
Abstract
Urinary tract infections (UTIs) are a major public health concern in both community and hospital settings worldwide. Uropathogenic Escherichia coli (UPEC) is the main causative agent of UTI and increasingly associated with antibiotic resistance. Herein, we report the draft genome sequence of 9 fluoroquinolone-resistant UPEC isolates from Brazil and examine selected major phenotypic features, such as antimicrobial resistance profile, phylogroup, serotype, sequence type (ST), virulence genes, and resistance marks. Besides the quinolone resistance, beta-lactams, ESBL production, aminoglycosides, and tetracycline resistance were observed. High prevalence of 20 virulence genes was detected in all isolates, such as those encoding type 1 fimbriae, acid tolerance system, and hemolysin E, particularly within E. coli B2 phylogroup, as ST131 and ST1193 strains, among other genomic analyses as genomic islands, resistance plasmids, and integron identification.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Urinary tract infections (UTIs) are one of the most common bacterial infections in humans, affecting ~ 150 million individuals across the globe each year [1, 2]. It is estimated ~ 50% of women and 5% of men will develop a UTI in their lifetime, and UTI accounts for > 1 million hospitalizations and ~ $3.5 billion in medical expenses each year in the USA [3]. UTI usually starts as a bladder infection (cystitis) but can develop to acute kidney infection (pyelonephritis) and lead to severe sequelae such as sepsis [1,2,3].
Uropathogenic Escherichia coli (UPEC) are the primary cause of UTI, and responsible for ~ 75% of uncomplicated and ~ 65% of complicated infections [1,2,3]. UPEC are also associated with increasing antibiotic resistance, including last-line treatments [4]. Over the last 10 years, UPEC belonging to ST131 has emerged and disseminated globally [5]. Genomic epidemiology has described the phylogeny of ST131 and identified a dominant fluoroquinolone-resistant sub-lineage defined as clade C (or H30) [6,7,8]. Further analysis also revealed that ST131 strains containing the extended spectrum beta-lactamase (ESBL) blaCTX-M-15 allele comprised a subset of strains within this sub-lineage referred to as clade C2 (or H30-Rx). An increased incidence of resistance to fluoroquinolones has also been described for E. coli ST1193 [9] and ST410 [10].
In Brazil, ESBL-producing E. coli strains (including strains containing the blaCTX-M-15 allele) have been reported since 2007 [11]. Concern around the inappropriate use of cephalosporins and last-resort carbapenems led to a ban in the use of antimicrobials without a prescription in 2010 by the National Health Surveillance Agency (ANVISA). However, the incidence of infections in the community caused by ESBL-producing E. coli remains high, demonstrating the complexity of this problem [12,13,14,15].
This study reports the draft genome sequence and analysis of selected features of 9 fluoroquinolone resistant UPEC strains isolated from the urine of patients with community-acquired UTI in São Paulo State, Brazil.
Material and methods
Strains used in this study
E. coli isolates were obtained from UTI in outpatients at teaching hospital located in the city of Botucatu, Brazil. The whole collection has 77 UPEC isolates obtained in 2015, predominantly from female patients. Isolates were cultured from urine samples by plating onto MacConkey agar using sterile 1 μL calibrated wire loops. After aerobic incubation at 37 °C for approximately 24 h, a single bacterial colony was selected from cultures that contained > 105 CFU /mL. Bacterial identification was initially performed by VITEK® automated v2.0 system (bioMérieux). All isolates resistant to ciprofloxacin were selected to be detailed studied; randomly from this selection, nine strains were selected to be sequenced and further characterized.
