
Abstract
The development of an efficient transformation system is essential to enrich the genetic understanding of Trichoderma atroviride. To acquire an additional homologous selectable marker, uracil auxotrophic mutants were generated. First, the pyr4 gene encoding OMP decarboxylase was replaced by the hph marker gene, encoding a hygromycin phosphotransferase. Then, uracil auxotrophs were employed to determine that 5 mM uracil restores their growth and conidia production, and 1 mg ml−1 is the lethal dose of 5-fluoroorotic acid in T. atroviride. Subsequently, uracil auxotrophic strains, free of a drug-selectable marker, were selected by 5-fluoroorotic acid resistance. Two different deletions in pyr4 were mapped in four auxotrophs, encoding a protein with frameshifts at the 310 and 335 amino acids in their COOH-terminal. Six auxotrophs did not have changes in the pyr4 ORF even though a specific cassette to delete the pyr4 was used, suggesting that 5-FOA could have mutagenic activity. The Ura−1 strain was selected as a genetic background to knock out the MAPKK Pbs2, MAPK Tmk3, and the blue light receptors Blr1/Blr2, using a short version of pyr4 as a homologous marker. The ∆tmk3 and ∆pbs2 mutants selected with pyr4 or hph marker were phenotypically identical, highly sensitive to different stressors, and affected in photoconidiation. The ∆blr1 and ∆blr2 mutants were not responsive to light, and complementation of uracil biosynthesis did not interfere in the expression of blu1, grg2, phr1, and env1 genes upregulated by blue light. Overall, uracil metabolism can be used as a tool for genetic manipulation in T. atroviride.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ascomycota and Basidiomycota fungi are primarily the causative agents for diseases in an extensive variety of crops, but modern crop management can control them; however, a high risk of developing resistance to the fungicides exist [1, 2]. As an alternative, Trichoderma species are widely used as biofungicides and biofertilizers in agriculture; they improve crop quality and production, and the use of agrochemicals can be decreased [3, 4]. Nevertheless, the effects of fungi as biocontrol can be slow as compared with chemical control. For this reason, a deep understanding of Trichoderma species’ biology would improve their biotechnological application.
Trichoderma atroviride is a filamentous fungus antagonist of phytopathogenic fungi. Its mycoparasitic capacity is attributed to competition for nutrients, the production of cell wall degrading enzymes, and antibiosis [4, 5]. In addition, it is considered an excellent model to understand asexual reproduction, as well as light and injury responses [6,7,8]. Thus, it is necessary to develop effective strategies for gene studies in T. atroviride in order to expand its industrial and agricultural applications.
The use of antibiotics, including fungicides, for selection is high priced and may interfere with the regular function of the targeted gene [9, 10]. One way to avoid the use of antibiotics is to generate auxotrophic mutants as a genetic tool, in which the same affected gene is utilized as a selectable marker, and its manipulation is less expensive [9]. Several auxotrophic strains have been reported in Trichoderma species. The arg2 gene, which encodes the small subunit of carbamoyl phosphate synthetase, was isolated and used to complement an arginine auxotroph strain in Trichoderma virens [11]. In Trichoderma reesei, the argB gene of Aspergillus nidulans can restore the arginine synthesis [12]. Adenine auxotrophs of T. reesei lacking the ade2 gene, encoding a phosphoribosylaminoimidazole carboxylase required for production of purines, can be easily detected by selection of red colonies [13]. In T. reesei and Trichoderma hypoxylon, uracil auxotrophs selected by 5-fluoro-orotic acid (5-FOA) were used successfully as a genetic tool for the disruption of a single gene [14,15,16].
The pyrimidine analogue 5-FOA is generally used for positive selection of uracil auxotrophic mutants and is catalyzed by orotate phosphoribosyl transferase (OPRTase) and orotidine-5′-monophosphate decarboxylase (OMP decarboxylase) to obtain 5-fluorouridine 5′-monophosphate (5-FUMP) and 5-fluorouracil (5-FU) [17]. The 5-FU inhibits thymidylate synthase activity, consequently leading to thymine nucleotide depletion and affecting DNA synthesis [17,18,19]. Therefore, the toxic effect of 5-FOA on wild-type strains facilitates its selection, whereas mutants deficient in OPRTase or OMP decarboxylase, essential for uracil synthesis, can grow on 5-FOA medium supplemented with uridine/uracil [20, 21].
In order to obtain an alternative transformation system in Trichoderma atroviride, we generated uracil auxotrophic strains that are unable to grow in uracil-deficient media. Physiological and molecular results indicate that the pyr4 gene can be used as a selectable marker in a uracil auxotrophic background. Four different genes related to stress and light responses in T. atroviride were effectively replaced in a uracil auxotroph strain, and the mutants showed identical phenotypes in comparison with knockouts generated by resistance to hygromycin B.
Methods
Strains and culture conditions
In this study, T. atroviride IMI206040 was used as the wild-type strain (WT) and as the parent strain for gene replacement experiments. The ∆pbs2–7 and ∆tmk3–13 strains were previously reported [8]. All strains were propagated on potato dextrose agar (PDA; DIFCO) at 27 °C. Transformants were selected on minimal medium (MM) containing per liter: 20 g glucose, 0.2 g MgSO4.7H2O, 0.9 g K2HPO4, 0.2 g KCl, 1 g NH4NO3, 2 mg FeSO4.7H2O, 2 mg ZnSO4.7H2O, 2 mg MnCl2.7H2O, and 15 g agar, and pH was adjusted to 5.5 with KOH. MM was supplemented with uracil 5 mM according to Gruber et al. [14]. When necessary, 100 μg ml−1 hygromycin B (Invitrogen) or 5-FOA was added to the medium. Escherichia coli DH5α strain was propagated in a Luria-Bertani (LB) medium with appropriate antibiotics for cloning and plasmid DNA isolation purposes.
