
Abstract
Hudaira Drain of Pakistan is one of the largest storm water drains which is a potential threat to the environment and biodiversity. It is considered as the second largest water polluting body in Pakistan. The major polluting contributors are toxic industrial wastes, chemical fertilizers and poultry or animal waste. The objective of this study was to analyze the microbial diversity of wastewater from Hudiara Drain using culture-independent and culture-dependent techniques. For culture-independent analysis, the 16S rRNA gene-based Illumina sequencing technique was used. Metagenomic sequence analysis revealed a total of 20 different phyla in the source drain water samples. Bacterial phyla Actinobacteria, Proteobacteria, Bacteroidetes, Firmicutes, Acidobacteria, Chloroflexi, Planctomycetes, and Fusobacteria and archaeal phylum Thaumarchaeota were commonly identified from all the wastewater samples. A total of 282 genera of bacteria and archaea were identified in this study, with Bacillus, Nitrospira, Nitrososphaera, Gemmatimonas, Pseudomonas, Stenotrophomonas, Myxococcus, Azospirilum, Skermanella, Nitrososphaera, Pontibacter and Aquicella being the dominant genera. Using culture-dependent techniques, 32 bacterial strains were identified and characterized for heavy metal and antibiotic resistance. The common genera were Bacillus, Aeromonas and Enterobacter. These results suggest that the microbial diversity from Hudiara Drain wastewater samples is a potential source of clinically and industrially important bacterial and archaeal strains.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hudiara Drain originates in Batala, Gurdaspur District, India and enters Pakistan through the Laloo Village. It is located at the latitude 31° 23′ 45.7′′ north and longitude 74° 21′ 59.4′′ east. The drain runs for 44.2 km in Indian territory after which it extends for 54.4 km in Pakistan before it is disposed in River Ravi (Yamin and Ahmad 2007). The Hudiara Drain of Pakistan used to serve as a storm water drain about 40 years ago. Its water is also used for irrigating acres of agricultural land stretching around it. However, due to the burgeoning industrialization, Hudiara Drain now serves as an industrial wastewater drain, with contamination levels far above the World Wide Fund for Nature (WWF) Pakistan’s allotted safety limits. The major contaminants in the drain water have been reported to be fecal coliforms, industrial effluents and suspended solids (Afzal et al. 2000). Along its entire length, 120 industries have been set up, including tanneries, leather industries and paper industries. All of these industries dump their effluent-leaden waste untreated into the waters of Hudiara Drain. Having become more of a wastewater dump, the Hudiara Drain falls into River Ravi with a flux of about 5041 L, loaded with toxins and effluents (Shah et al. 2010).
The wastewater concentrations of heavy metals are slightly lower than the permissible concentrations (Thomson 2003), but the heavy metals such as cadmium (Cd), lead (Pb), mercury (Hg), nickel (Ni), chromium (Cr) and copper (Cu) are present nevertheless, and they accumulate in each organism feeding upon the contaminated source, accumulating up the trophic levels. The milk of the cows and buffaloes being fed to the infants along the sides of Hudiara Drain, the crops being cultivated in that area and the meat being eaten are all contaminated with heavy metal ions, known to be carcinogenic and disease-causing in nature. Yamin and Ahmad (2007) illustrated the heavy metal accumulation in crops cultivated with Hudiara Drain wastewater and its ultimate consequences on soil and crop quality. A global concern arises due to the fact that even minor concentrations of heavy metals bring about highly hazardous health consequences (Su 2016). The drain also serves as a major provenance of waterborne diseases such as cholera, hepatitis, dysentery and typhoid (Jabbar 2020).
Due to high contamination levels, microbial communities dwelling in wastewater drains are also affected. Changes in environmental factors such as pH and temperature, amount and types of pollutants, e.g., toxic organic compounds and heavy metal ions have great impacts on the structure and composition of microbial communities (Balcom et al. 2016; Hu et al. 2012; Meerburg et al. 2016). Organic effluents and heavy metals can change the function of microbial enzymes and other proteins and ultimately affect the metabolic activity of microorganisms living in the wastewater drains (Bettiol and Ghini 2011; Paruch et al. 2019). These microorganisms have the ability to degrade different effluents, accumulate heavy metals and metabolize inorganic and organic compounds that are present in such wastewater environments (Bettiol and Ghini 2011). The presence of different pollutants and limited energy resources play an important role in various biochemical pathways of these microorganisms and the evolution of novel genes that can be utilized for bioremediation of wastewater ecosystems (Ben-Dov et al. 2008).
