 7(6):1185–1188. https://doi.org/10.1111/j.1471-8286.2007.01825.x
Cabria MT, Gonzalez EG, Gomez-Moliner BJ, Michaux JR, Skumatov D, Kranz A, Fournier P, Palazon S, Zardoya R (2015) Patterns of genetic variation in the endangered European mink (Mustela lutreola L., 1761). BMC Evol Biol 15(1):141. https://doi.org/10.1186/s12862-015-0427-9
Carlsson NOL, Jeschke JM, Holmqvis N, Kindberg J (2010) Long-term data on invaders: when the fox is away, the mink will play. Biol Invasions 12(3):633–641. https://doi.org/10.1007/s10530-009-9470-z
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Direction Régionale de l’Environnement, de l’Aménagement et du Logement (DREAL), Groupe de Recherche et d’Investigation sur la Faune Sauvage (GRIFS), Cistude Nature, Office Français de la Biodiversité (OFB) (2021) Plan national d’actions en faveur du vison d’Europe (Mustela lutreola) 2021–2030, pp 174
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17(1):431–449. https://doi.org/10.1111/j.1365-294X.2007.03538.x
Earl DA, von Holdt BM (2012) Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Res 4(2):359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Fleming M, Ostrander E (1999) Cook J (1999) Microsatellite markers for American mink (Mustela vison) and ermine (Mustela erminea). Mol Ecol Notes 8:1352–1354
Fournier-Chambrillon C, Aasted B, Lie Perrot A, Pontier D, Sauvage F, Artois M, Cassiè JM, Chauby X, Dal Molin A, Simon C, Fournier P (2004) Antibodies to Aleutian mink disease parvovirus in free-ranging European mink (Mustela lutreola) and other small carnivores from Southwestern France. J Wildl Dis 40(3):394–402
Fournier-Chambrillon C, Bifolchi A, Mazzola-Rossi E, Sourice S, Albaret M, Bray Y, Ceña JC, Maya FU, Agraffel T, Fournier P (2010) Reliability of stained placental scar counts in farmed American mink and application to free-ranging mustelids. J Mammal 91(4):818–826. https://doi.org/10.1644/09-MAMM-A-297.1
Fraser EJ, Macdonald DW, Oliver MK, Piertney S, Lambin X (2013) Using population genetic structure of an invasive mammal to target control efforts—an example of the American mink in Scotland. Biol Conserv 167:35–42. https://doi.org/10.1016/j.biocon.2013.07.011
García K, Melero Y, Palazón S, Gosálbez J, Castresana J (2017) Spatial mixing of mitochondrial lineages and greater genetic diversity in some invasive populations of the American mink (Neovison vison) compared to native populations. Biol Invasions 19(9):2663–2673. https://doi.org/10.1007/s10530-017-1475-4
Genovesi P (2009) Invasive alien species in a changing world. Biodiversity 10(3):3–4. https://doi.org/10.1080/14888386.2009.9712838
Genovesi P, Carnevali L, Alonzi A, Scalera R (2012) Alien mammals in Europe: updated numbers and trends, and assessment of the effects on biodiversity. Integr Zool 7(3):247–253. https://doi.org/10.1111/j.1749-4877.2012.00309.x
Hammershøj M, Thomsen EA, Madsen AB (2004) Diet of free-ranging American mink and European polecat in Denmark. Acta Theriol 49(3):337–347
Hartl DL, Clark AG (1997) Principles of population genetics, 3rd edn. Sinauer Associates, Sunderland
Jacob Y, Capoulade M (2010) Prédation, compétition spatiale et dérangement interspécifique en baie de Morlaix. Actes du séminaire du LIFE in: Conservation de la sterne de Dougall en Bretagne. Penn Ar Bed 208:13–18
Jakobsson M, Rosenberg NA (2007) Clumpp: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23(14):1801–1806
Keenan K, Mcginnity P, Cross TF, Crozier WW, Prodöhl PA (2013) DiveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4(8):782–788. https://doi.org/10.1111/2041-210X.12067
Kidd AG, Bowman J, Lesbarrères D, Schulte-Hostedde AI (2009) Hybridization between escaped domestic and wild American mink (Neovison vison). Mol Ecol 18(6):1175–1186. https://doi.org/10.1111/j.1365-294X.2009.04100.x
