
Abstract
Clostridium chauvoei is the etiological agent of blackleg, an infectious disease affecting cattle and small ruminants worldwide. This disease can manifest as classical blackleg, a condition in which skeletal muscles are affected and visceral blackleg, which affects the heart, sublingual muscles, and the diaphragm. The pathogenesis of the visceral form of the disease is poorly understood. The objective of this study is to determine and analyze complete genomic sequences of six C. chauvoei strains, five isolates from skeletal muscle and one isolate from a visceral case of blackleg in Brazil, to provide insights into the differences in pathogenic profiles of strains causing the different forms of disease. The full genomes of the six C. chauvoei strains were sequenced and comparative analyses were performed among these genomes and the C. chauvoei reference strain JF4335. The results of this study revealed that the genomes of the C. chauvoei strains analyzed are highly conserved; no particular differences were noted that could be associated with the two different clinical manifestations of the disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Clostridium chauvoei is a Gram-positive, anaerobic, spore-forming rod found in the soil, feces, and the digestive tract of many animals [1]. Transmission of blackleg occurs through the ingestion of C. chauvoei spores from contaminated soil. The ingested spores, or those produced after germinative cycles in the gut, are transported from the intestine or lesions in the oral cavity to muscles and tissues by macrophages across Peyer’s patches [2, 3]. Once localized in the tissues, the spores remain dormant until specific conditions such as anaerobiosis lead to their germination, multiplication, and the consequent production of exotoxins [3].
Antibiotic sensitivity testing indicates susceptibility of C. chauvoei to penicillin, tylosin, metronidazole, florfenicol, and vancomycin [4, 5]. There are two clinical manifestations of blackleg in cattle, the classical and visceral forms, and both forms are usually fatal and treatment with antibiotics is discouraging [3, 6, 7]. In most cases of classical blackleg, acute neutrophil necrotizing myositis that affects skeletal muscle is observed. Visceral myonecrosis, which is rarely diagnosed, affects the heart, the sublingual muscles, and the diaphragm [6, 7]. The pathogenesis of cardiac lesions has not been successfully explained [8].
The relatively small, 2.8 million base-pair (bp) genome of C. chauvoei, is indicative of its adaptation to a restricted host range—cattle, sheep, and goats—where C. chauvoei is able to replicate and cause disease [9]. A recent study revealed a highly conserved genome of C. chauvoei in twenty strains isolated from around the world over more than half a century [10].
Whole genome sequencing is a powerful tool that greatly contributes to our understanding of bacterial intra-species diversity [11, 12]. To date, full genome sequence data are available only for 22 C. chauvoei strains including the type strain ATCC 10092 and the thoroughly studied classical blackleg-causing strain JF4335 (isolated from classical form blackleg-affected cattle in Switzerland), which serves as a reference strain for genomic comparisons [10, 13]. No genomic data are available, however, for strains that cause the visceral form of blackleg.
Considering the importance of the disease and in order to get insights into the molecular genetic profile of C. chauvoei, the complete genome sequences of six Brazilian C. chauvoei isolates were analyzed. The study particularly focuses on a C. chauvoei strain isolated from a case of visceral blackleg with unusual levels of cardiac involvement with the aim to understand the mechanisms that lead to the different clinical manifestations of the disease.
Materials and methods
Bacterial strains and necropsy of visceral case
Six C. chauvoei strains, five of skeletal and one of visceral origin, isolated from cases of blackleg in Brazil between 2002 and 2015 were used in this study. The strain of visceral origin was isolated from fragments of heart obtained during necropsy of a visceral case of blackleg. Samples were fixed in 10% neutral-buffered formalin and routinely processed for histopathological examination with hematoxylin and eosin (H&E). All strains were stocked in Reinforced Clostridial Medium (Oxoid™). Prior to genome sequencing, the isolates were confirmed as belonging to the species C. chauvoei by polymerase chain reaction (PCR) amplification of the fliC gene locus according to the protocol proposed by Sasaki [14] and by 16S rRNA gene sequencing.
Whole genome sequence assembly, annotation, and consensus comparison
The full genomes of the six C. chauvoei strains were sequenced by Illumina® technology HiSeq 2500/3000 (150 bp paired-end reads) by GATC-Biotech (Konstanz, Germany) according to the manufacturer’s protocols. Genome assembly was performed using the algorithms of the Geneious® software package (Biomatters Ltd. New Zealand). Annotation of the whole genomes was performed using the Geneious® annotation pipeline and Prokka [15].