According to previous studies, members of Proteobacteria and Actinobacteria were the most dominant as compared to Firmicutes, Bacteroidetes, Acidobacteria, Thaumarchaeota and Planctomycetes in wastewater drains and other polluted environments (Farzadkia and Bazrafshan 2014; Wang et al. 2012). Proteobacteria and Actinobacteria play an important role in the degradation of organic compounds originating from poultry, petroleum refineries, pharmaceutical industry and paper and pulp industry (Kang et al. 2020). The microbial communities studied from polluted drains may be used for biological treatment and bioremediation of various wastewater ecosystems (Balcom et al. 2016; Ben-Dov et al. 2008). Anaerobic members of Firmicutes and Bacteroidetes have been found abundantly in the wastewater drains, activated sludge and anaerobic bioreactors. 16S ribosomal RNA (rRNA) gene based metagenomic analyses are commonly used for the determination of microbial communities associated with polluted environments (Alma’abadi et al. 2015; Handelsman 2005). High throughput sequencing techniques have gained much interest of biologists worldwide because of their high accuracy and efficiency, allowing its widespread applications in microbial analysis of the soil samples, freshwater samples or the ecosystems of marine water (Nogales et al. 2011; Schuster 2007; Wang et al. 2012).
A number of previous studies have reported the structure and composition of microbial communities from various wastewater drains around the world (Ajmal et al. 2021; Balcom et al. 2016; Meerburg et al. 2016), but the current study is the first report about the microbial diversity analysis of Hudiara Drain wastewater by using culture-independent (Illumina sequencing) as well as culture-dependent approaches. We have also characterized bacterial strains with respect to heavy metals and antibiotics resistance.
Materials and methods
Sampling of wastewater from Hudiara Drain
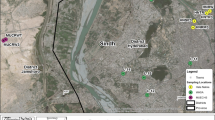
The sites chosen for sampling the wastewater of Hudiara Drain were around Gajju Matta Bridge, Lahore, Pakistan, where several other drains open and fall into the drain (Fig. 1A, B). The wastewater of Hudiara Drain was sampled from four sites with an average 5 feet depth (3 replicates from each site), each approximately 1 km apart from the other, along the length of the drain. For sample collection, grab sampling technique was followed and sterile ISO GL 45 Max 140 °C blue-capped bottles were used. The samples were immediately carried to Department of Life Sciences Laboratory at Forman Christian College University, Lahore, and filtered with sterile Whatman filter paper (11 µm pore size) to remove small rocks, dead leaves or other debris. The filtered water samples were then preserved at 4 °C until proceeded with further research work.

A map of sampling site (Hudiara Drain), B sampling site of Hudiara Drain at the Gajju Matta Bridge
Physicochemical properties of wastewater
The temperature and pH of wastewater samples were measured immediately after sampling. Biological oxygen demand (BOD) was calculated by means of a BOD meter over 5 days period and chemical oxygen demand (COD) was calculated using COD equation and simple titration method (Delzer and McKenzie 2003). The initial dissolved oxygen (DO) content was measured at 0 days post sampling. After 5 days, the DO content was measured again and BOD was calculated using the following equation.
In order to determine the COD of the samples, 50 ml of wastewater was added to refluxing flask of 500 ml and 1 g of HgSO4 was added along with several glass beads and 5 ml 98% concentrated sulfuric acid, and gently mixed. The solution was cooled down to room temperature and titrated against K2Cr2O7 with ferrous ammonium sulfate while ferroin was used as indicator. The color change from blue green to red was considered as end point. In the same way, a blank was also refluxed and titrated but with the same volume of distilled water as there was of wastewater sample (Lapage et al. 1970). The following equation was then used to calculate COD in mg/L:
where; A is the volume of ferrous ammonium sulfate used for blank, B is the volume of ferrous ammonium sulfate used for sample, M is the molarity of ferrous ammonium sulfate.
Metagenomic DNA isolation from Hudiara Drain wastewater
The metagenomic DNA from filtered wastewater samples, labelled as HD2D, HD7D, HD10D and HD14D, was isolated by using FastDNA® Spin Kit (MP Biomedicals) and the FastPrep® Instrument. The quality of DNA was determined by using 0.9% agarose gel and DNA quantity was measured by using Nanodrop technology (NanoDrop 200c Thermo Scientific, USA).
