
Abstract
In this paper, the effect of poly (ethylene oxide) (PEO) as an additive on the structure and properties of poly (m-phenylene dimethylene terephthalamide) (PMIA) fibers obtained by wet spinning was investigated. The tensile strength of the composite fibers was substantially enhanced compared to the pure PMIA fiber. This was due to the fact that the addition of PEO weakens the hydrogen bonding between PMIA molecular chains resulting in an improved orientation of the composite fibers. It was found that the optimum PEO addition was 2% and the tensile strength of the composite fiber was 4.74 cN/dtex, which was 76% higher compared to the pure PMIA fiber. However, the heat resistance and flame retardancy of the composite fibers were basically unchanged compared to the pure PMIA fiber. The modification method is simple, with low raw material cost and good stability, and has not only good academic value but also excellent industrial value.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
High-performance fiber is a new generation of synthetic fiber with high strength, high modulus and high temperature resistance. High-performance fibers have special properties that ordinary fibers do not have, and are mainly used in national defense and military industries [1]. Aromatic polyamide fibers are high temperature resistant fibers with is a kind of early and fast development, wide application, and large production, fast development of high temperature resistant fiber varieties, their total output ranked the second in the high-performance fibers. They are widely used in the fields of safety and protection, modern industry and environmental protection because of its good heat resistance, flame retardancy, chemical stability, electrical insulation and corrosion resistance [2,3,4,5,6,7,8,9,10,11,12]. Aromatic polyamide fibers mainly consist of poly(p-phenylene terephthalamide) (PPTA) fibers and poly(m-phenylene terephthalamide) (PMIA) fibers [13].
However, the molecular chains of PMIA are interconnected by benzene ring and amide bond by meta-position, thus the PMIA is a polymer with flexible chain structure [1]. Moreover, the C=O bonds and N–H bonds between PMIA molecular chains can form strong hydrogen bonding [14], unlike the weak force between ultra-high molecular weight polyethylene (UHMWPE) molecular chains, which makes PMIA molecular chains unable to be super-stretched and achieve highly oriented structure. Thus the mechanical properties of PMIA fibers are poor and the service life of their woven textiles is short, which limits their application in many fields. The mechanical properties of fibers are closely related to their orientation, and the fracture strength can generally be improved by increasing the orientation of fibers. Li et al. [2] used 2,4-diamino-benzenesulfonic acid (2,4-DABSA) as the third monomer to copolymerize with m-phenylenediamine and m-phthaloyl chloride in low-temperature solution, and the results showed that the introduction of 2,4-DABSA enhanced the hydrogen bonding between the molecular chains of the modified fibers and improved the fracture strength of the modified fibers. However, the cost of copolymerization modification is high, the operation is difficult, and the modification effect is average. Many other researchers have used inorganic nanoparticles to modify PMIA, such as graphene, carbon nanotubes, boron nitride, etc. Jiang et al. [15] prepared composite hollow fiber membranes by combining molybdenum disulfide oxide nanosheets and PMIA. The carbon nano tube and molybdenum disulfide nanosheets could effectively enhance the PMIA hollow fiber membranes. However, most inorganic nanoparticles are difficult to be well dispersed in the matrix [16, 17], which makes the effect of using inorganic nanoparticle modification method not very effective. Wang et al. [18] coated the PMIA surface with dopamine and then coated the fiber surface with a layer of silver, and the results showed that the conductivity of modified PMIA fibers was greatly improved. However, the mechanical properties of the fibers were hardly improved. The substances used for surface modification are generally easy to fall off, which makes the effect of surface modification short-lived. A common modification method that is simple, easy to operate and can be produced industrially is blend modification [8, 19, 20]. Many researchers strengthened the fibers by weakening the hydrogen bonds between molecular chains. Chen et al. [21] prepared composite fibers by adding Antarctic krill protein to chitosan solution during wet spinning process. The results showed that the Antarctic krill protein weakened the hydrogen bonding between the chitosan molecular chains, which led to the improvement of the fiber orientation and thus the mechanical properties. Lei et al. [22] stretched the chitosan hydrogel in water to make its molecular chains arranged regularly. Therefore, this research enhanced PMIA fibers by adding a certain amount functional components to PMIA solution through blend modification method. Poly (ethylene oxide) (PEO) is a very crystalline substance that is used as a solid electrolyte and forms hydrogen bonds with other substances easily [23]. Liu et al. [4] prepared PMIA/PEO solid electrolytes and found that PMIA could form good hydrogen bonding with PEO.