Comparative analyses were performed among the six C. chauvoei genomes and the C. chauvoei reference strain JF4335 (accession number NZ_LT799839.1). The assembled genomes were then examined for similarities to known sequences through BLASTx software using Blast2GO. Comparisons between genes and proteins were performed with the BLASTn and BLASTp programs (https://blast.ncbi.nlm.nih.gov/Blast.cgi). To obtain an overview of the genomes of the six C. chauvoei strains, the consensus sequences were extracted from the BAM files using SAMTools and visualized using the BLAST Ring Image Generator (BRIG) [16].
To analyze small differences among the genomes, the read files were evaluated for their quality by FastQC software (version 0.11.4 [http://www.bioinformatics.babraham.ac.uk/projects/fastqc]) after single-nucleotide polymorphism (SNP) extraction using the CFSAN SNP Pipeline with the JF4335 genome as reference [17]. The resulting SNP alignment file was used to generate a phylogenetic tree utilizing the SNAPP package from Beast 2 [18]. The phylogeny was visualized by FigTree v.1.4.3 and was rooted by Midpoint (http://tree.bio.ed.ac.uk/software/figtree/). SNP distance matrices were also obtained and used to quantify differences among the strains.
Minimal inhibitory concentration testing
Antibiotic susceptibility of the strains was determined using supplemented Brucella Broth using Sensititre™ ANO2B 96-well broth microdilution plates for anaerobic organisms (Trek Diagnostics Systems Ltd., England). The concentration ranges for antibiotics used in the minimal inhibitory concentration (MIC) testing are as follows: ampicillin/sulbactam 0.5/0.25–16/8 μg/ml, amoxicillin/clavulanic acid 0.5/0.25–16/8 μg/ml, cefotetan 4–64 μg/ml, penicillin 0.06–4 μg/ml, imipenem 0.12–8 μg/ml, meropenem 0.5–8 μg/ml, clindamycin 0.25–8 μg/ml, cefoxitin 1–32 μg/ml, metronidazole 0.5–16 μg/ml, chloramphenicol 2–64 μg/ml, ampicillin 0.5–16 μg/ml, piperacillin 4–128 μg/ml, tetracycline 0.25–8 μg/ml, mezlocillin 4–128 μg/ml, and piperacillin/tazobactam 0.25/4–128/4 μg/ml. The MIC testing and interpretation of the results were performed according to the manufacturer’s instructions. Clostridium septicum ATCC 8065 was used as the quality control strain.
Results
Gross necropsy and histological findings
Necropsy of the visceral case of blackleg revealed a small amount of blood covering the pericardial sac. These hemorrhagic areas were found to extend through the pleura of the adjacent pulmonary lobes and throughout the cranial mediastinum. On the epicardial surface, there were multiple areas of extensive focal hemorrhages (Fig. 1). In some areas of the myocardium, especially at the level of the papillary muscle, slight crepitation was noted after fixing in 10% formaldehyde, an evidence of cavitation due to the accumulation of gas. Bacteriological analysis of fragments of the heart revealed the presence of strong hemolytic bacteria identified as C. chauvoei. Likewise, causal C. chauvoei strains were isolated from affected skeletal muscles of five cases of classical blackleg. These six isolates were used for genomic sequencing, as previously described.

Opening of the thoracic cavity demonstrating hemorrhage on the epicardial surface extending through the pleura of the adjacent pulmonary lobes
Genome assembly and annotation
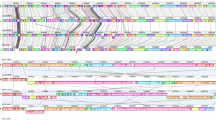
Genome assembly and circularization summary for the six complete genomes are represented in Table 1. The length of the circular genomes was found to range from 2,882,958 to 2,887,475 bp. Genome coverage varied from 102X to 569X. The consensus sequences of six C. chauvoei strains from Brazilian blackleg cases aligned against the reference sequence of C. chauvoei JF4335 [19] was illustrated in Supplementary Figure 1.