Amplification of 16S rRNA gene and Illumina sequencing
The V3-V4 region of the 16S rRNA gene was amplified by using primers Bakt_341F and Bakt_805R, which were linked with unique identifiers and adapter sequences (Table 1). The detailed PCR conditions for amplicon sequencing were the same as described previously by Herlemann et al. (2011). Amplified PCR products were purified with Agencourt AMPure beads (Beckman Coulter, Brea, CA). Finally, about 10 ng of DNA from each environmental sample was sequenced on the Illumina MiSeq platform by Macrogen Korea, (Seoul, South Korea).
Bioinformatics and statistical analyses
Sequences were processed and sorted using the default parameters in e QIIME 2 (V. 2018.8.0) software package (Caporaso et al. 2010). An offset of 10 nucleotides was set in order to remove the first 10 bases of each sequence, and high quality sequences with an average length of 350 bases were selected. Chimera Slayer software was used to check chimeric sequences (DeSantis et al. 2006). De novo OTU (operational taxonomic unit) picking was done to generate OTU files using UCLUST that is a default parameter of QIIME, with 97% sequence similarity and Ribosomal Database Project classifier was used to assign taxonomy (Wang et al. 2007). For each taxonomic level (phylum, class, order, family and genus) Good’s coverage was calculated by using 97% similarity cutoff (Good 1953).
Alpha and beta diversity were determined by using QIIME alpha_rerefaction.py and beta_diversity_through_plots.py commands respectively. Alpha diversity was calculated at a sequence depth of 82,152 reads per wastewater sample, as alpha diversity indices are correlated with the number of sequences. The selected maximum sampling depth corresponded to the minimum number of reads obtained from any of the remaining sequenced samples. Beta diversity was analyzed by using principal component analysis (PCA). A matrix was calculated using the weighted and unweighted UniFrac distances among samples at a sequence depth of 82,152 reads per water sample. This analysis showed the overall patterns of microbial diversity in different wastewater samples by using PAST 3.12 (Hammer et al. 2001).
To explain the differences in the composition of taxa inside the data matrix community, a heatmap (relative abundance matrix) was generated at class level using XLSTAT 7.0 software (Fahmy 2003). To detect the taxonomic classifications that were significantly abundant in wastewater samples, Wilkcoxon non-parametric rank-sum test and Linear Discriminant Analysis (LDA) using the LEfSe (LDA Effective Size) program was used (Segata et al. 2011). Retrieved 16S rRNA sequences datasets were deposited in NCBI GenBank under SRA accession numbers SRX9852485 to SRX9852488 (BioProject PRJNA563090).
Isolation of culturable bacteria
For the isolation of culture-dependent bacteria from the wastewater, four different media were used which were Nutrient agar (Lapage et al. 1970), Salmonella Shigella agar (Lennette and Balows 1985), Mannitol Salt Agar (Chapman 1943) and Eosin Methylene Blue agar (Levine 1918). About 100 µL of each dilution of wastewater ranging from 10–1 to 10–8 of each sample was spread on nutrient agar plates in triplicates and incubated at 30 °C for 24 h. For differential media, a two day enriched sample of Hudiara Drain water in nutrient broth was diluted from 10–1 to 10–8 and 100 µL of each dilution was spread on each of the differential media mentioned above. About 100 µL of non-enriched sample of the drain water was also spread on each agar medium. The plates were incubated at 30 °C for 24–48 h.
For isolation of heavy metal resistant bacteria, the drainwater was diluted from 10–1 to 10–8 and 100 µL inoculum was spread on 2, 5, 8 and 10 mM Pb2+ ion-rich nutrient agar (peptone 0.5%, beef extract 0.3%, NaCl 0.5%, agar 1.5%) in triplicates. The same amount of inoculum was spread on 1, 3, 5 and 8 mM Cr 3+ rich nutrient agar and on 5, 8, 10 and 20 mM AsO43+ ion rich nutrient agar.
Identification of bacterial isolates on the basis of 16S rRNA sequence
Genomic DNA was isolated from individual bacterial isolates (Winnepenninckx et al. 1993). For PCR amplification of 16S rRNA gene, universal forward primer FD1 (AGAGTTTGATCCTGGCTCAG) and universal reverse primer (rP1) (ACGGACTTACCTTGTTACGACTT) were used for the amplification. Denaturation temperature was 95 ℃ for 5 min followed by 35 cycles of 94 ℃ for 1 min, 57 ℃ for 1 min and 72 ℃ for 1 min and final extension at 72 ℃ for 10 min. A reaction mixture of 25 μL was prepared by using Taq buffer (10X) 2.5μL, MgCl2 (25 mM) 3 μL, Taq polymerase 1 μL, dNTPs (2.5 mM) 2 μL, 2 μL of forward and reverse primer (10 pmol) and 2 μL of the template DNA (> 50 ng/μL) (Tan et al. 1997). These PCR products were purified and sequenced commercially by using universal forward and reverse primers (Eurofins, Germany).