The strong hydrogen bonding between PMIA molecular chains makes it difficult to achieve a high degree of orientation. The drawability of PMIA fibers may be enhanced by adding substances that weaken the hydrogen bonding between PMIA molecular chains. Thus PEO can be added to PMIA as a functional component to improve the orientation of the fiber and thus the mechanical properties of the fiber.
In the present study, PEO was added to PMIA solution and prepared PEO/PMIA composite fibers by wet spinning. The structure and properties of the composite fibers were characterized in details.
Experiments
Materials
PMIA solution with a solid content of 18.5% was supported by X-Fiper New Material Co., Ltd. PEO powder with different weight average molecular weight (Mw) of 8000, 600,000, 4,000,000, and 5,000,000 were purchased from BASF Co., Ltd. N, N-Dimethylacetamide (DMAc) was purchased from Shanghai Lingfeng Chemical Reagent Co., Ltd.
Preparation of PEO/PMIA Blend Solution
The PMIA solution with a PMIA content of 18.5% was diluted with DMAc into PMIA concentration of 15% in a three-neck flask, then a corresponding mass of PEO was added into the solution and stirred at 70 °C for 8 h. The PEO additions were 0, 1%, 1.5%, 2%, 2.5% relative to PMIA, and such blend solutions and the prepared fibers were named PMIA, 1% PEO/PMIA, 1.5% PEO/PMIA, 2% PEO/PMIA and 2.5% PEO/PMIA, respectively.
Preparation of PEO/PMIA Composite Fibers via Wet-Spinning
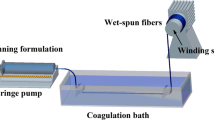
PEO/PMIA composite fibers were prepared by a laboratory-made wet spinning device. The PEO/PMIA/DMAc solution was extruded from a spinneret with 50 spinnerets of 0.08 mm diameter at 40 °C and subsequently entered into a Coagulation water bath containing 50% DMAc and solidified into fibers under the draw ratio of 0.5. Then the fibers entered into a water bath and stretched with different ratios under 80 °C, finally washed in a water bath at 60 °C. The collected fibers were soaked in a 60 °C water bath for 24 h with several water changes to remove residual solvent. After dried, the fibers were stretched 1.1 times in a heat pipe at 300 °C. The technological diagram of fibers preparation is shown in Fig. 1.

Technological diagram of preparation of PEO/PMIA composite fibers
Characterization
The steady-state and dynamic rheological behavior of the solutions were characterized by means of a rotational rheometer (Physica MCR 301, Anton Paar). The diameter of the plate used was 25 mm, the distance between two flat plates was 1 mm, the shear rate range for steady-state rheology was from 0.1 to 100 s−1, and the temperatures chosen were 40 °C, 50 °C, and 60 °C. The viscoelastic range was determined by dynamic tensile scans using a frequency of 6.3 rad/s. The strain 10% was in the linear viscoelastic range and the dynamic rheological strain was 10% with a frequency scan range of 100–0.1 rad/s.
Fourier transform infrared spectra (FTIR) of the composite fibers were characterized by Nicolet Nexus 670 spectrometer at wavenumber of 600–4000 cm − 1 with an attenuated total reflection (ATR) accessory.
The cross-section and surface morphology of the fibers were measured by a scanning electron microscope (SEM) (SU8010, Hitachi, Japan). Stick the broken fiber after mechanical performance test on conductive glue and spray gold to observe its morphology.