Replacement of pyr4 by the hph marker
To generate pyr4 mutants, its sequence was identified in the T. atroviride genome database (v2.0) of the Department of Energy Joint Genome Institute through a BLASTp search against the amino acid sequence of Trichoderma harzianum Pyr4 (AAA51865.1), which has been previously reported [22]. The pyr4 open-reading frame (ORF) was replaced by a hygromycin B resistance cassette based on the double-joint PCR method [23] with modified conditions as previously described [24]. Primers designed to replace the pyr4 ORF by the selectable marker are listed in Table 1. The 5′ and 3′ flanking regions of the pyr4 gene were amplified using the primers Ppyr4-F–PQpyr4-R and TQpyr4-F–Tpyr4-R, respectively. The hygromycin B resistance marker, hph, was amplified using the primers Hyg-F – Hyg-R. In a second PCR round, the three fragments were joined. The complementary sequences to hph were added to the chimeric primers (PQpyr4-R – TQpyr4-F). In a third PCR, the cassette was amplified using nested primers (N5pyr4-F – N3pyr4-R). The Platinum™ Taq DNA Polymerase High Fidelity (Invitrogen) was used to construct the cassette, and the PCR conditions were as follows: first step at 94 °C for 2 min, 35 cycles at 94 °C/15 s, 60 °C/15 s, 68 °C/1 min per kb, and a final extension at 68 °C for 5 min. The final amplicon was used to transform T. atroviride protoplasts.
Protoplasts isolation
Protoplasts of T. atroviride were prepared by modifying the method of Baek and Kenerley [11]. A conidial suspension of the WT strain (1 × 106 conidia ml−1) was cultivated in 100 ml of GYEC liquid medium (1.5% glucose, 0.3% yeast extract, 0.5% casein, and pH adjusted to 5.5 with KOH) and incubated in constant orbital agitation (160 rpm) at 27 °C for 18 h. Then, 0.2 g of mycelia were recovered by filtration and placed in a 50-ml centrifuge tube with a 7 ml of osmotic solution (50 mM CaCl2, 0.5 M mannitol, 50 mM MES, pH 5.5) and 0.1 g of lyophilized lysing enzymes from T. harzianum (Sigma-Aldrich). Mycelia were incubated in orbital agitation (120 rpm) at room temperature for 2 h for the formation of protoplasts. Protoplasts were harvested through sterile Miracloth and carefully transferred into 1.5-ml microcentrifuge tubes. After centrifuging at 8000 rpm for 10 min at 4 °C, the supernatant was discarded, and the protoplast pellet was resuspended in osmotic solution at a concentration of 1 × 107–8 ml−1.
Protoplast transformation by the polyethylene glycol (PEG)–CaCl2 method
Transformation was conducted with 250 μl protoplasts and 100 μl DNA (50 μl PCR product plus 50 μl osmotic solution), and mixture remained on ice for 20 min. Afterwards, 350 μl of PEG (40% PEG 6000 dissolved in osmotic solution) at 42 °C was added and incubated at room temperature for 30 min. To select transformants using a hph marker, transformation was mixed with 7–10-ml soft PDA (PDB, 0.7% bacteriological agar, 1 M sorbitol and 100 μg.ml−1 hygromycin B) and poured on selective PDA plates (PDA, 1 M sorbitol and 100 μg.ml−1 hygromycin B). To select transformants using the pyr4 marker, the transformation reaction was mixed with 7–10-ml soft MM (MM, 0.7% bacterial agar and 1 M sorbitol) and poured on MM plates (MM and 1 M sorbitol). Cultures were incubated at 27 °C for 4–5 days.
Assay to identify uracil auxotrophy
Transformants resistant to hygromycin were subjected to three rounds of single-spore isolation in MM plus 5 mM uracil and 100 μg ml−1 hygromycin B. To identify auxotrophic mutants, drops of 500 conidia (5 μl of a 1×105 conidia per ml suspension) of the WT and transformants were inoculated on MM plates with 0.5% Triton X-100, with or without 5 mM uracil, and incubated for 4 days at 27 °C. Transformants unable to grow in uracil-deficient medium were selected, and the pyr4 ORF replacement was confirmed by PCR using specific primers flanking the recombined sequences. Genomic DNA of the WT and mutant strains were prepared according to the procedure described by Raeder & Broda [25]. The DreamTaq DNA Polymerase (Thermo Scientific) was used for PCR reactions, and the conditions were as follows: first step at 95 °C for 3 min, 35 cycles at 95 °C/30 s, 60 °C/30 s, 72 °C/1 min per kb, and a final extension at 72 °C for 5 min.
Uracil assays to restore auxotrophy
The minimal amount of uracil required for the auxotrophic mutants to restore their growth was determined using MM with different concentrations of uracil. Drops of 500 conidia of the WT and mutant strains were inoculated on MM plates plus 0.5% Triton X-100 and with different uracil concentrations (0, 1, 2.5, 5, and 10 mM) for 4 days at 27 °C. To restore growth, mycelial plugs were taken from colonies grown on MM plates with 5 mM uracil (0.5 mm of diameter of the colony front) and inoculated on MM plates supplemented with uracil (0, 1, 2.5, 5, and 10 mM). Cultures were incubated at 27 °C during 72 h, and pictures were taken for radial growth measurements with ImageJ software (https://imagej.nih.gov/ij/). The experiment was conducted in triplicate.
5-FOA sensitivity assays
To establish the lethal dose of 5-FOA in T. atroviride, drops of 500 conidia of the WT and auxotrophic mutants were inoculated on MM plates with various concentrations of 5-FOA (0.5, 1, 2, 3, and 4 mg ml−1), plus 5 mM uracil and 0.5% Triton X-100, and incubated during 4 days at 27 °C. The experiment was conducted in triplicate.
Transformation based on a positive selection by use of 5-FOA
To obtain uracil auxotrophic strains free of an antibiotic-resistance marker, a co-transformation was carried out using a free-marker cassette created by joining the 5′ and 3′ flanking regions of the pyr4 gene of T. atroviride, 5-FOA for positive selection [15, 26], and the plasmid pCB1004 [24], which is autoreplicative in T. atroviride (Esquivel-Naranjo and Herrera-Estrella, under review). Primers listed in Table 1 were designed to amplify the 5′ (Ppyr4-F – Q5pyr4-R) and 3′ (Q3pyr4-F – Tpyr4-R) flanking regions of pyr4, excluding the complete ORF. In a second PCR, 5′ and 3′ regions were joined through 22 complementary nucleotides added on the 5′-end of the chimeric primers (Q5pyr4-R - Q3pyr4-F). In a third PCR, the cassette was amplified using nested primers N5pyr4-F – N3pyr4-R and purified by QIAGEN columns. The Platinum™ Taq DNA Polymerase High Fidelity was used to construct the free-marker cassette, and the PCR conditions were as mentioned above. T. atroviride protoplasts were co-transformed with the free-marker cassette and pCB1004 in a 2:1 M ratio. Transformants were selected on MM supplemented with 5 mM uracil, 0.5 mg ml−1 5-FOA (sublethal dose), and 100 μg ml−1 hygromycin B. Then, one round of single-spore isolation was performed in MM plus 5 mM uracil, 2 mg ml−1 5-FOA, and 100 μg ml−1 hygromycin B and the next two rounds in MM plus 5 mM uracil, 0.5 mg ml−1 5-FOA, and without hygromycin B. Auxotrophy tests were carried out as previously described. The pyr4 gene deletion was confirmed by PCR using primers flanking the coding region and sequencing. The DreamTaq DNA Polymerase was used for PCR reactions, and the conditions were as detailed above.