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191. https://doi.org/10.1111/1755-0998.12387
Korablev MP, Korablev NP, Korablev PN (2018) Genetic polymorphism and population structure of the introduced American mink (Neovison vison Schreber, 1777) in the center of European Russia based on microsatellite loci. Russ J Genet 54(10):1179–1184. https://doi.org/10.1134/S1022795418100083
Krawczyk AJ, Bogdziewicz M, Czyz MJ (2013) Diet of the American mink Neovison vison in an agricultural landscape in western Poland. Folia Zool 62(4):304–310. https://doi.org/10.25225/fozo.v62.i4.a8.2013
le Roux J, Wieczorek AM (2009) Molecular systematics and population genetics of biological invasions: towards a better understanding of invasive species management. Ann Appl Biol 154(1):1–17. https://doi.org/10.1111/j.1744-7348.2008.00280.x
Lecis R, Ferrando A, Ruiz-Olmo J, Mañas S, Domingo-Roura X (2008) Population genetic structure and distribution of introduced American mink (Mustela vison) in Spain, based on microsatellite variation. Conserv Genet 9(5):1149–1161. https://doi.org/10.1007/s10592-007-9428-6
Léger F, Steinmetz J, Laoué E, Maillard JF, Ruette S (2018) L’expansion du vison d’Amérique en France, période 2000–2015. Faune Sauvage 318:23–31
Lockwood JL, Hoopes MF, Marchetti MP (2007) Invasion ecology, 1st edn. Wiley-Blackwell, New York, p 466
Lorvelec O, Riallin S, Boisson PY, Bredin M, Deniau A, Dutouquet L, Guiguen S, Le Hervé Q, Le Quilliec P, Primas O, Provost P (2024) L’apport de la génétique pour comprendre la colonisation de l’île Tomé (Côtes-d’Armor, France) par le vison d’Amérique, Mustela vison Schreber, 1777: Conséquences pour sa gestion. Naturae 2:13–30. https://doi.org/10.5852/naturae2024a2
Melero Y, Cornulier T, Oliver MK, Lambin X (2018) Ecological traps for large-scale invasive species control: predicting settling rules by recolonising American mink post-culling. J Appl Ecol 55(4):1769–1779. https://doi.org/10.1111/1365-2664.13115
Melero Y, Palazón S, Bonesi L, Gosàlbez J (2008) Feeding habits of three sympatric mammals in NE Spain: the American mink, the spotted genet, and the Eurasian otter. Acta Theriol 53(3):263–273. https://doi.org/10.1007/BF03193123
Mezzetto D, Dartora F, Mori E (2021) Feeding plasticity and temporal behaviour of the alien American mink in Europe. Acta Oecol. https://doi.org/10.1016/j.actao.2020.103700
Michaux JR, Hardy OJ, Justy F, Fournier P, Kranz A, Cabria M, Davison A, Rosoux R, Libois R (2005) Conservation genetics and population history of the threatened European mink Mustela lutreola, with an emphasis on the west European population. Mol Ecol 14(8):2373–2388. https://doi.org/10.1111/j.1365-294X.2005.02597.x
Mora M, Medina-Vogel G, Sepúlveda MA, Noll D, Álvarez-Varas R, Vianna JA (2018) Genetic structure of introduced American mink (Neovison vison) in Patagonia: colonisation insights and implications for control and management strategies. Wildl Res 45(4):344–356. https://doi.org/10.1071/WR18026
Mucha A, Zatoń-Dobrowolska M, Moska M, Wierzbicki H, Dziech A, Bukaciński D, Bukacińska M (2021) How selective breeding has changed the morphology of the American mink (Neovison vison)—a comparative analysis of farm and feral animals. Animals 11(1):1–13. https://doi.org/10.3390/ani11010106
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in excel. Population genetic software for teaching and research—an update. Bioinformatics 28(19):2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Põdra M, Gómez A, Palazón S (2013) Do American mink kill European mink? Cautionary message for future recovery efforts. Eur J Wildl Res 59(3):431–440. https://doi.org/10.1007/s10344-013-0689-8