The thermal properties of the composite fibers were tested by a thermogravimetric analysis (TG209F1, Netzsch, Germany). The measurements were carried out under high purity nitrogen atmosphere with a temperature range from 50 to 800 ºC at a heating rate of 20 ºC/min.
Fiber tensile tester (XQ-1, China) was used to test the mechanical properties of fibers. The tensile rate chosen for the test was 30 mm/min, the clamping distance was 20 mm. Each sample was measured 30 times and the averaged value was got.
According to GB-T5454 use the limit oxygen index tester to test the limit oxygen index of the fiber (JF-3, China). Take 1 m fiber separately. Fix one end of the fiber, pinch the other end by hand, twist the fiber with a twist number of 60, then fold it into a strand along the middle, twist the strand 30 times again by the same method, fold it into a strand, then twist it 10 times again, then fold it, and tie the head of the strand to prevent the strand from falling apart, the strand was the test sample.
The thermodynamic mechanical behavior of fibers was recorded on the dynamic mechanical analysis (DMA) system (Q800, America) at temperatures ranging from 30 to 370 °C. The frequency was 1.0 Hz and the heating rate was 4 ºC/min.
The SCY-3 type sound velocity orientation meter was used to measure the sonic orientation of fiber (fs). The experiment was repeated 5 times to get the average value. fiber orientation factor was calculated according to Eq. (1).
where C and Cu are the sound velocity of fiber sample and Polypropylene amorphous fibers, respectively.
X-ray diffraction (XRD) patterns were recorded on an X-ray diffractometer (D2 Phaser), by using Cu Ka radiation at 40 kV and 30 mA with a diffraction angle ranged from 5 to 60º.
Wide-angle X-ray diffraction (WAXD) patterns of the composite fibers were collected using this XRD machine with an imaging plate. The Herman orientation factor (fc) is acquired by Eq. (2). The orientation function of fibers is usually gained using Eq. (3).
where, (cosφ)2 is orientation function, I is diffraction peak intensity, φ is azimuth angle.
Fiber thermal shrinkage test process was as follows: take 30 cm long fiber, place them in high temperature oven under loose state for 30 min, and accurately measure the fiber length (L, cm), the temperature used were 280, 300 and 320 ºC, respectively (Fig. 2). The fiber thermal shrinkage (s) was calculated according to the Eq. (4).

Photos of 2% PEO solutions (a, b) and 2% PEO/PMIA blend solutions (c, d) under different temperatures (PMIA concentration = 15%, PEO Mw = 600,000)
Results and Discussion
Figure 3 shows the shear rheology curves of different blend solutions at different temperatures. From Fig. 3a, it could be seen that the viscosity of the blend solution tends to increase with the addition of PEO, which was because the solid content of the whole solution increases with the addition of PEO, and the higher molecular weight of the added PEO resulted the viscosity of the whole solution increase. With the increase of PEO addition in the blend solution, the phenomenon of shear thinning of the blend solution with the increase of shear rate was more obvious. This was because the addition of PEO weakens the hydrogen bonding between the molecular chains of PMIA, which resulted in the decrease in the entanglement of the molecular chains in the solution, and then reduced the intermolecular forces in the whole solution, thus the solution viscosity decrease faster as the shear rate increases [24]. From Fig. 3b and c, it could be seen that the viscosity of both pure PMIA solution and 2% PEO/PMIA solution tended to decrease as the temperature increases, which was due to the increase in the molecular chain movement ability with the increase of temperature, thus the whole molecular chain interaction became weaker and solution viscosity decreased. However, the viscosity of the 2% PEO/PMIA blend solution decreased more obviously with the increase of temperature. Because the addition of PEO weakened the intermolecular force between the PMIA molecular chains, which made it easier to destroy the intermolecular force with the increase of temperature.