The pyr4 gene as a selectable marker for gene replacement
In order to explore of uracil auxotrophic mutants as a genetic tool, deletion cassettes to replace the blr1, blr2, tmk3, and pbs2 genes by the pyr4 gene were generated. The Ura−1 strain of T. atroviride was used as a parental strain. In a first PCR, the 5′ and 3′ flanking regions were amplified using primers for tmk3 (Ptmk3-F – pPQtmk3-R and pTQtmk3-F – Ttmk3-R), pbs2 (Ppbs2-F – pPQpbs2-R and pTQpbs2-F – Tpbs2-R), blr1 (Pblr1-F – pPQblr1-R and pTQblr1-F – Tblr1-R), and blr2 (Pblr2-F – pPQblr2-R and pTQblr2-F – Tblr2-R) genes, and the smPyr4-F - smPyr4-R primers were used to amplify 1247 bp of pyr4 gene. In a second PCR, 5′ and 3′ flanking regions of each gene were joined to pyr4 gene. In a third PCR, the cassettes were amplified using nested primers for tmk3 (N5tmk3-F – N3tmk3-R), pbs2 (N5pbs2-F – N3pbs2-R), blr1 (N5blr1-F – N3blr1-R), and blr2 (N5blr2-F – N3blr2-R) genes, and the transformation proceeded as described above. MM plates plus sorbitol 1 M were used as the selection medium. The gene replacement was determined by PCR with primer flanking the recombined 5′- and 3′-end sequences of tmk3, pbs2, blr1, and blr2 genes. The DreamTaq DNA Polymerase was used for PCR reactions, and the conditions were as detailed above.
Cellular stress assays
Drops of 500 conidia of the WT, ∆pbs2, and ∆tmk3 mutant strains were placed on PDA plates with 0.5% Triton X-100 plus KCl (0, 50 and 100 mM) for osmotic stress response; for heavy metal tolerance, cadmium (0, 400, and 800 mM CdCl2); for oxidative stress tolerance, menadione (0, 200, and 400 mM); or Congo red (0, 50, and 100 mM) to challenge cell wall integrity. To analyze osmotic stress tolerance in mycelia, precultures were generated on PDA plates with 2 μl of conidia of the WT, ∆pbs2, and ∆tmk3 strains and allowed to grow for 40 h at 27 °C in darkness. Mycelial plugs from precultures were inoculated on PDA with NaCl (0 and 0.2 M) for osmotic stress. The cultures were incubated at 27 °C during 4 days, and pictures were taken. The experiments were carried out in triplicate.
Analysis of conidiation induced by light and injure
Light response assays were performed as previously described [8]. Briefly, mycelial plugs from WT, ∆blr1, and ∆blr2 precultures strains were inoculated on PDA plates and incubated for 40 h at 27 °C in darkness. To analyze conidiation stimulated by light, plates were exposed to blue light for 5 min (152.4 μmol m−2) and incubated for 48 h at 27 °C in darkness. To analyze conidiation stimulated by injury, colonies grown on darkness for 40 h were cut with a sterile scalpel to induce conidiation and incubated 48 h at 27 °C in darkness. The experiments were carried out in triplicate.
Analysis of gene expression
Expression of blu1, grg2, env1, and phr1 genes was determined in the WT strain, ∆pbs2, ∆tmk3, ∆blr1, ∆blr2 mutants generated with the pyr4 marker, and ∆pbs2–7; ∆tmk3–13 mutants were selected with the hph marker. Total RNA from mycelia exposed to blue light for 30 min (914.4 μmol m−2) or in darkness was extracted using TRIzol. DNase I and RNase-free (Thermo Scientific) were used to eliminate genomic DNA. Then, complementary DNA was synthesized following the manufacturer’s recommendations (RevertAid Reverse Transcriptase; Thermo Scientific). To estimate transcript levels of genes regulated by light, 1 μl cDNA was used as template, and the DreamTaq DNA Polymerase was used for PCR reactions, following the conditions detailed above.
Statistical analysis
Statistical analysis and graphs were made with GraphPad Prism (version 5). The presented graph shows the average of three different experiments, and standard deviation is indicated. The data were analyzed through a one-way analysis of variance (ANOVA) with a Tukey-Kramer post-test.
Results
Functional analysis of the T. atroviride pyr4 gene encoding an OMP decarboxylase
The pyr4 gene identified in the T. atroviride genome encodes an OMP decarboxylase of 379 amino acids, with 91.6%, 67.3%, 56.0%, and 46.0% amino acids identities with OMP decarboxylases from T. reesei (ETS06629.1), Neurospora crassa (CAD21085.1), A. nidulans (AAB66359.1), and Saccharomyces cerevisiae (AAB64498.1), respectively. Trichoderma atroviride pyr4 does not contain introns similar to those of T. reesei and N. crassa pyr4 genes unlike pyrG of A. nidulans and Aspergillus niger which contains two introns [27], suggesting that also T. atroviride is more closely related to N. crassa than to the two Aspergillus species.
