
Abstract
Involvement of the central nervous system (CNS) is commonly seen in patients with systemic sarcoidosis; however, an isolated case of neurosarcoidosis is a substantially infrequent scenario. Neurosarcoidosis may present with numerous nonspecific clinical manifestations and radiological features, which makes it difficult to diagnose. Correct interpretation of magnetic resonance imaging (MRI) findings incorporating sarcoidosis as a possible diagnosis is important for timely management of the disease. We report the unusual case of a 47-year-old female with brain lesions and a sudden appearance of spinal lesions mimicking CNS demyelination and metastases. Biopsy provided a definitive diagnosis of neurosarcoidosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sarcoidosis is a multisystem, non-caseating, granulomatous disease of unknown etiology [1,2,3,4,5]. An isolated impairment of the CNS, termed neurosarcoidosis, is a rare condition with a wide spectrum of nonspecific clinical symptoms and radiological presentations. Findings include headache, weakness, paresthesias, dysphagia, dysarthria, cranial nerve palsies, seizures, increased intracranial pressure, and endocrine abnormalities due to hypothalamic/pituitary involvement, as well as spinal manifestations due to cord myelopathy and pachymeningeal and leptomeningeal sarcoidosis. Probable diagnosis may be achieved through MR imaging and exclusion of other inflammatory, infectious, and granulomatous diseases, as well as disease processes such as demyelination/multiple sclerosis, tuberculosis (TB), lymphoma, and metastases [2, 5]. Definitive diagnosis requires biopsy for histopathology [6,7,8]. We present a case of unusual progression of isolated neurosarcoidosis, whereby lesions were limited to the brain parenchyma and meninges, mimicking demyelination, with sudden spread to the spinal cord in a manner mimicking metastatic disease.
Case Report
A 47-year-old female with no past medical history presented with perceived balance and gait disturbance after the resolution of concussive symptoms stemming from a fall injury. MRI of the head was initially performed and revealed several small (up to 9 mm), deep, and periventricular white matter T2 hyperintense lesions in the right temporal and occipital lobes, right basal ganglia, and corpus callosum. Combined with neurological assessment, this led to a working diagnosis of multiple sclerosis (MS), followed by treatment with Avonex, an interferon beta-1a agent. There was some clinical improvement in the patient’s balance while on treatment; however, the diagnosis of MS was retracted due to a lack of recognized McDonald criteria for MS, and the treatment was discontinued. The patient’s fall was determined to be accidental; however, upon close review of history, there were also two fall-related injuries the previous year requiring emergency room visits. In retrospect, the falls may have been early signs of illness.
After MS treatment was discontinued, the patient reported return of balance and gait disturbance, accompanied by new symptoms of left upper and lower extremity weakness, headaches, and memory difficulties. The patient did not experience any constitutional symptoms or symptoms suggestive of infection such as fever or fatigue. However, due to the unusual nature of the illness, referral was made to an infectious disease specialist and infectious disease as an etiology was subsequently ruled out. Further MRI studies, over the period of a year, demonstrated interval progression of existing brain lesions and revealed new lesions in both cerebral hemispheres. MRI of the entire spine exhibited preserved morphology and normal signal intensity of the cord, without evidence for myelopathy, intramedullary lesion, abnormal enhancement, or syrinx. Series of X-rays and computed tomography (CT) of the chest failed to reveal any lung abnormalities such as infection, lymphadenopathy, calcified nodes or nodules, and specifically, there were no signs of granulomatous disease. Given progressive neuroimaging abnormalities along with the development of further clinical symptoms, the decision for resection and biopsy of the right temporal lobe lesion via craniotomy was elected by the patient. This was chosen over another less invasive but less specific testing such as lumbar puncture, as a means to secure a diagnosis by histology. The duration of the illness at the time of craniotomy was 4 years.