Sequences of 16S rRNA gene were analyzed using Chromas 2.6 software (Technelysium Pty Ltd. Australia). Using National Center for Biotechnology Information, Basic Local Alignment Search Tool (NCBI BLAST), these sequences were compared with related sequences that were already deposited in the GenBank database. Sequences of 16S rRNA gene were aligned by using Clustal W 2.2 software and phylogenetic tree was constructed by using MEGA7 software with neighbor joining method (Kumar et al. 2016). All the parameters for phylogenetic tree construction were used according to Mukhtar et al. (2016). The sequences were submitted to NCBI GenBank under the accession numbers MN371798-MN371807.
Antibiotic resistance analysis
Antibiotic resistance analysis was done according to Kirby-Bauer disk diffusion method (Bauer et al. 1966; El-Sayed et al. 2016). Each strain was tested for resistance against four antibiotics including ofloxacin (OFX), trimethoprim (TMP), gentamicin (GN) and ciprofloxacin (CIP) by using disc diffusion method. The antibiotics were chosen based on the most used in the area. Each disc contained 5 µg of the antibiotic. About 100 μL of overnight grown bacterial culture was spread on nutrient agar. All antibiotic discs were placed on each of the seeded plates at appropriate distances from one another and plates were incubated at 30 °C for 24 h. The bacterial strains were classified as sensitive or resistant by the presence or absence of inhibition zone of growth around antibiotic discs.
Results
Physicochemical properties of wastewater
The pH and temperature of the wastewater were measured immediately after sampling from the site and recorded. The temperature range was found to be 33.45–34.27 ºC and the pH 7.42–7.98. The BOD of the wastewater samples ranged from 172 to 212 mg/L, whereas the COD of the Hudiara Drain wastewater samples ranged from 311 to 347 mg/L (Table S1).
Composition and diversity of microbial communities from wastewater of Hudiara Drain
The Illumina sequence analysis showed that a total of 62,942 sequences of 16S rRNA gene were obtained from 4 wastewater samples including HD2D, HD7D, HD10D and HD14D from Hudiara Drain. About 98.83% average sequences were related to domain Bacteria with highest number of sequences identified from the wastewater HD7D (98.8%) and the lowest from the wastewater HD10D (96.1%) and the average 2.17% sequences belonged to domain Archaea with highest number of sequences identified from the wastewater HD7D (3.8%) and the lowest from the wastewater HD10D (1.2%) (Fig. S1A). The maximum number of phyla (17) were identified from the wastewater sample HD10D and the minimum number of phyla (15) were detected from the wastewater sample HD14D. At the class level, RDP results suggested that a total of 48 bacterial and archaeal classes were detected in this study. The maximum number of classes (41) were identified from the wastewater sample HD2D and the minimum number of classes (36) from the wastewater sample HD14D. The results of sequence analysis at the genus level indicated that the maximum number of genera (183) were identified from the wastewater sample HD7D and the minimum number of genera (118) were detected from HD14D sample (Fig. S1B).
Diversity and richness analyses
The results for observed OTUs and alpha diversity pattern for microbial communities identified from the wastewater HD7D were variable as compared to other wastewater samples. In all samples, the Chao1 values were greater than the observed OTUs, indicating that more OTUs could be retrieved by additional sequencing (Fig. 2A, B). Beta diversity was analyzed on the basis of canonical corresponding analysis. Maximum dissimilarity coefficients were estimated between the microbial communities identified from the wastewater HD2D and HD10D while the lowest dissimilarity coefficient was observed between HD2D and HD14D (Fig. 2C).

A Alpha diversity analysis by calculating number of observed OTUs and B Shannon index. All the wastewater samples showed a significant difference after a Mann–Whitney test with a confidence level of 99% (p ≤ 0.05) in each index and C Beta diversity index of the bacterial and archaeal communities identified from all the wastewater samples. Beta diversity was analyzed on the basis of unweighted PCoA UniFrac distances
Microbial diversity analysis at the phylum and class level
The OTUs from all the wastewater samples were assigned to 17 bacterial and 2 archaeal phyla. The bacterial phyla including Proteobacteria (21.45–28.29%), Actinobacteria (20.11–27.71%), Firmicutes (4.85–7.93%) and Bacteroidetes (2.15–5.27%) were found to be abundant in all wastewater samples (Fig. 3). Sequences related to Thaumarchaeota, Chloroflexi, Gemmatimonadetes, Fusobacteria, Deinococcus-Thermus, Acidobacteria, Nitrospirae, Planctomycetes, Cyanobacteria/Chloroplast and Euryarchaeota were relatively less abundant but detected from all the wastewater samples. Sequences belonging to Euryarchaeota were detected only from the wastewater sample HD10D (Fig. 3). Microbial communities identified from the wastewater sample HD10D showed a great variation as compared to microbial communities identified from other wastewater samples.