In order to generate uracil auxotrophic mutants of T. atroviride, a knockout cassette to replace the pyr4 gene by the hph selectable marker was constructed and used to transform T. atroviride protoplasts. Transformants were selected in MM plus 5 mM uracil and 100 μg ml−1 hygromycin B. Eight uracil auxotrophic mutants were identified and were able to grow only on MM supplemented with uracil (Fig. 1a). The gene replacement in auxotrophic strains was confirmed by PCR (Fig. S1). To evaluate the minimal amount of uracil required to supply the uracil starvation, the WT and mutant strains were cultured on MM with different uracil concentrations, and the growth rate was determined. The auxotrophic strains restored their growth with 5 mM uracil added to the MM (Fig. 1b), and conidiation was induced by light and injury at a similar level as occurred in the WT strain (Fig. S2).

Uracil auxotrophic strains lacking pyr4 gene and uracil concentrations to restore auxotrophy. a Auxotrophy test for mutant identification. Drops of 500 conidia of WT, ∆pyr4–1, − 2, − 3, − 4, − 5, − 6, − 7, and − 8 strains (from top-left to bottom-right) were inoculated on MM plates with or without 5 mM uracil and incubated at 27 °C for 4 days. b Recover of uracil auxotrophy. Comparison among the mycelial growth rates of WT, ∆pyr4–3, − 4, and − 5 strains. Mycelial plugs were inoculated on MM plates plus different uracil concentrations. After 72 h, colony diameter was measured. Lines on bars represent standard errors. All experiments were carried out in triplicate
5-FOA has commonly been used in fungi to select uracil auxotrophic strains without drug-resistance markers [20, 21, 27]. In order to establish a positive selection, a conidial suspension of uracil auxotrophs and parental strain was inoculated on MM plates with 0.5% Triton X-100, 5 mM uracil, and different concentrations of 5-FOA (0.5, 1, 2, 3, and 4 mg ml−1). All auxotrophic strains were resistant to 4 mg ml−1 5-FOA, whereas the WT strain showed a strong reduction of growth in 0.5 mg ml−1 of 5-FOA and was unable to grow at 1 mg ml−1 or even higher concentrations of 5-FOA (Fig. 2a). Interestingly, all auxotrophic strains were still able to produce conidia in 0.5 mg ml−1 5-FOA, but at higher concentrations, conidiation was severally inhibited, thus suggesting a relationship between conidiation and 5-FOA.

Lethal dose of 5-FOA. a Drops of 500 conidia of WT, ∆pyr4–1, − 2, − 3, − 4, − 5, − 6, − 7, and − 8 strains (from top-left to bottom-right) were inoculated on MM plates plus 5 mM uracil and the indicated 5-FOA concentrations. b Effect of uracil concentrations on the sensitivity to 5-FOA. Drops of 500 conidia of strains indicated in Fig. 2a were inoculated on MM plates plus 2 mg ml−1 5-FOA and different uracil concentrations. Plates were incubated at 27 °C for 4 days. All experiments were performed in triplicate
Metabolic pathways can be subject to negative feedback inhibition, in which the final product decreases enzymatic activity or gene expression. For the above-mentioned reason, we wanted to examine if the selected uracil concentration has an effect on sensitivity to 5-FOA. A conidial suspension was inoculated on MM plus 2 mg ml−1 5-FOA and different uracil concentrations. Parental and auxotrophic strains showed the same phenotype, without changes in 5-FOA toxicity (Fig. 2b). Altogether, 5 mM uracil is the optimal amount to restore uracil auxotrophy of T. atroviride properly, and 2 mg ml−1 5-FOA can be used for a positive selection approach to select uracil auxotrophic strains in this fungus.
Generation of uracil auxotrophy by positive selection
In fungi, uracil auxotrophy has been widely explored to use ura5/pyr4/pyrG or ura3/pyr2 genes as selectable marker [15, 26, 28,29,30]. In order to create uracil auxotrophic strains using positive selection with 5-FOA, a free-marker cassette was constructed to eliminate the T. atroviride pyr4 gene. Given the fact that the plasmid pCB1004 acts as auto-replicative in T. atroviride (Esquivel-Naranjo and Herrera-Estrella, under review), we co-transformed protoplasts with the free-marker cassette and pCB1004 (2:1). To assure the auxotrophic mutants selection, transformants were recovered on MM plus 0.5 mg ml−1 5-FOA, 5 mM uracil, and 100 μg ml−1 hygromycin B. Trichoderma atroviride protoplasts are multinucleated cells harboring transformed and non-transformed heterokaryotic nuclei. For this reason, we chose a 5-FOA sublethal concentration to select transformants. To obtain homokaryons, transformants were subjected to three rounds of single-spore isolation with modifications. Firstly, the selective medium comprised 5 mM uracil, 2 mg ml−1 5-FOA, and 100 μg ml−1 hygromycin B. Secondly, the colonies were grown on MM supplemented with 5 mM uracil and 0.5 mg ml−1 5-FOA to produce conidia. Ten stable mutants were identified by an auxotrophy test and designated Ura−1-10 (Fig. 3a). All were unable to grow on uracil-deficient MM and sensitive to hygromycin B, indicating that the pCB1004 plasmid was lost in all auxotrophic strains when this antibiotic was not added to the MM. The auxotrophic strains were then analyzed in their mycelium growth, production of conidia stimulated by light, and injury on PDA plates plus 5 mM uracil. All uracil auxotrophic mutants were stable and showed a similar behavior to the parental strain, suggesting that these strains can be used as a reliable receiver strain for genetic manipulation (data not presented). Unexpectedly, when we amplified the coding region of the pyr4 gene, the amplicons of six auxotrophic strains showed an electrophoretic mobility similar to that of the WT strain, whereas the amplicon of four auxotrophic strains (Ura−1, Ura−4, Ura−9, and Ura−10) was slightly shorter (Fig. S3). In order to identify mutations in the coding region of the pyr4 gene, it was amplified from WT and Ura−1, − 2, − 3, − 4, − 5, − 6, − 7, − 9, and − 10 strains and sequenced. As expected, the Ura−2, − 3, − 5, − 6, and − 7 strains did not have changes in the pyr4 nucleotide sequences and were identical to WT strain, whereas Ura−1, − 4, and − 10 had a deletion of 139 bp and Ura−9 deletion of 23 bp at the positions 928 bp and 997 bp, respectively (Fig. S4), considering the start codon. The deletion caused a frameshift, where the Ura−1, − 4, and − 10 strains encoded a polypeptide of 351 amino acids of which 309 were identical, and Ura−9 encoded 345 amino acids of which 334 were identical. Both of these were shorter than the Pyr4 protein (379 amino acids) of the WT strain (Fig. 3b–c). Given that, all produced auxotrophic strains showing at least two different mutations, and our results suggest that 5-FOA could be a mutagenic chemical or it could promote genomic instability, provoking mutagenesis. Furthermore, these data shows that the last 45 amino acids in the COOH-terminal of Pyr4 are essential for uracil biosynthesis.