Rheological properties of a PEO/PMIA blend solutions with different contents of PEO at 40 ºC, b PMIA solution at different temperatures, c 2% PEO/PMIA blend solution at different temperatures (PMIA concentration = 15%, PEO Mw = 600,000)
Figure 4 shows the dynamic rheological results of the blend solutions with different amounts of PEO. From Fig. 4a, and b, it could be seen that the storage modulus and the loss modulus of the blend solution increased as the amount of PEO in the blend solution increased. This was because the addition of PEO increased the solid content of the whole solution. The energy storage modulus and loss modulus of the solution were closely related to its concentration, and since the concentration of the whole solution increased after the addition of PEO, the energy storage modulus and loss modulus both increased. From Fig. 4c, it could be seen that the loss factor of the blend solution decreased, and the more the loss factor decreased with the increase of PEO addition. The loss factor of the solution was closely related to its structure, and the loss factor can reflect the ratio of solution viscosity and elasticity. Because the addition of PEO weakened the hydrogen bonding between the molecular chains of PMIA, the intermolecular forces in the solution reduced and the entanglement between the molecular chains reduced, which resulted the viscosity of the whole solution decrease and the elasticity ratio increase.

Angular frequency dependence of shear storage modulus and loss modulus and loss factor Tanθ at 40 ºC of PEO/PMIA blend solutions with different contents of PEO (PMIA concentration = 15%, PEO Mw = 600,000)
Figure 5 shows the FTIR spectra of the composite fibers with different PEO additions. The FTIR spectra of the composite fibers were similar to that of pure PMIA fiber, and no new absorption peaks appeared. The infrared spectrum of PEO showed a distinctive absorption peak at 1280 cm−1 for carbon oxide–carbon, which indicated that there was no new chemical bonding between the two. However, there was no PEO characteristic absorption peak inside the composite fiber. This was because the added amount of PEO was too small or because PEO was removed during the washing pro.

Fourier transform infrared spectrum of composite fibers (PEO Mw = 600,000)
To investigate the reinforcing effect of different molecular weight of PEO, 1% PEO/PMIA composite fibers were prepared by adding different Mw of PEO to PMIA solution. Figure 6 shows the effect of PEO Mw on the mechanical properties of the composite fibers. PEO was soluble in water [25], and the lower the molecular weight, the better its solubility in water. As the added PEO molecular weight was 8000, the added PEO would be washed away during water bath stretching and washing process, their reinforcement effect would become weak. Besides, as the PEO Mw is low, too much PEO chain ends would become the defects of the fibers and the mechanical properties of the fibers would be worse. When the molecular weight of PEO added was 5,000,000, they have fewer chain ends and basically no PEO would be washed away, so its reinforcing effect was the best. However, the added PEO had a negative effect on the heat resistance and flame retardancy of the fiber. When the molecular weight of PEO added was 600,000, the mechanical properties of the composite fiber can be greatly improved, so PEO with a Mw of 600,000 was chosen as the additive in this study.

Fiber mechanical properties of 1% PEO/PMIA fibers with different molecular weight of PEO(Mw = 600,000), F1-8000, F2-600,000, F3-4,000,000, F4-5,000,000
Table 1 shows the maximum water bath draw ratio of the composite fibers with different PEO additions. It could be seen that the maximum water bath draw ratios of PEO/PMIA composite fibers were higher than that of pure PMIA solution, and the water bath draw ratio of 2% PEO/PMIA could reach up to 5 times. Before the addition of 2%, as the addition of PEO increased, the maximum water bath draw ratio increased. This was because the increase in PEO content caused more hydrogen bonding between PMIA molecular chains to be destroyed, thus the drawability of PMIA fibers in water bath became better and higher draw ratio was obtained. However, when the added amount of PEO was 2.5%, too many hydrogen bonding between PMIA molecular chains were destroyed, which resulted in the slippage of fiber chains in water bath drawing process and the drawability of fibers became decreased.
Figure 7 shows the mechanical properties of the composite fibers before hot stretching and after hot stretching. From Fig. 7a, it could be seen that the tensile strength of the composite fibers after water bath stretching were increased compared with that of pure PMIA fiber, and the tensile strength of 2% PEO/PMIA fibers could reach 3.61 cN/dtex. However, the fiber strength decreased a little as the amount of added PEO continued to increase. This was because the water bath draw ratio of the composite fibers increased after added PEO, the higher the water bath drawing ratio, the higher the orientation of fibers and the better the mechanical properties. 2% PEO/PMIA had the highest maximum water bath draw ratio, so its tensile strength was the highest. As the added PEO content increased to 2.5%, the maximum water bath draw ratio decreased to 4.7, and the fiber strength decreased a little.