Pohle AK, Zalewski A, Muturi M, Dullin C, Farková L, Keicher L, Dechmann DKN (2023) Domestication effect of reduced brain size is reverted when mink become feral. R Soc Open Sci. https://doi.org/10.1098/rsos.230463
Pritchard JK, Stephens M, Donnell P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):949–959
R Development Core Team (2008) R: A language and environment for statistical computing. R foundation for statistical computing, Vienna. http://www.R-project.org
Reid F, Schiaffini M, Schipper J (2016) Neovison vison. The IUCN red list of threatened species 2016: e.T41661A45214988. https://doi.org/10.2305/IUCN.UK.2016-1.RLTS.T41661A45214988.en. Accessed on 15 Dec 2023
Romero R (2015) Depredación de visón americano sobre desmán ibérico en Galicia. Galemys 27:13–22
Sakai AK, Allendorf FW, Holt JS, Lodge DM, Molofsky J, With KA, Baughman S, Cabin RJ, Cohen JE, Ellstrand NC, Mccauley DE, Oneil P, Parker IM, Thompson JN, Weller SG (2001) The population biology of invasive species. Annu Rev Ecol Evol Syst 32:305–332
Savouré-Soubelet A, Aulagnier S, Haffner P, Maille A, Moutou F, Richard-Hansen C, Ruette S, Veron G (2024) Atlas des mammifères sauvages de France, volume 3: Carnivores et Primates. Muséum National d’Histoire Naturelle, Paris; OFB, Vincennes
Selkoe KA, Toonen RJ (2006) Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett 9(5)6:15–629. https://doi.org/10.1111/j.1461-0248.2006.00889.x
Shimatani Y, Fukue Y, Kishimoto R, Masuda R (2010) Genetic variation and population structure of the feral American mink (Neovison vison) in Nagano, Japan, revealed by microsatellite analysis. Mamm Study 35(1):1–7. https://doi.org/10.3106/041.035.0101
Sidorovich VE, Polozov AG, Zalewski A (2010) Food niche variation of European and American mink during the American mink invasion in north-eastern Belarus. Biol Invasions 12(7):2207–2217. https://doi.org/10.1007/s10530-009-9631-0
Torres J, Miquel J, Fournier P, Liberge M, Fons R, Feliu C (2008) Helminth communities of the autochthonous mustelids Mustela lutreola and M. putorius and the introduced Mustela vison in south-western France. J Helminthol 82:349–355. https://doi.org/10.1017/S0022149X08046920
Vada R, Illanas S, Acevedo P, Adriaens T, Apollonio M, Belova O, Blanco-Aguiar JA, Csányi S, Body G, Fernández-De-Mera IG, Ferroglio E, Jansen PA, Jeschke JM, Keuling O, Palazón S, Plis K, Podgórski T, Rickowski F, Scandura M, Vicente J et al (2023) Feral American mink Neogale vison continues to expand its European range: time to harmonise population monitoring and coordinate control. Mamm Rev 53(3):158–176. https://doi.org/10.1111/mam.12315
Vahedi SM, Salek Ardestani S, Banabazi MH, Clark F (2023) Epidemiology, pathogenesis, and diagnosis of Aleutian disease caused by Aleutian mink disease virus: a literature review with a perspective of genomic breeding for disease control in American mink (Neogale vison). Vir Res. https://doi.org/10.1016/j.virusres.2023.199208
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4(3):535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Zaleska-Wawro M, Szczerba-Turek A, Szweda W, Siemionek J (2021) Seroprevalence and molecular epidemiology of Aleutian disease in various countries during 1972–2021: a review and meta-analysis. Animals 11(10):2975. https://doi.org/10.3390/ani11102975
Zalewski A, Michalska-Parda A, Bartoszewicz M, Kozakiewicz M, Brzeziński M (2010) Multiple introductions determine the genetic structure of an invasive species population: American mink Neovison vison in Poland. Biol Conserv 143(6):1355–1363. https://doi.org/10.1016/j.biocon.2010.03.009