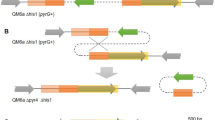
Analysis of auxotrophic mutants selected by resistance to 5-FOA. a Drops of 500 conidia of WT, Ura−1, − 2, − 3, − 4, − 5, − 7, − 8, and − 9 strains (from top-left to bottom-right) were inoculated on MM plates plus different uracil concentrations and incubated at 27 °C for 4 days. This assay was carried out in triplicate. b Schematic representation of pyr4 deletions. Deletions and positions were determined by alignment of the nucleotide sequence using the Clustal V method. c Protein alignment conducted among Pyr4 proteins from WT, Ura−1, and Ura−9 strains using the Clustal V method. Arrows indicate the end of the conserved amino acid sequence for the indicated strain
The Ura−1 and Ura−9 strains were selected for complementation tests due to their truncated pyr4 gene. The coding region of the WT pyr4 gene, including 1372-bp upstream and 1548-bp downstream, was amplified using the primers Ppyr4-F and Tpyr4-R (4060 bp), cloned into a pCR2.1-TOPO vector (Invitrogen) and used to complement uracil auxotrophic strains. In both strains, the pyr4 gene transformed the uracil auxotrophy to prototrophy (Fig. S5), indicating that the uracil auxotrophy was due to a truncation in the pyr4 gene, as it was demonstrated by pyr4 sequencing analysis.
Deletion of genes encoding the MAPK Tmk3 and MAPKK Pbs2 using pyr4 as selectable marker
To evaluate if the uracil auxotrophic mutants can be used as a genetic tool for gene replacement and to validate if pyr4 restores the uracil starvation, we designed cassettes to replace the tmk3 and pbs2 genes with the pyr4 gene. The tmk3 and pbs2 genes encode for MAPK Tmk3 and MAPKK Pbs2, respectively, related to light and stress responses [8]. These genes were selected to compare our results with those that have been previously reported in mutants selected by hph gene (Δtmk3–13:hph and Δpbs2–7:hph mutants). To avoid gene replacement at the pyr4 locus and favor the targeted knockouts, the Ura−1 strain was chosen because the truncation is bigger than in Ura−9. To construct the deletion cassettes, the pyr4 ORF was amplified with smPyr4-F and smPyr4-R primers to generate a selectable marker of 1247 bp (71-bp upstream—coding region—36-bp downstream). In this sense, the pyr4 marker was positioned under the control of the promoter and terminator of the deleted genes.
Transformation was carried out, and eight out of 24 transformants (33.3%) were mutants lacking the tmk3 gene (Δtmk3:pyr4 mutants), and three out of 20 (15%) were ∆pbs2:pyr4 mutants (Fig. S6–S7), all able to grow on MM without uracil. Conidiation stimulated by light in ∆tmk3:pyr4 and ∆pbs2:pyr4 strains was analyzed, and in all mutants, strains conidiation was reduced to 80%, similar to data obtained with ∆tmk3:hph and ∆pbs2:hph strains (Fig. 4a–b). As reported before [8], all mutants were sensitive to osmotic stress, Congo red, CdCl2, and menadione (Fig. 4c–d). There was no difference between mutants selected by hph or pyr4 markers, indicating that the Ura−1 strain was not affected neither in conidia production stimulated by light nor cellular stress responses.

Validation of pyr4 gene as a selectable marker in T. atroviride for gene deletion. a Comparison between WT and mutant strains selected with the hph marker (∆tmk3–13 and ∆pbs2–7) and pyr4 marker (∆tmk3–4 and ∆pbs2–5) grown on PDA plates in constant white light (0.586 μmol m−2 s−1) for 7 days at 27 °C. b Conidial production of the WT strain and mutants selected with the hph marker (∆tmk3–13 and ∆pbs2–7) and the pyr4 marker (∆pbs2–5, − 15, − 18, ∆tmk3–4, − 12, and − 16) grown on PDA plates in constant white light for 7 days at 27 °C. c Stress sensitivity in conidia of ∆tmk3 and ∆pbs2 strains. Drops of 500 conidia of ∆tmk3–4, − 12, − 16, − 13, WT, ∆pbs2–7, − 5, − 15, and − 18 strains (from top-left to bottom-right) were inoculated on PDA plates plus different concentrations of the indicated stressors. They were then, incubated in constant white light or darkness for 4 days at 27 °C. d Osmotic stress sensitivity in mycelia of ∆tmk3 and ∆pbs2 strains. Mycelial plugs of the indicated strains were inoculated on PDA plates with or without 200 mM NaCl and incubated for 4 days at 27 °C. All experiments were carried out in triplicate
Deletion of genes encoding the blue-light receptor Blr1/2 using the pyr4 marker
To acquire more evidence on the application of Ura−1 strain as a genetic background, cassettes were designed to replace the blr1 and blr2 coding regions by the pyr4 gene marker. Eight out of 20 transformants (40%) were blr1 mutants, and ten out of 18 transformants (55.5%) were blr2 mutants (Fig. S8–S9). The ∆blr1:pyr4 and ∆blr2:pyr4 mutants were exposed to a blue-light pulse (152.4 μmol m−2). As expected, the ∆blr1 and ∆blr2 strains were not photoresponsive, and the phenotype in light was the same as the control strain kept in darkness. Thus, mutant strains could not form a conidia ring in response to light on PDA medium (Fig. 5a–b), but they conidiated in response to injury as described before [31].