All six strains were found to possess a 3941 bp cryptic plasmid. The sequenced cryptic plasmids have a G+C content of 27%, without any displayed variability between strains. Sequence analysis revealed that the cryptic plasmids from the strains JF5835, JF5837, JF5839, JF5840, and JF5842 contains four open reading frames (ORFs), while JF5834 cryptic plasmid has three ORFs. All plasmids have an ORF encoded to plasmid stabilization protein, and the other ORFs encoded to hypothetical proteins.
Comparative analysis of the genomes indicated a high degree of identity. The differences were carefully scrutinized and genes in these regions were noted; differences were observed in the presence or absence of an insertion sequence (IS). Classical Clostridium virulence factors, mainly exoenzymes and toxins, were screened by BLASTp to search factors exclusive for the visceral blackleg strain. However, the exclusive ORFs identified were hypothetical proteins, pseudo genes, or transposase genes, which, currently, have no association with virulence or pathogenesis profile. Between one to eight, such exclusive coding sequences were found in the genomes of these six strains when compared with the JF4335 reference genome that contains 2624 predicted coding sequences [10] (Table 1). Genome sequence comparisons between the C. chauvoei strain of visceral origin (JF5840) and strains isolated from skeletal muscle sources revealed no particular differences in the genes encoding known proteins, as well as in the gene sequences, so that there is an entire gene identity among the genes. Hence, it can be concluded that differences in the pathogenicity profile of C. chauvoei strains causing classical and visceral blackleg cannot be accounted for by differences in genome sequences.
To analyze the small differences among these strains, the SNPs were identified using the C. chauvoei JF4335 genome as a reference sequence. This resulted in an alignment containing 893 variable sites among the genomes of the six strains and that of the reference; 158 (17.7%) of these are located in the non-coding regions. The least difference was noted at 72 variable sites between strains JF4335 and JF5839; the greatest difference was noted at 210 variable sites between strains JF5837 and JF5839. At least 65 SNPs were detected between the C. chauvoei strain of visceral origin, JF5840, and strain JF5842; and 190 SNPs were detected between JF5840 and JF5837, strains JF5842 and JF5837 being isolates from the infected skeletal muscle (Supplementary Table 1). The visceral strain showed 37 unique SNPs, 30 of these in coding region with 27 non synonymous mutations, two of them generated truncated proteins at positions 820,486 nc (hypothetical protein) and 2,821,039 nc (Rrf2, family transcriptional regulator).
The phylogenetic tree constructed based on SNPs from all six C. chauvoei strains and 18 C. chauvoei genomes available (access numbers SRR5429430-SRR542934, SRR542936-SRR542947, and JF4335) used in this study and that from the reference strain shows the degree of separation of each strain. As shown in Fig. 2, the visceral strain does not take up a specific place in the phylogeny.

Phylogenetic analysis based on SNPs among the six Clostridium chauvoei genomes analyzed in this study and the reference strain JF4335 (NZ_LT799839)
Minimal inhibitory concentration
The minimal inhibitory concentrations (MIC) of drugs tested for their ability to inhibit C. chauvoei strains are shown in Table 2. Testing with metronidazole revealed an MIC50 of 4 μg/ml and an MIC90 of 8 μg/ml, whereas that with penicillin revealed an inhibition of three out of the six strains at all drug concentrations tested. All strains were found to be sensitive to every other antimicrobial tested in this study.
Discussion
The pathogenesis of the classical form of blackleg in cattle is relatively well understood. In contrast, the pathogenesis of visceral lesions, particularly in unusual cases where only cardiac lesions are observed, has not yet been satisfactorily explained [8]. We have performed genomic analysis of six Brazilian C. chauvoei strains, five isolated from cases of classical blackleg and one isolated from an uncommon manifestation of visceral blackleg in the myocardium, in order to determine if genetic differences among C. chauvoei strains could account for the manifestation of visceral blackleg as opposed to the classical form.
The six strains analyzed are genetically very similar (Supplementary Figure 1). Comparative analysis did not reveal specific virulence determinants that could explain the differences of pathogenicity between the visceral and classical C. chauvoei strains. In effect, our observations with these six strains are in agreement with the findings of Richener et al. [10] that indicate a high level of homogeneity and conservation over a long period of time among C. chauvoei strains, which is in contrast to the genomes of other species of Clostridium such as C. difficile that display higher rates of mutation and consequently greater genetic variability [20].