Relative abundance of bacterial and archaeal phyla from all the wastewater samples
A class-level analysis, sequence analysis revealed a total of 41 diverse classes (Fig. S2). A total of 52 classes were identified in this study with the highest abundance of Actinobacteria (29.58%) followed by Alphaproteobacteria (12.07%), Bacilli (6.59%) and Betaproteobacteria (5.47%). Sphingobacteriia, Deltaproteobacteria, Gammaproteobacteria, Nitrososphaeria, Planctomycetia, Acidobacteria_Gp4, Acidobacteria_Gp6 and Clostridia were the less dominant classes identified from all the wastewater samples (Fig. S2).
Microbial diversity analysis at genus level
On genus level, a total of 279 diverse genera were obtained from all four wastewater samples. There were 49 phylotypes that were commonly detected from all the wastewater samples and 7, 34, 21 and 11 phylotypes exclusive to microbial diversity identified from wastewater samples HD2D, HD7D, HD10D and HD14D respectively (Fig. 4A). The relative abundance of bacterial and archaeal genera from all the wastewater samples were compared in the form of a heatmap. Bacterial genera included Bacillus, Aridibacter, Nitrospira, Gemmatimonas, Pseudomonas, Aquicella, Steroidobacter, Stenotrophomonas, Ramlibacter, Polyangium, Myxococcus, Geminicoccus, Azospirilum, Skermanella, Nitrososphaera, Ochrobactrum, Pontibacter, Pirelulla and Zavarzinella were commonly found in all wastewater samples (Fig. 4B).

A Venn diagram showing the numbers of bacterial and archaeal genera. B Abundance of bacterial genera identified from wastewater samples of Hudiara Drain
Enumeration of culturable bacterial population
The value of colony forming units (CFU) was higher for bacteria isolated on nutrient medium as compared to bacterial isolates grown on differential media. Using nutrient medium, the highest CFU was observed in 0DE-Hudiara Drain water sample, whereas using differential media, the highest CFU was calculated in 5 mM Cr3+ supplemented nutrient agar (Table 2; Fig. S3 and S4).
Antibiotic resistance profile of bacterial isolates
More than 40% of bacterial isolates showed resistance against gentamicin and trimethoprim (Figs. 5 and S5). Two Enterobacter isolates 7D2C3 and 3D2C3 and one Bacillus isolate MSA2 showed positive results for ofloxacin and one Enterobacter isolate 7D2C3 and one Klebsiella isolate 14D1C2 showed resistance against ciprofloxacin (Fig. S5).

Antibiotic resistance ability of bacterial isolates from the Hudiara Drain wastewater using disc diffusion method, TMP trimethoprim, OFX ofloxacin, CIP ciprofloxacin, GN gentamicin
Phylogenetic analysis of culturable bacterial isolates
Based on heavy metal and antibiotics resistance, 10 bacterial isolates were selected for molecular characterization and identification through 16S rRNA gene sequence analysis. Three isolates EMB2, 7D2C3 and 3D2C3 were identified as Enterobacter ludwigii and isolate 7D2C2 was identified as Enterobacter cloacae (Fig. 6). Two bacterial isolates MSA2 and MSA3 were related to Bacillus licheniformis and isolate Pb1 was belonging to Bacillus pumilus. Isolate EMB1 was identified as Salmonella enterica, 14D1C2 was identified as Klebsiella pneumoniae and Cr1 was identified as Serratia liquefaciens (Fig. 6).

Phylogenetic tree based on 16S rRNA gene sequences of bacterial isolates from the Hudiara Drain wastewater samples. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches
Discussion
Microbial diversity associated with water systems is a key indicator to the impact of water pollutants. These microbial communities have evolved distinct biochemical and genetic properties that help them to survive under polluted environments. The diversity of bacterial and archaeal communities in the wastewater is owed to the presence of various industrial and municipal effluents (Haller et al. 2011). The main objective of the current study was to analyze microbial communities associated with Hudiara Drain wastewater by using culture-independent (Illumina® MiSeq sequencing) as well as culture-dependent approaches. Heavy metals and antibiotic resistance abilities of bacterial isolates were also analyzed.