Zalewski A, Michalska-Parda A, Ratkiewicz M, Kozakiewicz M, Bartoszewicz M, Brzeziński M (2011) High mitochondrial DNA diversity of an introduced alien carnivore: comparison of feral and ranch American mink Neovison vison in Poland. Divers Distrib 17(4):757–768. https://doi.org/10.1111/j.1472-4642.2011.00767.x
Zalewski A, Virtanen JME, Zalewska H, Sironen T, Kołodziej-Sobocińska M (2023) Asymptomatic viral infection is associated with lower host reproductive output in wild mink populations. Sci Rep. https://doi.org/10.1038/s41598-023-36581-8
Zhang L, Hua Y, Wei S (2021) High genetic diversity of an invasive alien species: comparison between fur-farmed and feral American mink (Neovison vison) in China. Animals 11(2):1–11. https://doi.org/10.3390/ani11020472
Acknowledgements
We would like to thank all the American mink collectors involved in this study: Associations Départementales des Piégeurs Agréés de Charente, de Dordogne, de Gironde et de Lot-et-Garonne; Associations Charente Nature et Cistude Nature; Association d’Insertion AI17; Conseils Généraux de Charente, de Dordogne, des Landes, des Pyrénées-Atlantiques et du Gers; CPIE des Pays de Seignanx et de Baïgorry; Conservatoires d’Espaces Naturels d’Aquitaine et de Poitou-Charentes; Eleveurs de Vison d’Amérique de Charente, Dordogne et des Pyrénées-Atlantiques; Fédération Aude Claire; Fédérations Départementales des Chasseurs de l’Aude, de Charente, de Dordogne, du Gers, de Gironde, des Landes, des Hautes-Pyrénées, et du Morbihan; Fédérations Départementales des Groupements de Défense contre les Organismes Nuisibles de Charente, de Dordogne, des Landes et de Lot-et-Garonne; GREGE; Ligue pour la Protection des Oiseaux; Lycée Agricole et Forestier de Bazas; MIFEN; Office National de la Chasse et de la Faune Sauvage (Services départementaux); Parc National des Pyrénées; Parcs Naturels Régionaux du Périgord-Limousin et des Landes de Gascogne; Piégeurs Agréés de l’Aude, des Landes et du Gers; Pisciculteurs de Corrèze, du Gers, de Gironde et des Landes; Réserves Naturelles de l’Etang Noir, de l’Etang de la Mazière, des Marais d’Orx, et du Courant d’Huchet; SEPANLOG; SFEP; Syndicat Intercommunal d’Aménagement Hydraulique du Son et de la Sonnette et de la Charente amont; Syndicat Intercommunal d’Aménagement Hydraulique de la Tude; Syndicat Mixte d'Etude et d'Aménagement du Pays des cantons de Ribérac-Verteillac-Montagrier; Syndicat Mixte d'Aménagement et de Gestion des Eaux du bassin de la Dronne.
Funding
This study was funded by DREAL Nouvelle-Aquitaine, GREGE, and the conservation Genetics laboratory of the University of Liège. Carcasses were collected for studies funded by the Conseil Regional d’Aquitaine, the Ministère de l’Ecologie et du Développement Durable, and The Union Européenne. Author P. van Leeuwen received financial support from NSERC CREATE grant, ReNewZoo. Johan Michaux benefited from FRS-FNRS grants (“directeur de recherches”). Lise-Marie Pigneur benefited from a FRS-FNRS grant (“chargée de recherches”).
Author information
Authors and Affiliations
Contributions
J.M., P.F. and C.F.C. conceived and planned the study. P.F, C.F.C. and E.I.L. carried out the sampling. L.M.P. contributed to sample processing. L.M.P., P.V.L. C.F.C. and J.M. contributed to the analysis and interpretation of the results. P.V.L. took the lead in writing the manuscript. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Additional information
Handling editor: Laura Iacolina.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Van Leeuwen, P., Pigneur, LM., Fournier-Chambrillon, C. et al. Population genetic structure of the invasive American mink (Mustela vison) in France: evidence of a high genetic diversity and the existence of multiple genetic lineages. Mamm Biol (2024). https://doi.org/10.1007/s42991-024-00435-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42991-024-00435-1