Mechanical properties of composite fibers: a before hot stretching, b after hot stretching, F0-PMIA, F1-1% PEO/PMIA, F2-1.5% PEO/PMIA, F3-2% PEO/PMIA, F4-2.5% PEO/PMIA (PEO Mw = 600,000)
Figure 7b shows the mechanical properties of the fibers after hot stretching at 300 °C. Comparing with Fig. 7a, it could be found that the mechanical properties of the fibers were substantially improved after the hot stretching. This was because the water or solvent molecules acted as the plasticizer for the water bath stretching of fibers, which made the structure of the fibers not very compact, and larger defects would be generated inside the fiber after solvent was washed away and the water was removed after dried. During hot stretching process at 300 °C, which was much higher than the glass transition temperature of PMIA, PMIA molecular chains segment could move under the stretching force and the structure of the composite fibers became more compact at certain stretching multipliers. The tensile strength of 2% PEO/PMIA fibers reached 4.71 cN/dtex, which was 76% higher than that of pure PMIA fiber. In short, the addition of PEO weakened the hydrogen bonding between the PMIA molecular chains so that the PMIA molecules can be arranged in a more regular manner during the stretching process, as illustrated in Fig. 8. The regular molecular chain arrangement made the tensile strength of the fiber be improved.

The schematic presentation of PMIA of chemical structure transition during stretching
From the X-ray diffraction pattern in Fig. 9, it could be seen that PEO was a very crystalline substance, and the PMIA fibers without hot stretching were less crystalline. Although PEO is highly crystalline, the crystalline characteristic absorption peak of PEO did not appear inside the composite fiber. This was partly due to the fact that the amount of PEO added was too small, and PEO was soluble in water, and part of PEO would be removed during water bath stretching and washing.

XRD patterns of PEO/PMIA composite fibers after water stretching: a 5–60°, b 10–30°
The crystallinity of different fibers are listed in Table 1. It could be seen that the crystallinity of the composite fibers increased compared with that of pure PMIA fiber, and the highest crystallinity of 2% PEO/PMIA reached 34.89%. This was probably due to the increase in the water bath drawing ratio of the composite fibers, and the larger drawing ratio was beneficial to the crystallization of molecular chains. Higher crystallinity of the fibers contributed to their higher mechanical properties.
Figure 10 shows the cross-sectional view of the thermally stretched fibers after pulling off. It could be seen that the cross sections of the PEO/PMIA composite fibers with different content of PEO exhibited much tough structures after stretching. Pure PMIA fiber and 1% PEO/PMIA fiber had a relatively regular cross-sectional structure with no obvious hairline structure. However, 2% PEO/PMIA and 2.5% PEO/PMIA fibers had more hairy filaments in their fractured surface. The higher the orientation of the fibers, the more regular the arrangement of their molecular chains, which lead to a fibrious structure during the stretching process. It was the higher orientation of the composite fiber that led to a higher tensile strength and resulted in a more hairy structure in its fractured surface.

The photos of tensile fractured surface of hot-drawn fibers: a PMIA, b 1% PEO/PMIA, c 1.5% PEO/PMIA, d 2% PEO/PMIA, e 2.5% PEO/PMIA
The fiber orientation of different fibers are measured by sonic velocity and XRD method, the orientation factors are listed in Table 1. The mechanical properties of the fibers were closely related to their orientation, and the greater the orientation of the fiber, the higher its tensile strength. From Table 1, it can be seen that the orientation factors of composite fibers changed with the same trend as that of pure PMIA fiber. Composite fibers had higher orientation factor than that of pure PMIA fiber, which was due to their increased water bath draw ratio. So the fracture strength of composite fibers was increased compared with that of PMIA fiber. 2% PEO/PMIA composite fiber have the maximum orientation factor, which resulted in the highest tensile strength for 2% PEO/PMIA composite fiber.