The total genomes of the Brazilian C. chauvoei strains share greater identity with the reference JF4335 strain from Switzerland [19] than with the German C. chauvoei strain 12S0467 described by Thomas et al. [13]. The cryptic plasmid from Brazilian strains, however, showed 100% identity with the 12S0467 plasmid [13] and 95% identity with the plasmid found in JF4335 [10]. Cryptic plasmids generally do not have genes that contribute to the phenotype of the host cell. They usually have genes to self-replicate; however, no genes of interest to the host bacteria are identified. The plasmids sequenced in the six Brazilian C. chauvoei strains contain four ORFs that are unrelated to virulence genes or other known biological functions, as also seen with the other C. chauvoei cryptic plasmids described [9, 10, 13].
In contrast to the high overall identity among the genomes, differences were observed in the presence or absence of an insertion sequence (IS). An IS element from the IS256 family was observed in the genomes of four strains, but absent from the genomes of strains JF5842 and JF5840, which were isolated from nearby cities (Table 3). This IS has 1239 bp of length encode a poorly characterized transposase [21] and is present in all other C. chauvoei genome available. However, as no information about in transposase is available, we could not infer any correlation with its presence or absence. Interestingly, C. chauvoei JF5840 is the visceral blackleg isolate from the animal with clinical signs in the myocardium. Comparison of the IS element deletion in JF5842 with its presence in previously isolated strains is indicative that this IS element is still active and able to transposition [10]. Therefore, if active, this transposable element could serve as crossover points for homologous recombination events, which could be relevant to the pathogenicity profile of C. chauvoei in general.
The SNP analyses of the complete genomes presented in this study showed that the isolates have high identity among them with relatively few variable sites, showing that these isolates diverged very slowly, but enough to allow a phylogenetic differentiation among the strains (Fig. 2). The presence of only a few differences among the skeletal and visceral strains indicates that they are very similar irrespective of their origin; it is not possible to affirm how these unique differences contribute in the form of the disease [10].
To perform a more robust analysis, we have included 18 C. chauvoei genomes to the phylogeny with the SNPs identified (data nor shown). However, all six strains studied here are clusterized to the same clade. For a better understanding of the small differences among the visceral and skeletal strains, the phylogeny contains only the sequences of this study (Fig. 2).
We performed MIC testing to establish the antimicrobial susceptibility profile of the C. chauvoei strains. Strain JF5840 showed strikingly high MIC values 8 μg/ml for metronidazole and 0.12 μg/ml for penicillin. This susceptibility profile could, however, not be explained at the genomic level. The generally observed susceptibility of C. chauvoei strains to antibiotics could be reflective of the fact that blackleg is not typically treated with antibiotics owing to the acute course of the disease. For the same reasons stated above, it is difficult to compare these results with the MICs of other members of the Clostridium genus.
Overall, the total genome sequencing data were not indicative of genetic traits that could distinguish the visceral strain JF5840 from the other C. chauvoei strains. Our study reveals that the genomes of the Brazilian C. chauvoei strains are highly conserved, as are strains of C. chauvoei from other parts of the world.