Metagenomic analysis of the 16S rRNA gene described that overall 97.83% of retrieved sequences were assigned to the domain Bacteria and 2.17% sequences to the domain Archaea. At the phylum level, microbial communities from wastewater HD10D (17 phyla) were found to be more diverse as compared to microbial communities associated with other wastewater samples of Hudiara Drain. Bacterial phyla Actinobacteria, Proteobacteria, Bacteroidetes, Firmicutes, Acidobacteria, Chloroflexi, Planctomycetes, and Fusobacteria and archaeal phylum Thaumarchaeota were commonly identified from all the wastewater samples. Previous studies on microbial diversity from sewage and wastewater samples of drains also reported that Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes were the most dominating phyla (Zhao et al. 2014; Balcom et al. 2016; Nascimento et al. 2018).
The highest number of 16S rRNA sequences identified in this study were observed to belong to Proteobacteria that are Gram-negative and constitute major human pathogens as well as heavy metal resistant bacteria. Gammaproteobacteria, the most abundant class in Proteobacteria, comprise medically and ecologically important bacteria such as members of Enterobacteriaceae and Pseudomonadaceae (Yang et al. 2011). Bacterial genera including Pseudomonas, Enterobacter, Azospirillum and Serratia identified in this study have also been reported previously to be abundant polluted environments. These bacteria have the potential for plant growth promotion and have an important role in bioremediation of contaminated environments including wastewaters and polluted soils (Ali et al. 2021; Kang et al. 2020).
Among Alphaproteobacteria, various genera including plant symbionts such as Bosea, Rhizobium and Rhodobacter, phototrophs, several arthropod endosymbionts and a number of intracellular pathogens were also detected in this study. A number of bacterial pathogenic groups, such as Rickettsiaceae, Anaplasmataceae and Midichloriaceae were also previously identified from polluted water and soils (Da Silva et al. 2015; Stiborova et al. 2015). Members of Betaproteobacteria such as Alcaligenes, Achromobacter, Delftia, Massilia, Comamonas, Polynucleobacter, Sphaerotilus, Variovorax, Acidovorax, Rubrivivax, Rhodoferax, Nitrosomonas, Sterolibacter and Neisseria identified from Hudiara Drain water were mostly human pathogenic bacteria and underground water dwellers (Lang et al. 2018). Deltaproteobacteria, such as Nitrospina, Desulfocaldus, Nautilia, Thioreductor and Desulfarculus are characterized by two terminally distinct groups of obligate anaerobes, that are known to grow on sulfur-rich soils, and obligate aerobes consisting of bdellovibrios capable of predation of other prokaryotes (Cheng et al. 2014).
The second highest number of sequences belonged to the phylum Actinobacteria, a phylum characterized by its aquatic or terrestrial members that are Gram-positive in nature. Actinobacteria including Arthrobacter, Streptomycetes and Zavarzinella were abundant in HD2D and HD7D drain samples. Arthrobacter is an obligate aerobic genus predominating soil and wastewater with ability to accumulate chromium and lead (Albuquerque et al. 2011). Streptomyces is a high GC-content genus attributed to be the largest genus of Actinobacteria and is distinguished because of its “earthy” odor as it is predominantly found in decaying vegetation and soil. Streptomycetes are known to contain a complex system of secondary metabolism and are industrially important because of their production of almost 2/3rd of clinically important antibiotics including ampicillin, cypemycin, streptomycin and neomycin (Kieser et al. 2000). Actinobacteria are capable of digesting polysaccharides like cellulose, by which they become a crucial part of the carbon cycle (Meerbergen et al. 2017; Płaza et al. 2021).
The 3rd highest number of sequences belonged to Firmicutes especially members of bacterial genera Bacillus and Clostridium. Bacilli have the ability to produce phytohormones and biocontrol agents as part of their action to promote plant growth under heavy metal stressed environments (Ali et al. 2021; Nascimento et al. 2018). Clostridia are classified as obligate anaerobes and some are potentially pathogenic in nature (Cheng et al. 2014).