Figure 11 shows the thermal properties of PMIA fiber and PEO/PMIA composite fibers, Table 2 shows temperature at maximum decomposition rate (Tmd) of PMIA fiber and PEO/PMIA composite fibers. It could be seen that the heat resistance of the composite fiber and PMIA fiber were almost the same, and the temperature of starting decomposition and the temperature of maximum decomposition rate of the composite fibers were very close to that of PMIA. This was probably because the PEO content added was too small and part of the PEO would be removed during water bath drawing and washing process, so the amount of PEO contained in the composite fiber was too small. The Tmd of 1% PEO/PMIA and 1.5% PEO/PMIA composite fibers was increased compared with that of pure PMIA fiber, while 2% PEO/PMIA and 2.5% PEO/PMIA composite fibers had a lower Tmd. This was because 1% PEO/PMIA and 1.5% PEO/PMIA fibers had increased molecular chain orientation than pure PMIA fiber, while 2% PEO/PMIA and 2.5% PEO/PMIA fibers had increased molecular chain orientation and contained more PEO than pure PMIA fiber. Overall, the heat resistance of the composite fiber did not deteriorate significantly, indicating that the composite fibers could still meet the basic requirements of protective clothing applications.

TGA curves (a) and DTG curves (b) of PMIA fiber and PEO/PMIA composite fibers
Figure 12 shows the DMA curves of the composite fibers, the glass transition temperature (Tg) of different fibers are listed in Table 2. As can be seen from Fig. 12b, the temperature at which the energy storage modulus of the composite fiber began to drop was relatively close to that of pure PMIA fiber, which indicated that the heat resistance of the composite fiber remains essentially unchanged. The Tg of the composite fibers increased compared with that of pure PMIA fiber, and the Tg of 2% PEO/PMIA was the highest. The Tg was closely related to the orientation and crystallinity of fibers, and the higher the orientation and crystallinity of fibers, the higher the glass transition temperature. It may be because that higher orientation and higher crystallinity resulted in much compact structure of the composite fibers, thus the glass transition temperature of the fibers became higher. 2% PEO/PMIA fiber had the highest orientation and highest crystallinity (see Table 1), and its Tg was also the highest. The Tg of the material determines its service temperature range, and it could be seen that the increase in the glass transition temperature of the composite fiber makes the application range of the composite fiber larger.

DMA curves of PEO/PMIA composite fibers. a Tanθ-Temperature curves, b Storage modulus-Temperature curves
The limit oxygen index (LOI) of the composite fibers was also tested and listed in Table 2. The LOI of the material determines whether it could be used for flame retardant protection, and the higher the LOI value, the better the flame retardant performance. From Table 2, it can be seen that the LOI of the composite fiber decreased a little bit, but it was still a flame retardant fiber, and the LOI decreased as the PEO content in the composite fiber increased. This was because PEO was a flammable polymer, with the increase of PEO content inside the composite fiber, the fiber became more flammable. PEO was a water-soluble polymer, part of PEO would be removed during water bath stretching and washing process, which resulted in the decrease in the PEO content inside the composite fiber, so that the LOI of the composite fiber would not change significantly. Overall, the composite fibers were still flame-retardant fibers and can still meet the application needs in the field of flame retardant protection.
Table 3 shows the thermal shrinkage results for different fibers at temperatures between 280 and 320 °C. One of the main reference factors for the application of fibers in the field of flame retardant protection is their high temperature thermal shrinkage. From Table 3, it can be seen that the thermal shrinkage of the composite fibers was about the same as that of pure PMIA fiber at 280 °C and 300 °C. The thermal shrinkage of 1% PEO/PMIA fiber at 280 °C had a little decrease, and at other temperatures the thermal shrinkage of the fiber showed a tendency to become larger as the PEO content inside the composite fiber increases. This was because the PEO content in the 1% PEO/PMIA composite fiber was little, but its structure was denser than PMIA fiber, which induced its thermal shrinkage rate decreased. When the surrounding temperature was higher as 320 °C, the thermal shrinkage of the composite fibers became obviously higher than that of pure PMIA fiber. This was because that PEO was a kind of polymer which was not resistant to high temperature and it was easy to shrink at high temperature, as the PEO content in the composite fiber increased, the thermal shrinkage rate of the composite fibers also increased. Overall, the heat shrinkage of the composite fibers does not change significantly compared with that of pure PMIA fiber, and it can still meet the application requirements in the field of flame retardant protection.