Phenotypes of T. atroviride WT, ∆blr1, and ∆blr2 strains. a Mycelial plugs of WT, ∆blr1– 14, − 17, − 18, − 19, and − 20 strains or b ∆blr2–5, − 9, − 10, − 15, and − 16 strains were inoculated on PDA plates and incubated for 40 h at 27 °C. Then, a blue-light pulse (152.4 μmol m−2) was applied for light-response assays or the mycelia were injured with a sterile scalpel for wound response. Pictures were taken 48 h later. Strains kept in darkness are shown as control. All experiments were performed in triplicate
Selection by pyr4 did not affect the expression of genes regulated by light
All mutants selected by the pyr4 marker showed the same phenotype in stress tolerance and photoconidiation, such as previously described using hph marker [8, 31]. However, gene expression epistasis has been associated with the use of auxotrophic strains in S. cerevisiae [32]. In order to discard changes in gene expression generated by the selectable marker, the expression of four light-regulated genes was examined in the ∆blr1:pyr4, ∆blr2:pyr4, ∆tmk3:hph, ∆tmk3:pyr4, ∆pbs2:hph, and ∆pbs2:pyr4 strains. As reported previously [8, 31, 33], blu1, grg2, phr1 (CPD-photolyase), and env1 (photoreceptor for blue-light tolerance) gene expression was not detected in darkness; however, after a blue-light pulse, their transcript levels were induced in the WT strain. In the ∆tmk3 and ∆pbs2 strains, expression of blu1 and grg2 genes was not induced, whereas phr1 and env1 genes were responsive to blue light at a similar level as the WT strain. Under the same conditions, the blue-light-regulated genes presented similar patterns in the ∆tmk3 and ∆pbs2 mutants obtained with pyr4 and hph markers (Fig. 6). In the ∆blr1 and ∆blr2 strains, blu1, grg2, phr1, and env1 genes were not induced by blue-light (Fig. 6), indicating a preponderant function of Blr1/2 blue-light receptors on light responses in T. atroviride. Altogether, these data provide evidence that the T. atroviride Ura−1 strain can be used as a background for genomic functional studies.

Expression of light-responsive genes in ∆pbs2, ∆tmk3, ∆blr1, and ∆blr2 strains. An RT-PCR was performed to compare the expression of blu1, grg2, phr1, and env1 genes in WT and mutants selected with the hph marker (∆pbs2–7 and ∆tmk3–13) and the pyr4 marker (∆pbs2–15, ∆tmk3–4, ∆blr1–14, and ∆blr2–15). The strains were kept in darkness for 48 h. Then, the strains were exposed to blue light for 30 min (914.4 μmol m−2). After the blue-light induction, total RNA was extracted with TRIzol. The gpd gene was used as control, and this experiment were carried out in duplicate obtaining practically the same results. Control WT: control without RT. D: darkness. L: light.
Discussion
The use of auxotrophic mutants as a genetic tool is an option to improve our knowledge of gene function in a low-cost way. Here we describe our transformation system in T. atroviride to obtain uracil auxotrophs free of drug-selectable markers, based on the 5-FOA positive selection. The pyrG/pyr4 gene in T. reesei, T. harzianum, and T. hypoxylon encodes orotidine-5′-monophosphate decarboxylase [15, 22, 27]. The absence of pyr4 in T. reesei can be restored by the pyrG gene of A. niger and the pyr4 gene of N. crassa [14], indicating that pyr4 is highly conserved in fungi. Furthermore, it is possible to use uracil auxotrophs in T. atroviride to establish homologous and heterologous transformation system.
To determine growth conditions, uracil auxotrophs were generated using resistance to hygromycin B. The results demonstrate that the knockout of pyr4 in T. atroviride leads to a uracil-deficient strain. Uracil in the medium restores growth of mutants, and the Δpyr4 strains were able to grow in media containing 5-FOA. This drug has already been used in Trichoderma species to select strains mutagenized through UV-light irradiation [14] and knockout [15, 26]. The 5-FOA lethal dose was determined to begin at 1 mg ml−1, and high uracil concentrations have no effect on 5-FOA toxicity. Together, data obtained were used to establish conditions for a markerless transformation in T. atroviride.
Techniques have been developed to eliminate the pyr4 gene without a drug-selectable marker in T. reesei and T. hypoxylon [15, 26]. In T. reesei, a genetic background lacking a non-homologous end-joining (NHEJ) repair system was used to efficiently eliminate the coding region of pyr4, but in T. hypoxylon, the WT strain was employed. Although a free-marker cassette was designed as previously described [15, 26], with the promoter and terminator fragments joined, excluding the ORF, our results showed that the mutant strains were generated by a different mechanism. Analysis of pyr4 deletion strains by PCR and sequencing revealed that the pyr4 ORF of four uracil auxotrophs (Ura−1, Ura−4, Ura−9, and Ura−10) was truncated by at least two independent events. Even though the pyr4 gene was not completely deleted, the produced alleles encoded a non-functional Pyr4 protein that was unable to produce uracil, which was demonstrated by auxotrophy, and were successfully complemented with the WT pyr4 gene. On the other hand, six auxotrophic strains showed bands with electrophoretic mobility comparable with WT pyr4 locus, suggesting that the mutations occurred outside pyr4 coding region as confirmed by sequencing in Ura−2, − 3, − 5, − 6, and − 7 auxotrophic strains. The auxotrophs without changes in pyr4 ORF could be explained by a mutation in a cis or trans element necessary for pyr4 expression. Another possibility that able to explain our results is that a mutation took place in another gene involved in pyrimidine biosynthesis, such as the ura3/pyr2 gene encoding OPRTase. These results suggest that 5-FOA may be mutagenic or a stress condition caused by genomic instability during transformation process. In this sense, 5-FU and FdUMP, toxic molecules produced by 5-FOA metabolism, can be incorporated into RNA or DNA matching 5-FU:A or 5-FU:G and induce genomic instability [18, 19]. Saccharomyces cerevisiae mutants lacking Apn1, the major abasic site endonuclease, showed a pronounced sensitivity to 5-FU, suggesting that abasic sites formed during DNA repair are more toxic, which can provoke DNA strand breaks [34]. However, 5-FOA has not been documented to cause DNA deletions. More data supporting this hypothesis comes from experiments in Candida albicans. When C. albicans was exposed to 5-FOA, its chromosomes suffered alteration [35, 36], presumably as an adaptive mutation under the stress conditions by 5-FOA that caused genetic changes. The above can explain the mutation that occurred in T. atroviride, as well as the partial loss of the pyr4 ORF by 5-FOA added in the culture medium.