Members of Chloroflexi constitute a functionally diverse group that are aerobic thermophiles or anoxygenic phototrophs (Brenner et al. 2005). Nitrososphaera was identified as the most abundant genus belonging to archaea that is responsible for oxidizing ammonia (Tourna et al. 2011). Acidobacteria Gp6 and Gp3, subgroups of Acidobacteria were also observed to be present in the samples. These subgroups of Acidobacteria are known to dominate grassland soils (Naether et al. 2012; Wang et al., 2012). Gp3 is the only Acidobacterial subgroup that holds strains that are culturable whereas Gp6 is among the genera of Acidobacteria which can proliferate in low pH environments along with Gp1 and Gp4 (Naether et al. 2012). Gemmatimonas is a phosphate solubilizing strain that has been previously reported from various environments and sinks for reduction of nitrous oxide, as they contain the enzymes nitrous oxide reductases (Naether et al. 2012).
Microbial diversity analysis through culture-dependent approaches showed that bacterial genera including Enterobacter, Bacillus, Salmonella, Klebsiella and Serratia were found to be dominant in all the wastewater samples. A number of previous studies have already reported that Bacillus, Staphylococcus, Enterococcus, Salmonella and Klebsiella were dominant bacterial genera isolated from contaminated soils and wastewaters and supported the theory that large numbers of bacteria remain uncultured (Ajmal et al. 2021; Atashgahi et al. 2015; Płaza et al. 2021).
More than 49% of bacterial isolates showed resistance against heavy metals such as Cr and Pb. Some previous studies also reported that microbial diversity from polluted environments such as wastewater, contaminated soils and activated sludge samples included a variety of heavy metal resistant bacterial and archaeal strains (Balcom et al. 2016; Dickinson et al. 2019; Mukhtar et al. 2018). Most bacterial isolates showed resistance against gentamicin, trimethoprim and ofloxacin but only two isolates Enterobacter 7D2C3 and one Klebsiella 14D1C2 showed resistance against ciprofloxacin. Antimicrobial activity of bacterial isolates (Pseudomonas, Enterobacter, Klebsiella and Staphylococcus) from polluted environments revealed that these bacteria have resistance to different antibiotics, such as streptomycin, penicillin, tetracycline, kanamycin, vancomycin, erythromycin and chloramphenicol (Ali et al. 2021; Irawati et al. 2016).
Conclusion
In the presented study, microbial diversity of Hudiara Drain recovered using culture-dependent protocols was substantially lower than that obtained with the cultivation-independent method; thus, a combination of two approaches is required to provide a deeper knowledge of the microbial communities associated with habitats such as Hudiara Drain. The metagenomic analysis of 16S rRNA gene revealed that Proteobacteria, Actinobacteria, Firmicutes and Bacteroidetes were dominant phyla and a large fraction of unclassified bacteria. Dominant groups (classes and genera) detected in this study included α, β, γ and δ-Proteobacteria, Clostridia, Bacilli, Flavobacteria, Bacteroidia, Sphingobacteria, Cytophagia, Arthrobacter, Acidobacteria Gp3, Gp6, Gemmatimonas, Stenotrophomonas, Microvirga, Zavarzinella, Mesorhizobium and Gaiella. Majority of the bacterial genera were found to have a profound bioremediative capacity, heavy metal tolerance and pesticide/chemical degradation. These bacteria also have the ability to resist against different heavy metals and antibiotics. Heavy metals including Pb and Cr and antibiotics such as gentamicin, ofloxacin and trimethoprim resistant bacteria belonging to Enterobacter, Bacillus, Klebsiella and Serratia were found to be dominant in all the wastewater samples. This study revealed that Hudiara Drain is a source of diverse bacterial population and a promising source of efficient bacterial strains capable of assisting the research on bioremediation and sustainable agriculture.