Conclusions
A novel technique for PEO reinforced PMIA fibers was proposed in the present study. The effects of PEO addition on structure and properties of PMIA fibers were investigated in details. The optimum PEO addition was 2%, which increased the tensile strength from 2.68 cN/dtex of pure PMIA fiber to 4.74 cN/dtex of the composite fiber, with increased Tg. Meanwhile, the heat resistance, flame retardancy and thermal stability of composite fibers did not change significantly. The higher degree of fiber orientation greatly enhanced the fiber strength. This modification technology will provide a highly oriented, high-strength PMIA composite fiber that retains its excellent heat and flame resistance.
References
García JM, García FC, Serna F, de la Peña JL. High-performance aromatic polyamides. Prog Polym Sci 2010;35:623.
Li N, Zhang X-K, Yu J-R, Wang Y, Zhu J, Hu Z-M. Increased hydrogen-bonding of Poly(m-phenylene isophthalamide) (PMIA) with sulfonate moiety for high-performance easily dyeable fiber. Chin J Polym Sci 2020;38:1230–8.
Li Y, Ma X, Deng N, Kang W, Zhao H, Li Z, Cheng B. Electrospun SiO2/PMIA nanofiber membranes with higher ionic conductivity for high temperature resistance lithium-ion batteries. Fibers Polym 2017;18:212.
Liu L, Mo J, Li J, Liu J, Yan H, Lyu J, Jiang B, Chu L, Li M. Comprehensively-modified polymer electrolyte membranes with multifunctional PMIA for highly-stable all-solid-state lithium-ion batteries. J Energy Chem 2020;48:334.
Mazzocchetti L, Benelli T, Maccaferri E, Merighi S, Belcari J, Zucchelli A, Giorgini L. Poly-m-aramid electrospun nanofibrous mats as high-performance flame retardants for carbon fiber reinforced composites. Compos B Eng 2018;145:252.
Ouyang S, Wang T, Yu Y, Yang B, Yao J, Wang S. From trans to cis conformation: further understanding the surface properties of poly(m-phenylene isophthalamide). ACS Omega 2017;2:290.
Wang X, Si Y, Wang X, Yang J, Ding B, Chen L, Hu Z, Yu J. Tuning hierarchically aligned structures for high-strength PMIA-MWCNT hybrid nanofibers. Nanoscale 2013;5:886.
Wang Z, Li T, Yu J, Hu Z, Zhu J, Wang Y. Constructing flexible and CuS-coated meta-aramid/polyacrylonitrile composite films with excellent coating adhesion. Ind Eng Chem Res 2019;58:17965.
Yang M, Zhao C, Zhang S, Li P, Hou D. Preparation of graphene oxide modified poly(m-phenylene isophthalamide) nanofiltration membrane with improved water flux and antifouling property. Appl Surf Sci 2017;394:149.
Zhai Y, Wang N, Mao X, Si Y, Yu J, Al-Deyab SS, El-Newehy M, Ding B. Sandwich-structured PVdF/PMIA/PVdF nanofibrous separators with robust mechanical strength and thermal stability for lithium ion batteries. J Mater Chem A 2014;2:14511.
Zhang H, Zhang Y, Xu T, John AE, Li Y, Li W, Zhu B. Poly(m-phenylene isophthalamide) separator for improving the heat resistance and power density of lithium-ion batteries. J Power Sources 2016;329:8.