Draft genome sequencing and assembly
The selected UPEC isolates were grown overnight aerobically at 37 °C on Luria–Bertani broth (LB), and cells were harvested for genomic DNA extraction using the GenElute™ Bacterial Genomic DNA kit (Merck KGaA, Darmstadt, Germany). Paired-end DNA libraries were prepared using the Nextera XT kit (Illumina, San Diego, CA, USA) in accordance with the manufacturer’s instructions and previously described [16]. Sequencing was performed on the Illumina NextSeq 500 (Australian Centre for Ecogenomics, University of Queensland), generating reads with 150 bp in length. Bacterial genomes were assembled through a multi-reference assisted de novo approach. Initially, raw reads served as input for A5 pipeline (August 2016 version) [17, 18], software specially designed for Illumina sequencing data. This pipeline started trimming the reads with Trimmomatic [19], which performed quality filtering removing Illumina adaptor sequences, low-quality bases (phred score quality < 28), and reads shorter than 35 bp. Trimming was followed by read error correction by SGA k-mer-based algorithm [20]. Contig assembly was performed using the IDBA-UD algorithm [21], which produced contigs that were scaffolded with SSPACE [22] using the error-corrected paired reads. After scaffold quality control checking for misassemblies (A5QC), SSPACE used the broken scaffolds, followed by a final round of scaffolding, to finish the A5 pipeline, set to –end = 5. Nonetheless, this pipeline did not achieve closed genomes or L50 scores of at least 3. Hence, reference-assisted scaffolding in iteration was performed using MeDuSa 1.6, set to default mode [23], which exploited information from a set of closely related genomes to guide the previously scaffolded contigs in the correct order and orientation. Based on a previous in silico Multi-Locus Sequence Typing (MLST 2.0) analysis [24] of the raw reads, seven complete reference genomes were chosen for the MeDuSa scaffolding guidance. They were six UPEC strains (536, CFT073, EC958, IAI39, UMN026, and UTI89) and the E. coli K-12 strain MG1655. After at least three rounds of iteration, a single scaffold representing the draft chromosome was obtained for each isolate. The final scaffolds were passed through Gapfiller 1.10 [25], which used the error-corrected reads to fill gaps between the contig sequences. Gapfiller was programmed to perform up to 20 iterations, which was enough to achieve an L50 contig count of 1 for eight of the assemblies and 2 for the other three assemblies. Library file used by Gapfiller was set with 800 as expected insert size between paired reads, 0.5 as minimum allowed error in insert size, read orientation as FR, and bwasw as aligner method (other settings were operated as -m 30 -o 2 -r 0.7 -n 10 -d 50 -t 10 -g 0 -i 20).
Assembled genomes in silico and their main features
The sequence type of each strain was determined in silico with MLST 2.0 [24]. In silico serotyping was performed using SerotypeFinder [26]. The 9 draft genomes were deposited in GenBank, as listed in Supplementary Table 1, and annotated by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) [27]. A BLAST Atlas was created with all the Brazilian UPEC strains described here plus the same representative E. coli strains used for the phylogeny analysis through GView [28]. To evaluate the evolution distance between the sequenced genomes and Escherichia coli representative strains, it was built a neighbor-joining phylogenetic tree with the ezTree algorithm and ggTree R package [29]. The Genbank file generated by PGAP was used as input for SeqWord Gene Island Sniffer (SWGIS) [30] for genomic island prediction, as the fasta files were the input for the software Integron Finder for identification of integrons and their associated resistance genes [31], and posteriorly, the whole genomes were submitted to INTEGRALL platform for search and recognition (http://integrall.bio.ua.pt/) [32]
Identification of virulence genes
Virulence genes were identified using the Virulence Finder software (https://bitbucket.org/genomicepidemiology/virulencefinder/src/master/) [33] and manual interrogation of the genomes using UPEC CFT073, UTI89, and EC958 as reference strains. Sequence comparisons were performed using the BLASTn tool.
Antimicrobial resistance profile
Antibiotic resistance genes were identified in silico using ResFinder [34]. Antimicrobial susceptibility testing was performed using the disk diffusion method according to Clinical and Laboratory Standards Institute (CLSI) [35] guidelines and using commercial disks obtained from Cefar, Brazil. The antimicrobial disks used were as follows: ampicillin (AMP, 10 μg), piperacillin/tazobactam (PPT, 100/10 μg), ampicillin/sulbactam (ASB, 10/10 μg), amoxicillin/clavulanic acid (AMC, 20/10 μg), cephalothin (CFL, 30 μg), cefuroxime (CRX, 30 μg), ceftriaxone (CRO, 30 μg), ceftazidime (CAZ, 30 μg), cefotaxime (CTX, 30 μg), cefepime (CPM, 30 μg), imipenem (IPM, 10 μg), ertapenem (ETP, 10 μg), meropenem (MER, 10 μg), gentamicin (GEN, 10 μg), amikacin (AMI, 30 μg), tobramycin (TOB, 10 μg), chloramphenicol (CLO, 30 μg), tetracycline (TET, 30 μg), trimethoprim/sulfamethoxazole (SUT, 1.25/23.75 μg), nitrofurantoin (NIT, 300 μg), and fosfomycin (FOS, 200 μg). Extended-spectrum β-lactamase (ESBL) production was investigated using ceftazidime (30 μg) and cefotaxime (30 μg) with and without clavulanic-acid (10 μg). An increase of 5 mm or more in the zone diameter observed in the presence of clavulanic acid was considered positive for ESBL production. The Quinolone Resistance Determinant Region (QRDR) mutations to GyrA, ParC, and ParE were also evaluated. The minimum inhibitory concentration (MIC) for ofloxacin and ciprofloxacin was also determined to all 9 selected strains, when necessary, the strains have had the MIC assessed to cephalothin and ceftazidime by Etest® gradient strips (bioMérieux, Marcy-l’Étoile, France) according to the manufactures’ recommendations. Briefly, the turbidity of the bacterial inoculum was adjusted using the 0.5 McFarland standard as a reference, and then, the Mueller–Hinton agar plates (Bio-Rad, Marnes-la-Coquette, France) were inoculated with a swab. Etest® strips were applied to the inoculated agar, and the plates incubated at 37° C for 24 h. MIC were defined as the lowest drug concentration at which the border of the elliptical inhibition intercepted the scale of the Etest® strip. In addition, the MIC for norfloxacin was also assessed by VITEK® 2 Compact (bioMérieux) protocol as indicated.