Close postoperative MR follow-up imaging of the head with and without gadolinium demonstrated significant size progression of abnormal signal intraparenchymal lesions, as well as multiple, new intracranial enhancing lesions involving the supratentorial and infratentorial brain, hypothalamus, brainstem, and along the meninges and arachnoid compartment, measuring up to 2.3 cm. Progressive dural enhancement was also seen, along with worsening hydrocephalus (Figs. 1, 2, and 3). MRI of the cervical and thoracic spine showed diffuse abnormal signal intensity of the entire spinal cord, associated with irregular linear and nodular enhancement of the overlying meninges and intrathecal nodular enhancing foci (Fig. 4). This new, sudden expansion and enhancement pattern of CNS lesions mimicked the appearance of a widespread metastatic process, including spinal drop metastasis, which was therefore included in the differential diagnosis along with a granulomatous disease, such as sarcoidosis and TB.

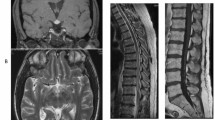
Axial fluid-attenuated inversion recovery (FLAIR)-weighted magnetic resonance images of the head depict multiple hyperintense lesions in the subcortical and periventricular white matter, and brainstem, as well as moderate hydrocephalus

Axial T1-weighted magnetic resonance gadolinium-enhanced images of the head demonstrate multiple enhancing foci in the leptomeningeal space

Sagittal T1-weighted magnetic resonance gadolinium-enhanced images of the head and upper cervical spine demonstrate multiple nodular, linear-shaped, and mass-like enhancing lesions in the leptomeningeal and pachymeningeal compartments of the lower supratentorial brain, posterior fossa, and along the upper cervical cord

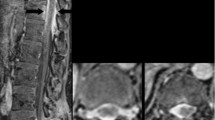
Sagittal T1-weighted magnetic resonance gadolinium-enhanced images of the cervical and thoracic spine exhibit innumerable nodular and linear-shaped enhancing lesions in the extramedullary intradural space
Biopsy results confirmed the diagnosis of neurosarciodosis. Specifically, pathological examination identified granulomatous inflammation with the presence of Langhans and foreign body type giant cells. The background specimen revealed normal brain parenchyma. There was no polarizable material identified. Ziehl-Neelson, periodic acid-Schiff diastase (PAS-D), and Gomori’s methenamine silver (GMS) histochemical stains were negative for acid fast bacilli and fungal elements, and immunostains glial fibrillary acidic protein (GFAP) and S-100 were also negative. There was no evidence for malignancy.
Discussion
Sarcoidosis is a multisystem inflammatory disease, characterized by the occurrence of non-caseating granulomas [1, 3, 4, 9]. The etiology of the disease is unknown, typically affecting young adults, women more commonly than men (6:4), with an incidence of 15–20 cases per 100,000 people [1, 2, 4, 7]. Sarcoidosis can involve essentially any body system, with lung involvement being the most common, seen in up to 90% of patients [2, 9, 10]. Chest X-ray is abnormal in 85–95% of patients with sarcoidosis [1, 6, 10]. Spontaneous remission within 5 years of disease onset is seen in approximately 70% of patients; however, disease progression must be carefully monitored with imaging [1, 10].
Involvement of the peripheral or central nervous system in patients with systemic sarcoidosis is not infrequent and characteristically presents as part of the disease course progression, generally manifesting within 2 years of systemic sarcoidosis onset [3, 4]. Isolated neurosarcoidosis (without systemic disease) is rare, comprising approximately 1–15% of all cases [2, 3, 6, 7, 10]. Neurological symptoms are commonly the first clinical manifestation in such patients, and initial neuroimaging is often performed without the diagnosis of systemic sarcoidosis having yet been made. On the contrary, not all patients with imaging findings of neurosarcoidosis are symptomatic [1,2,3, 9].
Clinical findings are various and non-specific depending on the disease location and whether intracranial or spinal structures are compromised. These findings include headaches and meningismus due to the involvement of the pachymeninges and leptomeninges, increased intracranial pressure, hydrocephalus, cranial nerve neuropathies, seizures, gait disturbance, cognitive alterations, dysarthria/dysphagia, weakness, and paresthesias. Endocrine dysfunctions such as diabetes insipidus, syndrome of inappropriate antidiuretic hormone secretion (SIADH), hypothyroidism, hypoadrenalism, and hyperprolactinemia may occur due to hypothalamic and pituitary gland sarcoidosis [2, 5]. Spinal cord myelopathy and meningeal involvement cause pain, motor or sensory deficits, abnormal reflexes, difficulty walking, and impaired urine/bowel control [6, 7, 10].