Zhang X, Tang X, Wang R, Wang R, Yan X, Shi M. Thermal degradation behaviors and fire retardant properties of poly(1,3,4-oxadiazole)s (POD) and poly(m-phenylene isophthalamide) (PMIA) fibers. Fibers Polym 2017;18:1421.
Trigo-López M, Miguel-Ortega Á, Vallejos S, Muñoz A, Izquierdo D, Colina Á, García FC, García JM. Intrinsically colored wholly aromatic polyamides (aramids). Dyes Pigm 2015;122:177.
Wang L, Zhang M, Yang B, Ding X, Tan J, Song S, Nie J. Flexible, robust, and durable aramid fiber/CNT composite paper as a multifunctional sensor for wearable applications. ACS Appl Mater Interfaces 2021;13:5486.
Jiang Q, Tian H, Zhang K. Enhanced performance of poly(m-phenylene isophthalamide) (PMIA) composite hollow fibre ultrafiltration membranes by O-MoS2 nanosheets modification. Desalin Water Treat 2019;166:245.
Malafeev KV, Moskalyuk OA, Yudin VE, Morganti P, Ivan’kova EM, Popova EN, Elokhovskii VY, Vaganov GV. Study of physicomechanical properties of composite fibers based on polylactide and modified chitin nanofibrils. Polym Sci Ser A 2020;62:249.
Yan M, Shi J, Tang S, Liu L, Zhu H, Zhou G, Zeng J, Zhang H, Yu Y, Guo J. Strengthening and toughening sodium alginate fibers using a dynamically cross-linked network of inorganic nanoparticles and sodium alginate through the hydrogen bonding strategy. New J Chem 2021;45:10362.
Wang Z, Li T, Yu J, Hu Z, Zhu J, Wang Y. General bioinspired, innovative method for fabrication of surface-nickeled meta-aramid fibers. Ind Eng Chem Res 2019;58:9458.
Barani H, Bahrami SH. Preparation of polyacrylonitrile and cellulose acetate blend fibers through wet-spinning. J Appl Polym Sci 2000;2007:103.
Wu K, Yu J, Yang J, Chen S, Wang X, Zhang Y, Wang H. Properties and phase morphology of cellulose/aromatic polysulfonamide alloy fibers regulated by the viscosity ratio of solution. Cellulose 2017;25:903.
Chen J, Guo J, Zhao M, Zhang R, Guan F. Hydrogen bonding in chitosan/Antarctic krill protein composite system: study on construction and enhancement mechanism. Int J Biol Macromol 2020;142:513.
Lei X, Ye D, Chen J, Tang S, Sun P, Chen L, Lu A, Du Y, Zhang L. Customizable multidimensional self-wrinkling structure constructed via modulus gradient in chitosan hydrogels. Chem Mater 2019;31:10032.
Xu Z, Yang T, Chu X, Su H, Wang Z, Chen N, Gu B, Zhang H, Deng W, Zhang H, Yang W. Strong lewis acid-base and weak hydrogen bond synergistically enhancing ionic conductivity of poly(ethylene oxide)@SiO2 electrolytes for a high rate capability li-metal battery. ACS Appl Mater Interfaces 2020;12:10341.
Zare Y, Rhee KY. Modeling of viscosity and complex modulus for poly (lactic acid)/poly (ethylene oxide)/carbon nanotubes nanocomposites assuming yield stress and network breaking time. Compos B Eng 2019;156:100.
Kupka V, Dvorakova E, Manakhov A, Michlicek M, Petrus J, Vojtova L, Zajickova L. Well-blended PCL/PEO electrospun nanofibers with functional properties enhanced by plasma processing. Polymers (Basel). 2020;12:1403.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51473031).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors state that there are no conflicts of interest to disclose.
Rights and permissions
About this article

Cite this article
Song, Q., Wu, W., Wang, Y. et al. The Structure and Properties of Polyethylene Oxide Reinforced Poly(Metaphenylene Isophthalamide) Fibers. Adv. Fiber Mater. 4, 436–447 (2022). https://doi.org/10.1007/s42765-021-00110-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42765-021-00110-x