Results
Gene conservation through the E. coli species
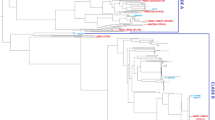
The E. coli MG1655 strain was used as reference against all the strains reported in this study and also the representative genomes to build a BLAST Atlas comparing the gene conservation between all these strains as if the MG1655 could be considered most close as a core genome (Fig. 1). There is a high conservation level of the MG1655 genes through the genomes herein reported, with similar gaps also presented on the others representatives genomes assessed (Fig. 1). The B2 phylogroup strains (Sup. Mat. Figure 1S ) share a gap region around 1450 kbp (Fig. 1).

BLAST Atlas of the 9 strains partial genomes sequenced and described herein and important E. coli representatives compared to the MG1655 strain
A range of different E. coli STs were associated with fluoroquinolone resistance
The complete collection of 77 E. coli isolates compromised 56 ciprofloxacin-resistant isolates (72%). From these, 9 isolates were randomly selected for genome sequencing, with the sequences deposited in GenBank (NCBI) as draft genomes under the accession numbers shown in the Sup. Mat. Table 1S, and their genome compared among them, other UPEC prototype strains and non-pathogenic E.coli MG1655 (Fig. 1). Among these 9 strains, we detected only one ST131 strain (BR43-DEC); two of the strains were ST2179, and one strain was from each of ST224, ST2509, ST1193, ST410, ST641, and ST617 (Table 1). Clinical details showed the age distribution was between 25 and 86 years old, with most strains isolated from female patients.
Phylogroup analysis
The 9 isolates were placed in the context of the E. coli phylogeny using a set of reference strains belonging to the different E. coli phylogroups sensu stricto (A, B1, B2, C, D, E, and F), as previously described [36]. Based on this analysis, two strains were from phylogroup B2 (BR14-DEC and BR43-DEC), one from phylogroup A (BR32-DEC), and the remainder were from phylogroup B1 (Sup. Mat. Figure 1S). In silico typing revealed the strains possessed a diversity of O antigen (O) and flagella (H) serotypes and many UPEC virulence factors (Table 1).
Virulence factors associated with UPEC
UPEC possess a range of 20 distinct virulence genes, these genes encoding diverse virulence factors were found at varying frequencies in the 9 UPEC strains, a finding consistent with their location in genomic islands frequently associated with pathogenesis, with detailed prediction and virulence features described (Table 1, Sup. Mat. Table 4S).
Antibiotic resistance profiles of clinical isolates and resistance genes
The full quinolone profile revealed strains were also non-susceptible to ofloxacin (OFX), nalidixic acid (NAL), norfloxacin (NOR), and levofloxacin (LVX); the only exception was BR02-DEC, which has presented an intermediate resistance only to norfloxacin (Table 1). Ciprofloxacin and ofloxacin MIC were also evaluated (Table 1). Most strains contained the GyrA, ParC, and ParE mutations, known to cause non-susceptibility to fluoroquinolones, with the exceptions to the ParE mutations in the BR02-DEC, BR07-DEC, and BR25-DEC, the same strains that have shown lower Ciprofloxacin MIC, respectively 6, 6, and 8 μg/mL (Table 1). Screening for sensitivity to other classes of antibiotics revealed a range of profiles (Table 1), with the following notable features: 11.1% (1/9) of isolates were non-susceptible to gentamicin (GEN10), 11.1% (1/9) to cefepime (CPM), 77.7% (7/9) to ampicillin (AMP), 55.5% (5/9) to sulfamethoxazole trimethoprim (SUT), 55.5% (5/9) to chloramphenicol (CLO30), and 66.6% (6/9) to tetracycline (TET 30). Furthermore, 11.1% (1/9) of isolates exhibited a profile consistent with extended spectrum beta-lactamase (ESBL) production (Table 1).