Contrast-enhanced MRI is the imaging modality of choice for assessing patients for possible neurosarcoidosis [3, 6, 7, 10, 11]. There are a number of radiologic findings characteristic of the disease, with the most common being leptomeningeal thickening. This is observed in approximately 40% of cases [2, 3, 6, 8,9,10,11]. Pachymeningeal (dural) thickening is also commonly seen [6,7,8,9,10,11]. Meningeal contrast enhancement may be focal or diffuse, smooth or nodular, with predominant distribution around the basal aspect of the brain and circle of Willis; however, this can involve the entire length of the spinal cord [6]. Parenchymal involvement is one of the most common findings and presents as T2 hyperintense white matter lesions with perivascular and periventricular space predominance [3, 6]. Dorsal subpial enhancement of the spinal cord in the shape of a trident, known as the trident sign, has been described in the literature as a reliable marker of neurosarcoidosis. This is best seen on MRI T1-weighted gadolinium-enhanced axial views [12]. The case demonstrated similar dorsal crescent-shaped enhancing lesions along the thoracic spinal cord, but they were not in the characteristic trident shape (Fig. 5).

Axial T1-weighted magnetic resonance gadolinium-enhanced images of the thoracic spine depict crescent shaped enhancing lesions along the posterolateral, and to a lesser extent, anterior aspect of the extramedullary intradural space
Cranial nerves may be compromised as part of leptomeningeal disease or irrespectively. The facial and optic nerves are most commonly affected, resulting in facial nerve palsy or visual disturbance. Hypothalamic and pituitary involvement are also noted as part of extensive leptomeningeal sarcoidosis or may sometimes be encountered in isolation. Hydrocephalus may be present in severe cases of meningeal inflammation [2, 3, 6].
In our case, MR imaging demonstrated leptomeningeal, pachymeningeal, and extensive parenchymal involvement, as well as hydrocephalus, a likely explanation for the early symptoms of gait disturbance and progressive memory difficulties.
Intramedullary spinal cord disease is rare in sarcoidosis [7]. Lesions usually appear in a sporadic distribution, spanning several spinal segments [6, 7]. It has been reported that spinal cord involvement generally appears more severe on imaging than the symptoms manifested by the patient [6]. Meningeal spread of sarcoidosis along the spinal cord and cauda equina has been described [3, 6, 7]. In our case, there were extensive diffuse lesions with irregular margins along the entirety of the spinal cord associated with non-specific clinical findings such as gait disturbance, imbalance, and neuropathy of the extremities. Sudden appearance of extensive extramedullary intradural spinal involvement with linear and nodular lesions is most likely related to meningeal sarcoidosis.
Although the constellation of radiological findings is characteristic of neurosarcoidosis, some of the imaging features can be identified in other neurological disorders. T2 hyperintense periventricular and perivascular lesions as well as infratentorial and spinal intramedullary lesions are the usual presentation of demyelination and multiple sclerosis [2, 5]. A combination of intraparenchymal and meningeal disease findings overlap with granulomatous, infectious, or lymphoproliferative diseases including mycobacterial and fungal infections and lymphoma. Extensive extramedullary intradural spinal nodules can also mimic drop metastases [1, 10].
The recently published Definition and Consensus Diagnostic Criteria for Neurosarcoidosis [13] specifies that a definite diagnosis of neurosarcoidosis requires biopsy [6,7,8, 13]. This, however, may be postponed in instances where symptomatology is not severe and/or the surgery for obtaining the biopsy material may result in serious neurological deficit [3, 6, 11]. Clinical presentation and diagnostic evaluation suggestive of neurosarcoidosis are also required for diagnosis [13]. This was evident in the case, but complicated by additional radiologic findings suggestive of demyelination disease, as well as the sudden post-biopsy MRI findings of spinal cord lesions mimicking metastases. In our case, other etiologies of granulomatous disease, including infectious diseases and malignancy, had been ruled out clinically and radiologically. Additionally, the patient had declined lumbar puncture for CNS analysis, instead opting for biopsy as a means to obtain an accurate diagnosis despite the known risks and potential complications. Histopathology provided a timely and definitive diagnosis, without which, the clinicians might have pursued the deceptive appearance of brain and spinal cord lesions.