Furthermore, we demonstrated that the Ura−1 strain can be used as background to knockout genes in T. atroviride. The blr1, blr2, pbs2, and tmk3 genes previously characterized [8, 31] were successfully replaced by pyr4 in the Ura−1 strain. The efficiency ranged from 15 to 55% among the genes included in our analysis. Noticeably, expression of blr1 and blr2 genes has been described to be undetectable by Northern-blot analysis, indicating that those genes are little expressed, whereas higher transcript levels were detected in tmk3 and pbs2 genes [31, 37]. Despite that the pyr4 gene was under control of the promoter of these deleted genes, in all cases uracil auxotrophy was successfully complemented. The conidial production and gene expression in mutants obtained from the auxotroph strain were evaluated, and the results are comparable with those of mutants obtained with the hph marker. Although the average efficiency of gene replacement was 36%, it was similar using the hph marker [8], suggesting that the knockout cassettes were targeted mainly to the corresponding locus.
In S. cerevisiae, auxotrophies alter the genetic background which induces different transcriptional responses [32]. Therefore, we evaluated the level of expression of four light-regulated genes in mutants obtained with a uracil auxotrophic strain, and the analyzed genes did not exhibited alterations in transcript levels. This suggests that the behavior of T. atroviride is different from that described in S. cerevisiae, although we cannot discard epistasis in other genes not included in this work. Our results show that the Ura−1 strain serves as a reliable genetic background, given that transcriptional perturbations were not detected under our experimental conditions.
In conclusion, the Ura−1 strain is an alternative tool to disrupt or complement genes in T. atroviride. Despite the fact that the uracil auxotroph was not obtained by knockout as initially designed, we did not observe differences in phenotype in comparison with mutants obtained with the hph marker. It would be interesting to know if a heterologous pyr4 gene from other fungi can improve the gene replacement efficiency in T. atroviride.
References
Braun H, Woitsch L, Hetzer B, Geisen R, Zange B, Schmidt-Heydt M (2018) Trichoderma harzianum: inhibition of mycotoxin producing fungi and toxin biosynthesis. Int J Food Microbiol 280:10–16. https://doi.org/10.1016/j.ijfoodmicro.2018.04.021
Doehlemann G, Ökmen B, Zhu W, Sharon A (2017) Plant pathogenic fungi. Microbiol Spectr:703–726. https://doi.org/10.1128/microbiolspec.FUNK-0023-2016
Lee S, Yap M, Behringer G, Hung R, Bennett JW (2016) Volatile organic compounds emitted by Trichoderma species mediate plant growth. Fungal Biol Biotechnol 3(1):7. https://doi.org/10.1186/s40694-016-0025-7
Guzmán-Guzmán P, Porras-Troncoso MD, Olmedo-Monfil V, Herrera-Estrella A (2019) Trichoderma species: versatile plant symbionts. Phytopathology 109(1):6–16. https://doi.org/10.1094/PHYTO-07-18-0218-RVW
Brunner K, Zeilinger S, Ciliento R, Woo SL, Lorito M, Kubicek CP, Mach RL (2005) Improvement of the fungal biocontrol agent Trichoderma atroviride to enhance both antagonism and induction of plant systemic disease resistance. Appl Environ Microbiol 71(7):3959–3965. https://doi.org/10.1128/AEM.71.7.3959-3965.2005
Steyaert JM, Weld RJ, Mendoza-Mendoza A, Stewart A (2010) Reproduction without sex: conidiation in the filamentous fungus Trichoderma. Microbiology 156(10):2887–2900. https://doi.org/10.1099/mic.0.041715-0
Medina-Castellanos E, Esquivel-Naranjo EU, Heil M, Herrera-Estrella A (2014) Extracellular ATP activates MAPK and ROS signaling during injury response in the fungus Trichoderma atroviride. Front Plant Sci 5. https://doi.org/10.3389/fpls.2014.00659
Esquivel-Naranjo EU, García-Esquivel M, Medina-Castellanos E, Correa-Pérez VA, Parra-Arriaga JL, Landeros-Jaime F, Cervantes-Chávez JA, Herrera-Estrella A (2016) A Trichoderma atroviride stress-activated MAPK pathway integrates stress and light signals. Mol Microbiol 100(5):860–876. https://doi.org/10.1111/mmi.13355
Pronk JT (2002) Auxotrophic yeast strains in fundamental and applied research. Appl Environ Microbiol 68(5):2095–2100. https://doi.org/10.1128/AEM.68.5.2095-2100.2002
Narasipura SD, Ren P, Dyavaiah M, Auger I, Chaturvedi V, Chaturvedi S (2006) An efficient method for homologous gene reconstitution in Cryptococcus gattii using URA5 auxotrophic marker. Mycopathologia 162(6):401–409. https://doi.org/10.1007/s11046-006-0076-z
Baek JM, Kenerley CM (1998) The arg2 gene of Trichoderma virens: cloning and development of a homologous transformation system. Fungal Genet Biol 23(1):34–44. https://doi.org/10.1006/fgbi.1997.1025
Penttilä M, Nevalainen H, Rättö M, Salminen E, Knowles J (1987) A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei. Gene 61(2):155–164. https://doi.org/10.1016/0378-1119(87)90110-7
Jørgensen M, Skovlund D, Johannesen P, Mortensen UH (2014) A novel platform for heterologous gene expression in Trichoderma reesei (Teleomorph Hypocrea jecorina). Microb Cell Factories 13(1):33. https://doi.org/10.1186/1475-2859-13-33
Gruber F, Visser J, Kubicek CP, de Graaff LH (1990) The development of a heterologous transformation system for the cellulolytic fungus Trichoderma reesei based on a pyrG-negative mutant strain. Curr Genet 18(1):71–76. https://doi.org/10.1007/BF00321118
Liu H, Wang G, Li W, Liu X, Li E, Yin WB (2018) A highly efficient genetic system for the identification of a harzianum B biosynthetic gene cluster in Trichoderma hypoxylon. Microbiology 164(5):769–778. https://doi.org/10.1099/mic.0.000649