Overall, genome sequence analyses have identified the genome presence of the β-lactamase genes blaTEM-1A, blaTEM-1B, blaCTX-M-15, and blaCMY-2 as the sources of resistance to β-lactams (Table 1). Several strains also possessed genes encoding resistance to aminoglycosides (aadA1, aadA2, aadA5, strA, strB, aac3), chloramphenicol (catA1, cmlA1, floR), trimethoprim (dfrA8, dfrA12, dfrA17), sulfonamide (sul1,2,3), tetracycline (tetA, tetB, tetM) (Table 1).
Class 1 integrons were identified in 6 of the 9 isolates reported herein, all carrying antibiotic resistance genes (Sup. Mat. Table 2S). The INTEGRALL platform and integron finder classified them as three different types: In54 (BR02-DEC, BR07-DEC, and BR12-DEC), In640 (BR10-DEC and BR29-DEC), and In1002 (BR32-DEC). Interestingly, two among the three In54 were identified within two phylogenetic close-related isolates (BR02-DEC and BR07-DEC), similarly with both In640 (BR10-DEC and BR29-DEC) (Sup. Mat. Figure 1S and Table 2S).
Discussion
UPEC is a major bacterial pathogen associated with increasing antibiotic resistance. However, despite the global impact of UPEC infection, there is limited data on the genomic characterization of fluoroquinolone-resistant strains from Brazil. This work presents the draft genome sequence of 9 fluoroquinolone-resistant UPEC isolates from patients with community-acquired UTI in São Paulo State, Brazil, and describes their major virulence features and also resistance to different antibiotic classes.
The worldwide prevalence of fluoroquinolone-resistant UPEC is largely dominated by the ST131 clone, which is a major cause of UTI and bloodstream infections [37,38,39,40]. Other fluoroquinolone-resistant UPEC clones that have been documented in recent literature include ST1193 and ST410. Our analysis identified E. coli strains from diverse phylogroups and a range of STs that were resistant to ciprofloxacin, including well-characterized (ST131, ST1193, ST410) and less well-characterized clones (ST2179, ST2509, ST641, and ST617). Another recent and larger study from Brazil examined a collection of 324 UPEC strains from patients with community-acquired UTI, and showed that 61 strains (18.8%) were resistant to ciprofloxacin [41]. Strains from phylogroups A (42.6%) and B2 (29.5%) were most common among the ciprofloxacin-resistant isolates, with ST131 comprising 14.7% (9/61) of these strains. Taken together with our own findings, which provides detailed genomic analysis of 9/56 ciprofloxacin-resistant isolates (from a total collection of 77 UPEC isolates), it appears that there is a diversity of fluoroquinolone-resistant UPEC STs circulating in Brazil, highlighting the urgent need for more extensive epidemiological analyses together with genomic studies to understand this complex resistance profile. This is further highlighted by the fact that among the 9 isolates sequenced in this study, three were from ST2179 (n = 2) and ST224 (n = 1). The virulence genes were found in all strains, whereas they were more prevalent in B2 phylogroup members, more than 10 virulence genes; conversely, the A phylogroup strain has only shown 6 virulence genes. The B2 phylogroup includes the well-characterized ST131 and ST1193, both described as very concerning UPEC pathogens. Correlation between the serotype identification and phylogroups was not conclusive among these strains. Fluoroquinolone-resistant strains from both of these STs have previously been cultured from dairy buffalo [42], and it remains to be determined if isolates from these lineages are exchanged between animal and human reservoirs. The quinolone profile includes resistance to ciprofloxacin, ofloxacin, nalidixic acid, and levofloxacin, and is associated with previously described point mutations found in GyrA, ParC, and ParE, related to their considerable fitness with the QRDR mutations [43]. Interesting with these QRDR, the BR02-DEC, BR07-DEC, and BR25-DEC strain absence of ParE mutations may be directly linked to the lower ciprofloxacin MIC, where ParE mutations, particularly Ser458Ala, are previously associated to non-susceptibility and an increase in the MIC for fluoroquinolones in E. coli isolates [44]. Additional resistance to beta-lactams, ESBLs, aminoglycosides, macrolides, and tetracycline was observed. Most of these resistance phenotypes were associated with known acquired plasmidial and chromosomal genes (Table 1, Sup. Mat. Table 3S); nevertheless, other mechanisms including 16S RNA modifications and efflux pump over-expression may account for some resistance phenotypes.