References
Quinn PJ, Markey BK, Leonard FC, Fitz Patrick ES, Fanning S, Hartigan PJ (2011) Veterinary microbiology and microbial disease. Vol Second, Ames
Jubb KVF, Kennedy PC, Palmer N (1991) Pathology of domestic animals, vol 2, 4th edn, San Diego
Uzal F (2012) a. Evidence-based medicine concerning efficacy of vaccination against Clostridium chauvoei infection in cattle. Vet Clin N Am Food Anim Pract 28(1):71–77, viii. https://doi.org/10.1016/j.cvfa.2011.12.006
Salvarani FM, Silva ROS, Pires PS et al (2012) Antimicrobial susceptibility of Clostridium perfringens isolated from piglets with or without diarrhea in Brazil. Braz J Microbiol 43(3):1030–1033. https://doi.org/10.1590/S1517-83822012000300027
Silva ROS, D’Elia ML, Tostes Teixeira ÉP et al (2014) Clostridium difficile and Clostridium perfringens from wild carnivore species in Brazil. Anaerobe 28:207–211. https://doi.org/10.1016/j.anaerobe.2014.06.012
De Assis RA, Lobato FCF, Nascimento RAP et al (2010) Mionecroses clostridiais bovinas. Arq Inst Biol (Sao Paulo) 77(2):331–334
Casagrande RA, Sonne L, Neves MS et al (2015) Histopathological, immunohistochemical and biomolecular diagnosis of myocarditis due to Clostridium chauvoei in a bovine. Cienc Rural. https://doi.org/10.1590/0103-8478cr20141447
Abreu CC, Edwards EE, Edwards JF et al (2017) Blackleg in cattle: A case report of fetal infection and a literature review. J Vet Diagn Investig 29(5):612–621. https://doi.org/10.1177/1040638717713796
Frey J, Falquet L (2015) Patho-genetics of Clostridium chauvoei. Res Microbiol 166(4):384–392. https://doi.org/10.1016/j.resmic.2014.10.013
Rychener L, In-Albon S, Djordjevic SP et al (2017) Clostridium chauvoei, an evolutionary dead-end pathogen. Front Microbiol 8(June):1–13. https://doi.org/10.3389/fmicb.2017.01054
Binnewies TT, Motro Y, Hallin PF et al (2006) Ten years of bacterial genome sequencing: comparative-genomics-based discoveries. Funct Integr Genomics 6(3):165–185. https://doi.org/10.1007/s10142-006-0027-2
Tettelin H, Riley D, Cattuto C, Medini D (2008) Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol 11(5):472–477. https://doi.org/10.1016/j.mib.2008.09.006
Thomas P, Semmler T, Eichhorn I et al (2017) First report of two complete Clostridium chauvoei genome sequences and detailed in silico genome analysis. Infect Genet Evol 54(March):287–298. https://doi.org/10.1016/j.meegid.2017.07.018
Sasaki Y, Kojima A, Aoki H (2002) Phylogenetic analysis and PCR detection of Clostridium chauvoei, Clostridium haemolyticum, Clostridium novyi types A and B, and Clostridium septicum based on the flagellin gene. Vet Microbiol 86:257–267
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069. https://doi.org/10.1093/bioinformatics/btu153
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12. https://doi.org/10.1186/1471-2164-12-402
Davis S, Pettengill JB, Luo Y et al (2015) CFSAN SNP Pipeline: an automated method for constructing SNP matrices from next-generation sequence data. PeerJ Comput Sci 1:e20. https://doi.org/10.7717/peerj-cs.20
Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, Roychoudhury A (2012) Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis research article. 29(1998):1917–1932. https://doi.org/10.1093/molbev/mss086
Falquet L, Calderon-Copete SP, Frey J (2013) Draft genome sequence of the virulent Clostridium chauvoei reference strain JF4335. Genome Announc 1(4):1. https://doi.org/10.1093/nar/gkq
Didelot X, Eyre DW, Cule M, Ip CL, Ansari MA, Griffiths D, Vaughan A, O'Connor L, Golubchik T, Batty EM, Piazza P, Wilson DJ, Bowden R, Donnelly PJ, Dingle KE, Wilcox M, Walker AS, Crook DW, Peto TE, Harding RM (2012) Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol 13(12):R118. https://doi.org/10.1186/gb-2012-13-12-r118
Hennig S, Ziebuhr W (2010) Characterization of the transposase encoded by IS256, the prototype of a major family of bacterial insertion sequence elements. 192(16):4153–4163. https://doi.org/10.1128/JB.00226-10
Acknowledgments
We are grateful to Iveraldo Dutra for kindly providing isolates from São Paulo outbreaks of blackleg and Gustavo Sambrano for his technical support.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brasil (CAPES). The authors are thankful to the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS), and to the Research Foundation 3R (Reduction, Refinement and Replacement of Animal Experimentation) grant no. 136-13 to JF.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Responsible Editor: Agnes Figueiredo.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Fig. 1
Consensus sequences of six Brazilian strains aligned against the reference sequence JF4335 (Reference number: NZ_LT799839) using BLAST Ring Image Generator. (JPG 585 kb)
Supplementary Table 1
(DOCX 13 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Ziech, R.E., Siqueira, F.M., Cibulski, S. et al. Genomic comparison of Clostridium chauvoei isolates from classical and visceral clinical manifestation. Braz J Microbiol 51, 1327–1332 (2020). https://doi.org/10.1007/s42770-019-00177-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-019-00177-0