Schuster A, Bruno KS, Collett JR, Baker SE, Seiboth B, Kubicek CP, Schmoll M (2012) A versatile toolkit for high throughput functional genomics with Trichoderma reesei. Biotechnol Biofuels 5(1):1. https://doi.org/10.1186/1754-6834-5-1
Atomi H, Imanaka T, Fukui T (2012) Overview of the genetic tools in the Archaea. Front Microbiol 3. https://doi.org/10.3389/fmicb.2012.00337
Matuo R, Sousa FG, Escargueil AE, Soares DG, Grivicich I, Saffi J, Larsen AK, Henriques JAP (2010) DNA repair pathways involved in repair of lesions induced by 5-fluorouracil and its active metabolite FdUMP. Biochem Pharmacol 79(2):147–153. https://doi.org/10.1016/j.bcp.2009.08.016
Wyatt MD, Wilson DM (2009) Participation of DNA repair in the response to 5-fluorouracil. Cell Mol Life Sci 66(5):788–799. https://doi.org/10.1007/s00018-008-8557-5
Boeke JD, La Croute F, Fink GR (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197(2):345–346. https://doi.org/10.1007/bf00330984
Razanamparany V, Bégueret J (1986) Positive screening and transformation of ura5 mutants in the fungus Podospora anserina: characterization of the transformants. Curr Genet 10(11):811–817. https://doi.org/10.1007/BF00418527
Heidenreich EJ, Kubicek CP (1994) Sequence of the pyr4 gene encoding orotidine-5′-phosphate decarboxylase from the biocontrol fungus Trichoderma harzianum. Gene 147(1):151–152. https://doi.org/10.1016/0378-1119(94)90057-4
Yu JH, Hamari Z, Han KH, Seo JA, Reyes-Domínguez Y, Scazzocchio C (2004) Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet Biol 41(11):973–981. https://doi.org/10.1016/j.fgb.2004.08.001
Castellanos F, Schmoll M, Martínez P, Tisch D, Kubicek CP, Herrera-Estrella A, Esquivel-Naranjo EU (2010) Crucial factors of the light perception machinery and their impact on growth and cellulase gene transcription in Trichoderma reesei. Fungal Genet Biol 47(5):468–476. https://doi.org/10.1016/j.fgb.2010.02.001
Raeder U, Broda P (1985) Rapid preparation of DNA from filamentous fungi. Lett Appl Microbiol 1(1):17–20. https://doi.org/10.1111/j.1472-765X.1985.tb01479.x
Derntl C, Kiesenhofer DP, Mach RL, Mach-Aigner AR (2015) Novel strategies for genomic manipulation of Trichoderma reesei with the purpose of strain engineering. Appl Environ Microbiol 81(18):6314–6323. https://doi.org/10.1128/AEM.01545-15
Smith JL, Bayliss FT, Ward M (1991) Sequence of the cloned pyr4 gene of Trichoderma reesei and its use as a homologous selectable marker for transformation. Curr Genet 19(1):27–33. https://doi.org/10.1007/bf00362084
Ballance DJ, Buxton FP, Turner G (1983) Transformation of Aspergillus nidulans by the orotidine-5′-phosphate decarboxylase gene of Neurospora crassa. Biochem Biophys Res Commun 112(1):284–289. https://doi.org/10.1016/0006-291x(83)91828-4
van Hartingsveldt W, Mattern IE, van Zeijl CM, Pouwels PH, van den Hondel CA (1987) Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet 206(1):71–75. https://doi.org/10.1007/bf00326538
Bergès T, Barreau C (1991) Isolation of uridine auxotrophs from Trichoderma reesei and efficient transformation with the cloned ura3 and ura5 genes. Curr Genet 19(5):359–365. https://doi.org/10.1007/bf00309596
Casas-Flores S, Rios-Momberg M, Bibbins M, Ponce-Noyola P, Herrera-Estrella A (2004) BLR-1 and BLR-2, key regulatory elements of photoconidiation and mycelial growth in Trichoderma atroviride. Microbiology 150(11):3561–3569. https://doi.org/10.1099/mic.0.27346-0
Alam MT, Zelezniak A, Mülleder M, Shliaha P, Schwarz R, Capuano F, Vowinckel J, Radmaneshfar E, Krüger A, Calvani E, Michel S, Börno S, Christen S, Patil KR, Timmermann B, Lilley KS, Ralser M (2016) The metabolic background is a global player in Saccharomyces gene expression epistasis. Nat Microbiol 1(3):15030. https://doi.org/10.1038/nmicrobiol.2015.30
Rosales-Saavedra T, Esquivel-Naranjo EU, Casas-Flores S, Martinez-Hernandez P, Ibarra-Laclette E et al (2006) Novel light-regulated genes in Trichoderma atroviride: a dissection by cDNA microarrays. Microbiology 152(11):3305–3317. https://doi.org/10.1099/mic.0.29000-0
Seiple L, Jaruga P, Dizdaroglu M, Stivers JT (2006) Linking uracil base excision repair and 5-fluorouracil toxicity in yeast. Nucleic Acids Res 34(1):140–151. https://doi.org/10.1093/nar/gkj430
Wellington M, Rustchenko E (2005) 5-Fluoro-orotic acid induces chromosome alterations in Candida albicans. Yeast 22(1):57–70. https://doi.org/10.1002/yea.1191
Wellington M, Kabir MA, Rustchenko E (2006) 5-fluoro-orotic acid induces chromosome alterations in genetically manipulated strains of Candida albicans. Mycologia 98(3):393–398. https://doi.org/10.3852/mycologia.98.3.393
Esquivel-Naranjo EU, Herrera-Estrella A (2007) Enhanced responsiveness and sensitivity to blue light by blr-2 overexpression in Trichoderma atroviride. Microbiology 153(11):3909–3902. https://doi.org/10.1099/mic.0.2007/007302-0
Funding
This work was supported by CONACYT (Consejo Nacional de Ciencia y Tecnología, CB-2011-169,045) and FOFI-UAQ-2012 (Fondo para el fortalecimiento de la investigación—Universidad Autónoma de Querétaro, FNB-2012-10) grants to E.U. E-N. G. C-H is indebted to CONACYT for a doctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Responsible Editor: Derlene Attili Agellis.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 5456 kb).
Rights and permissions
About this article

Cite this article
Calcáneo-Hernández, G., Rojas-Espinosa, E., Landeros-Jaime, F. et al. An efficient transformation system for Trichoderma atroviride using the pyr4 gene as a selectable marker. Braz J Microbiol 51, 1631–1643 (2020). https://doi.org/10.1007/s42770-020-00329-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-020-00329-7