The integron investigation within these strains’ genomes found antibiotic resistance genes in 6/9 strains. The integron Finder program was unable to identify the In1002 integron within BR32-DEC genome. Conversely, the structure of all In54 integrons was lesser complex in INTEGRALL platform identification, which compared to the one done through Integron Finder missed qacE∆1 and the sulfonamide resistance gene sul1, both vastly associated with class 1 integrons [45]. Furthermore, there was a disagreement between INTEGRALL and Integron Finder in In640 integrons quaternary ammonium compound efflux SMR transporter coding gene characterization, as INTEGRALL identified as qacH meanwhile Integron Finder did as qacL. To solve this conflict, a BLAST search with the aminoacid translated sequence has shown the qacL gene, which corroborates with the previously NCBI’s PGAP annotation deposited. Therefore, herein, we report an interesting class 1 integron carrying the qacL gene in both BR10-DEC and BR29-DEC strains (Sup. Mat. Table 2S), which is not commonly found in clinical strains, especially in Brazil, but it has been previously reported in Stenotrophomonas maltophilia [46].
UPEC possess a range of virulence genes that distinguish them from diarrhoeagenic and commensal E. coli [47, 48]. Pathogenic E. coli are generally differentiated into specific pathovars based on the presence of distinct virulence genes, which are linked to the capacity to colonize certain host sites and cause disease [47]. For example, UPEC frequently carry genes encoding multiple chaperone-usher fimbriae (e.g., P, F1C fimbriae, and S fimbriae), siderophore systems for scavenging iron (e.g., enterobactin, salmochelin, aerobactin, and yersiniabactin), toxins (e.g., hemolysin, cytotoxic necrotizing factor-1, and the serine proteases vacuolating autotransporter toxin and secreted autotransporter toxin), and surface polysaccharides comprising distinct O antigens, serine protease autotransporters Sat and Vat [47,48,49,50], all of which enhance colonization of the urinary tract [47].
The prevalence of the 20 virulence genes among the 9 strains detailed examined in this study was highly variable, with only 3 virulence genes found in all strains; the fimH gene encoding the tip adhesin of type 1 fimbriae, the gad genes encoding the acid tolerance system, and the hlyE encoding hemolysin E. Similarly, we observed extensive differences in the set of antibiotic resistance genes circulating in a background of fluoroquinolone resistance, including several different ESBL genes. Overall, the draft genomes generated and analyzed in this work highlight an emerging public health concern and stress the urgent need to understand better the occurrence of fluoroquinolone-resistant UPEC in Brazil and Latin America, and further to place this in the context of global UPEC resistance.
References
Stamm WE, Norrby SR (2001) Urinary tract infections: disease panorama and challenges. J Infect Dis 183(Suppl 1):S1-4. https://doi.org/10.1086/318850
Becknell B, Schober M, Korbel L, Spencer JD (2015) The diagnosis, evaluation and treatment of acute and recurrent pediatric urinary tract infections. Expert Rev Anti Infect Ther 13(1):81–90. https://doi.org/10.1586/14787210.2015.986097
Foxman B (2014) Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect Dis Clin North Am 28(1):1–13. https://doi.org/10.1016/j.idc.2013.09.003
Johnson JR, Tchesnokova V, Johnston B, Clabots C, Roberts PL, Billig M, Riddell K, Rogers P, Qin X, Butler-Wu S, Price LB, Aziz M, Nicolas-Chanoine MH, Debroy C, Robicsek A, Hansen G, Urban C, Platell J, Trott DJ, Zhanel G, Weissman SJ, Cookson BT, Fang FC, Limaye AP, Scholes D, Chattopadhyay S, Hooper DC, Sokurenko EV (2013) Abrupt emergence of a single dominant multidrug-resistant strain of Escherichia coli. J Infect Dis 207(6):919–28. https://doi.org/10.1093/infdis/jis933