Treatment for neurosarcoidosis is almost always warranted, and therefore developing a treatment plan is vital. Treatment recommendations include a stepwise approach based on the severity of the disease. This typically begins with oral glucocorticoids and progresses to high-dose oral or intravenous glucocorticoids for severe or progressive disease [14]. The next line of treatment involves steroid-sparing agents, such as methotrexate or mycophenolate. For severe, unrelenting disease, treatment with TNF- α inhibitors such as infliximab is suggested. If patients are unable to take TNF- α inhibitors, or the efficacy is inadequate, other recommended agents include rituximab, a monoclonal antibody, and cyclophosphamide, a neoplastic agent.
Conclusion
In the above case, the clinical presentation, disease progression, and diagnostic imaging findings of a unique example of isolated neurosarcoidosis were reviewed. The bewildering variety of nonspecific symptoms and neuroradiological manifestations, combined with an unusual course of disease, may pose a significant clinical dilemma for the treating physician. Radiological features often mimic other serious neurological disorders, including demyelination and malignancy. Awareness of these similarities would assist in timely recognition of rare neurosarcoidosis findings and would therefore also contribute to improving patient health outcomes.
References
Nunes H, et al. Sarcoidosis. Orphanet J Rare Dis. 2007;2(1):46.
Smith JK, Matheus MG, Castillo M. Imaging manifestations of neurosarcoidosis. Am J Roentgenol. 2004;182(2):289–95.
Ungprasert P, Matteson EL. Neurosarcoidosis. Rheum Dis Clin N Am. 2017;43(4):593–606. https://doi.org/10.1016/j.rdc.2017.06.008.
Hughes BD, et al. Neurosarcoidosis. Contemp Neurosurg. 2007;29(3):1–7. https://doi.org/10.1097/00029679-200702150-00001.
Christoforidis GA, et al. MR of CNS sarcoidosis: correlation of imaging features to clinical symptoms and response to treatment. Am J Neuroradiol. 1999;20(4):655–69.
Tavee JO, Stren BJ. Neurosarcoidosis. Clin Chest Med. 2015;36(4):643–56.
Lower EE, Weiss KL. Neurosarcoidosis. Clin Chest Med. 2008;29(3):475–92.
Lacomis D. Neurosarcoidosis. Curr Neuropharmacol. 2011;9(3):429–36.
Shah R, Roberson GH, Curé JK. Correlation of MR imaging findings and clinical manifestations in neurosarcoidosis. Am J Neuroradiol. 2009;30(5):953–61.
Culver DA. Sarcoidosis. Immunol Allergy Clin N Am. 2012;32(4):487–511.
Segal BM. Neurosarcoidosis: diagnostic approaches and therapeutic strategies. Curr Opin Neurol. 2013;26(3):307–13.
Zalewski NL, et al. Central canal enhancement and the trident sign in spinal cord sarcoidosis. Neurology. 2016;87(7):743–4. https://doi.org/10.1212/wnl.0000000000002992.
Stern BJ, et al. Definition and consensus diagnostic criteria for neurosarcoidosis. JAMA Neurol. 2018;75(12):1546. https://doi.org/10.1001/jamaneurol.2018.2295.
Voortman M, Drent M, Baughman RP. Management of neurosarcoidosis: a clinical challenge. Curr Opin Neurol. 2019;32(3):475–83.
Acknowledgments
There is no funding source to declare.
Funding
There was no source of funding received for this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Ethics Review
All research conducted was done so in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. This research was reviewed by the Thunder Bay Regional Health Sciences Centre Research Ethics Board, and was provided a formal waiver for REB review.
Informed Consent
Informed consent from the patient described in the case report has not been obtained, as it was determined by the Thunder Bay Regional Health Sciences Centre Research Ethics Board that there was no personally identifiable information to necessitate obtaining consent.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Imaging
Rights and permissions
About this article

Cite this article
Domonkos, V., Shuster, A. Isolated Neurosarcoidosis Mimicking Demyelination and Metastases. SN Compr. Clin. Med. 1, 1004–1008 (2019). https://doi.org/10.1007/s42399-019-00171-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42399-019-00171-5