Petty NK, Zakour NLB, Stanton-Cook M, Skippington E, Totsika M, Forde BM, Phan M-D, Moriel DG, Peters KM, Davies M, Rogers BA, Dougan G, Rodriguez-Baño J, Pascual A, Pitout JDD, Upton M, Paterson DL, Walsh TR, Schembri MA, Beatson SA (2014) Global dissemination of a multidrug resistant Escherichia coli clone. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1322678111
Zakour NLB, Alsheikh-Hussain AS, Ashcroft MM, Nhu NTK, Roberts LW, Stanton-Cook M, Schembri MA, Beatson SA, Hultgren SJ, Chakraborty T, Chen S (2016) Sequential acquisition of virulence and fluoroquinolone resistance has shaped the evolution of Escherichia coli ST131. mBio 7:e00347. https://doi.org/10.1128/mBio.00347-16
Price LB, Johnson JR, Aziz M, Clabots C, Johnston B, Tchesnokova V, Nordstrom L, Billig M, Chattopadhyay S, Stegger M, Andersen PS, Pearson T, Riddell K, Rogers P, Scholes D, Kahl B, Keim P, Sokurenko EV (2013) The epidemic of extended-spectrum-β-lactamase producing Escherichia coli ST131 is driven by a single highly pathogenic subclone, H30-Rx. mBio 4(6). https://doi.org/10.1128/mBio.00377-13
Stoesser N, Sheppard AE, Pankhurst L, Maio ND, Moore CE, Sebra R, Turner P, Anson LW, Kasarskis A, Batty EM, Kos V, Wilson DJ, Phetsouvanh R, Wyllie D, Sokurenko E, Manges AR, Johnson TJ, Price LB, Peto TEA, Johnson JR, Didelot X, Walker AS, Crook DW, (MMMIG) MMMIG, Rasko DA, Keim PS (2016) Evolutionary history of the global emergence of the Escherichia coli Epidemic Clone ST131. mBio 7:e02162. https://doi.org/10.1128/mBio.02162-15
Tchesnokova V, Rechkina E, Larson L, Ferrier K, Weaver JL, Schroeder DW, She R, Butler-Wu SM, Aguero-Rosenfeld ME, Zerr D, Fang FC, Ralston J, Riddell K, Scholes D, Weissman SJ, Parker K, Spellberg B, Johnson JR, Sokurenko EV (2019) Rapid and extensive expansion in the United States of a new multidrug-resistant Escherichia coli Clonal Group, Sequence Type 1193. Clin Infect Dis 68(2):334–337. https://doi.org/10.1093/cid/ciy525
Roer L, Overballe-Petersen S, Hansen F, Schønning K, Wang M, Røder BL, Hansen DS, Justesen US, Andersen LP, Fulgsang-Damgaard D, Hopkins KL, Woodford N, Falgenhauer L, Chakraborty T, Samuelsen Ø, Sjöström K, Johannesen TB, Ng K, Nielsen J, Ethelberg S, Stegger M, Hammerum AM, Hasman H, Castanheira M (2018) Escherichia coli Sequence Type 410 Is Causing New International High-Risk Clones. mSphere. https://doi.org/10.1128/mSphere.00337-18
Cergole-Novella MC, Guth BEC, Castanheira M, Carmo MS, Pignatari ACC (2010) First description of blaCTX-M-14- and blaCTX-M-15-producing Escherichia coli isolates in Brazil. Microb Drug Resist. https://doi.org/10.1089/mdr.2010.0008
Peirano G, Asensi MD, Pitondo-Silva A, Pitout JD (2011) Molecular characteristics of extended-spectrum beta-lactamase-producing Escherichia coli from Rio de Janeiro. Brazil Clin Microbiol Infect 17(7):1039–1043. https://doi.org/10.1111/j.1469-0691.2010.03440.x
Kcd Silva, Lincopan N (2012) Epidemiologia das betalactamases de espectro estendido no Brasil: impacto clínico e implicações para o agronegócio. J Bras Patol Med Lab 48(2):91–99. https://doi.org/10.1590/S1676-24442012000200004
Rogers BA, Sidjabat HE, Paterson DL (2011) Escherichia coli O25b-ST131: a pandemic, multiresistant, community-associated strain. J Antimicrob Chemother 66(1):1–14. https://doi.org/10.1093/jac/dkq415
Campos ACC, Andrade NL, Ferdous M, Chlebowicz MA, Santos CC, Correal JCD, Lo Ten Foe JR, Rosa ACP, Damasco PV, Friedrich AW, Rossen JWA (2018) Comprehensive molecular characterization of escherichia coli isolates from urine samples of hospitalized patients in Rio de Janeiro. Brazil Front Microbiol 9:243. https://doi.org/10.3389/fmicb.2018.00243
Harris PNA, Ben Zakour NL, Roberts LW, Wailan AM, Zowawi HM, Tambyah PA, Lye DC, Jureen R, Lee TH, Yin M, Izharuddin E, Looke D, Runnegar N, Rogers B, Bhally H, Crowe A, Schembri MA, Beatson SA, Paterson DL (2018) Whole genome analysis of cephalosporin-resistant Escherichia coli from bloodstream infections in Australia, New Zealand and Singapore: high prevalence of CMY-2 producers and ST131 carrying blaCTX-M-15 and blaCTX-M-27. J Antimicrob Chemother 73(3):634–642. https://doi.org/10.1093/jac/dkx466
Tritt AEJ, Facciotti MT, Darling AE (2012) An integrated pipeline for de novo assembly of microbial genomes. PLoS One 7(9):e42304. https://doi.org/10.1371/journal.pone.0042304
Coil D, Jospin G, Darling A (2015) A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31(4):587–589. https://doi.org/10.1093/bioinformatics/btu661
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Simpson JT, Durbin R (2012) Efficient de novo assembly of large genomes using compressed data structures. Genome Res. https://doi.org/10.1101/gr.126953.111
Peng Y, Leung HCM, Yiu SM, Chin FYL (2012) IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics (Oxford, England) 28(11):1420–8. https://doi.org/10.1093/bioinformatics/bts174
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics (Oxford, England) 27(4):578–9. https://doi.org/10.1093/bioinformatics/btq683
Bosi E, Donati B, Galardini M, Brunetti S, Sagot M-F, Lio P, Crescenzi P, Fani R, Fondi M (2015) MeDuSa: a multi-draft based scaffolder. Bioinformatics (Oxford, England) 31(15):2443–51. https://doi.org/10.1093/bioinformatics/btv171
Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Sicheritz-Pontén T, Ussery DW, Aarestrup FM, Lund O (2012). Multilocus sequence typing of total-genome-sequenced bacteria. https://doi.org/10.1128/JCM.06094-11
Nadalin F, Vezzi F, Policriti A (2012) GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13(14):1–16. https://doi.org/10.1186/1471-2105-13-S14-S8
Joensen KG, Tetzschner AMM, Iguchi A, Aarestrup FM, Scheutz F, Carroll KC (2015). Rapid and easy in silico serotyping of escherichia coli isolates by use of whole-genome sequencing data. https://doi.org/10.1128/jcm.00008-15
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44(14):6614–6624. https://doi.org/10.1093/nar/gkw569
Petkau A, Stuart-Edwards M, Stothard P, Van Domselaar G (2010) Interactive microbial genome visualization with GView. Bioinformatics 26(24):3125–3126. https://doi.org/10.1093/bioinformatics/btq588
Yu G, Lam TT, Zhu H, Guan Y (2018) Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol Biol Evol 35(12):3041–3043. https://doi.org/10.1093/molbev/msy194
Bezuidt O, Lima-Mendez G, Reva ON (2009) SeqWord gene island sniffer: a program to study the lateral genetic exchange among bacteria. Int J Comput Inform Eng 3(10):2399–2404. https://doi.org/10.5281/zenodo.1071430
Cury J, Jove T, Touchon M, Neron B, Rocha EP (2016) Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res 44(10):4539–4550. https://doi.org/10.1093/nar/gkw319
Moura A, Soares M, Pereira C, Leitao N, Henriques I, Correia A (2009) INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics 25(8):1096–1098. https://doi.org/10.1093/bioinformatics/btp105
Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, Aarestrup FM, Carroll KC (2014) Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol. https://doi.org/10.1128/JCM.03617-13
Zankari E, Allesøe R, Joensen KG, Cavaco LM, Lund O, Aarestrup FM (2017) PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J Antimicrob Chemotherapy 72(10):2764–2768. https://doi.org/10.1093/jac/dkx217
CLSI (2020) M100Ed30 | Performance Standards for Antimicrobial Susceptibility Testing, 30th Edition. Thirtieth edn. CLSI_LabNews, https://clsi.org/standards/products/microbiology/documents/m100/
Clermont O, Christenson JK, Denamur E, Gordon DM (2013) The Clermont Escherichia coli phylo-typing method revisited: improvement of specificity and detection of new phylo-groups. Environ Microbiol Rep 5(1):58–65. https://doi.org/10.1111/1758-2229.12019
Day MJ, Hopkins KL, Wareham DW, Toleman MA, Elviss N, Randall L, Teale C, Cleary P, Wiuff C, Doumith M, Ellington MJ, Woodford N, Livermore DM (2019) Extended-spectrum β-lactamase-producing Escherichia coli in human-derived and foodchain-derived samples from England, Wales, and Scotland: an epidemiological surveillance and typing study. Lancet Infect Dis 19(12):1325–1335. https://doi.org/10.1016/S1473-3099(19)30273-7
Ludden C, Decano AG, Jamrozy D, Pickard D, Morris D, Parkhill J, Peacock SJ, Cormican M, Downing T (2020) Genomic surveillance of Escherichia coli ST131 identifies local expansion and serial replacement of subclones. Microb Genom. https://doi.org/10.1099/mgen.0.000352
Pitout JDD, DeVinney R (2017) Escherichia coli ST131: a multidrug-resistant clone primed for global domination. F1000Research 6:195. https://doi.org/10.12688/f1000research.10609.1
Johnson JR, Porter S, Thuras P, Castanheira M (2017) Epidemic emergence in the United States of Escherichia coli sequence type 131-H30 (ST131-H30), 2000 to 2009. https://doi.org/10.1128/AAC.00732-17
Moreira da Silva RCR, de Oliveira Martins Junior P, Goncalves LF, de Paulo Martins V, de Melo ABF, Pitondo-Silva A, de Campos TA (2017) Ciprofloxacin resistance in uropathogenic Escherichia coli isolates causing community-acquired urinary infections in Brasília, Brazil. J Glob Antimicrob Resist 9:61–67. https://doi.org/10.1016/j.jgar.2017.01.009
Aizawa J, Neuwirt N, Barbato L, Neves PR, Leigue L, Padilha J, Pestana de Castro AF, Gregory L, Lincopan N (2014) Identification of fluoroquinolone-resistant extended-spectrum β-lactamase (CTX-M-8)-producing Escherichia coli ST224, ST2179 and ST2308 in buffalo (Bubalus bubalis). J Antimicrob Chemother 69(10):2866–2869. https://doi.org/10.1093/jac/dku218
Johnson JR, Johnston B, Kuskowski MA, Sokurenko EV, Tchesnokova V (2015) Intensity and mechanisms of fluoroquinolone resistance within the H30 and H30Rx subclones of escherichia coli sequence type 131 compared with other fluoroquinolone-resistant E. coli. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.00673-15
Sorlozano A, Gutierrez J, Jimenez A, de Dios LJ, Martinez JL (2007) Contribution of a new mutation in parE to quinolone resistance in extended-spectrum-beta-lactamase-producing Escherichia coli isolates. J Clin Microbiol 45(8):2740–2742. https://doi.org/10.1128/JCM.01093-07
Deng Y, Bao X, Ji L, Chen L, Liu J, Miao J, Chen D, Bian H, Li Y, Yu G (2015) Resistance integrons: class 1, 2 and 3 integrons. Ann Clin Microbiol Antimicrob 14:45. https://doi.org/10.1186/s12941-015-0100-6
Huang YW, Hu RM, Lin YT, Huang HH, Yang TC (2015) The contribution of class 1 integron to antimicrobial resistance in Stenotrophomonas maltophilia. Microb Drug Resist 21(1):90–96. https://doi.org/10.1089/mdr.2014.0072
Kaper JB, Nataro JP, Mobley HL (2004) Pathogenic Escherichia coli. Nat Rev Microbiol 2(2):123–140. https://doi.org/10.1038/nrmicro818
Mann R, Mediati DG, Duggin IG, Harry EJ, Bottomley AL (2017) Metabolic adaptations of uropathogenic e. coli in the urinary tract. Front Cell Infect Microbiol 7:241. https://doi.org/10.3389/fcimb.2017.00241
Johnson JR, Stell AL (2000) Extended virulence genotypes of Escherichia coli strains from patients with urosepsis in relation to phylogeny and host compromise. J Infect Dis 181(1):261–272. https://doi.org/10.1086/315217
Schreiber HL 4th, Conover MS, Chou WC, Hibbing ME, Manson AL, Dodson KW, Hannan TJ, Roberts PL, Stapleton AE, Hooton TM, Livny J, Earl AM, Hultgren SJ (2017) Bacterial virulence phenotypes of Escherichia coli and host susceptibility determine risk for urinary tract infections. Sci Transl Med 9(382):eaaf1283. https://doi.org/10.1126/scitranslmed.aaf1283
Acknowledgements
We thank Cefar for providing the antimicrobial drug disks used in this study.
Funding
This study is funded by the CAPES, CNPq, and FAPESP Project 2014/06779–2 and 2019/03049-7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Responsible Editor: ANA LUCIA DA COSTA DARINI
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
da Silva, P., Lustri, B.C., Castilho, I.G. et al. Genome profiling of fluoroquinolone-resistant uropathogenic Escherichia coli isolates from Brazil. Braz J Microbiol 52, 1067–1075 (2021). https://doi.org/10.1007/s42770-021-00513-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-021-00513-3