References
Afzal S, Ahmad I, Younas M, Zahid MD, Khan MA, Ijaz A, Ali K (2000) Study of water quality of Hudiara drain. India-Pakistan. Environ. Int 26(1–2):87–96. https://doi.org/10.1016/S0160-4120(00)00086-6
Ajmal AW, Saroosh S, Mulk S, Hassan MN, Yasmin H et al (2021) Bacteria isolated from wastewater irrigated agricultural soils adapt to heavy metal toxicity while maintaining their plant growth promoting traits. Sustainability 13:7792
Albuquerque L, França L, Rainey FA, Schumann P, Nobre MF, da Costa MS (2011) Gaiella occulta gen. nov., sp. nov., a novel representative of a deep branching phylogenetic lineage within the class Actinobacteria and proposal of Gaiellaceae fam. nov. and Gaiellales ord. nov. Syst Appl Microbiol 34:595–599
Ali M, Waliat S, FarhanUlHaque M, Mukhtar S (2021) Antimicrobial activity of bacteria isolated from the rhizosphere and phyllosphere of Avena fatua and Brachiaria reptans growing in heavy-metal polluted environment. Environ Sci Pollut Res. https://doi.org/10.1007/s11356-021-15436-7
Alma’abadiGojoboriMineta ADTK (2015) Marine metagenome as a resource for novel enzymes. GPB 13:290–295
Atashgahi S, Aydin R, Dimitrov MR, Sipkema D, Hamonts K, Lahti L et al (2015) Impact of a wastewater treatment plant on microbial community composition and function in a hyporheic zone of a eutrophic river. Sci Rep 5:17284
Balcom IN, Driscoll H, Vincent J, Leduc M (2016) Metagenomic analysis of an ecological wastewater treatment plant’s microbial communities and their potential to metabolize pharmaceuticals. F1000Res 5:1881
Bauer AW, Kirby WMM, Sherris JC, Turck M (1966) Antibiotic susceptibility testing by a standardized single disk method. Am J Clin Pathol 45:493–496
Ben-Dov E, Shapiro OH, Gruber R, Brenner A, Kushmaro A (2008) Changes in microbial diversity in industrial wastewater evaporation ponds following artificial salination. FEMS Microbio Ecol 66:437–446
Bettiol W, Ghini R (2011) Impacts of sewage sludge in tropical soil: a case study in Brazil. Appl Environ Soil Sci. https://doi.org/10.1155/2011/212807
Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds) (2005) Bergey’s manual of systematic bacteriology, 2nd edn, vol 2 (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), Springer, New York
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Chapman GH (1943) Determination of the chromogenic property of Staphylococci. J Bacteriol 45:405
Cheng TW, Lin LH, Lin YT, Song SR, Wang PL (2014) Temperature-dependent variations in sulfate-reducing communities associated with a terrestrial hydrocarbon seep. Microbes and Environ 4:377–387. https://doi.org/10.1264/jsme2.ME14086
Da Silva MLB, Cantao ME, Mezzari MP, Ma J, Nossa CW (2015) Assessment of bacterial and archaeal community structure in swine wastewater treatment processes. Microb Ecol 70:77–87
Delzer GC, McKenzie SW (2003) Chapter A7. Section 7.0. Five-Day Biochemical Oxygen Demand (No. 09-A7. 0). US Geological Survey. https://doi.org/10.3133/twri09A7.0
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL et al (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Dickinson AW, Power A, Hansen MG, Brandt KK, Piliposian G, Appleby P, O’Neill PA, Jones RT, Sierocinski P, Koskella B, Vos M (2019) Heavy metal pollution and co-selection for antibiotic resistance: a microbial palaeontology approach. Environ Int 132:105117
El-Sayed MH, Arafat HH, Elsehemy IA, Basha M (2016) Optimization, purification and physicochemical characterization of curdlan produced by Paenibacillus sp. strain NBR-10. Biosci Biotechnol Res Asia 13(2):901–909
Fahmy T (2003) XLSTAT-Pro 7.0 (XLSTAT). Addinsoft, Paris
Farzadkia M, Bazrafshan E (2014) Lime stabilization of waste activated sludge. Health Scope 3:e16035
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrica 40:237–264
Haller L, Tonolla M, Zopfi J, Peduzzi R, Wildi W, Pote J (2011) Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (Lake Geneva, Switzerland). Water Res 45:1213–1228
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: Paleontological statistics software package for education and data analysis. Palaeont Electron 4:9–13
Handelsman J (2005) Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 69:195–195
Herlemann DP, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ et al (2011) Transitions in bacterial communities along the 2000km salinity gradient of the Baltic Sea. ISME J 5:1571–1579
Hu M, Wang X, Wen X, Xia Y (2012) Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour Technol 117:72–79
Irawati W, Yuwono T, Rusli A (2016) Detection of plasmids and curing analysis in copper resistant bacteria Acinetobacter sp. IrC1, Acinetobacter sp. IrC2, and Cupriavidus sp. IrC4. Biodiversitas 17:296–300
Jabbar M (2020) Spatial analysis of the factors responsible for waterborne diseases in rural communities located along the Hudiara Drain. Lahore. Pak Geo Rev 75(1):84–94
Kang SM, Asaf S, Khan AL, Lubna K, Mun BGA et al (2020) Complete genome sequence of Pseudomonas psychrotolerans CS51, a plant growth-promoting bacterium, under heavy metal stress conditions. Microorganisms 8:382
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces. Genetics, vol 291, John Innes Foundation, Norwich, p 397
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lang X-L, Chen X, Xu A-L, Song Z-W, Wang X, Wang H-B (2018) Variation of bacterial and archaeal community structures in a full-scale constructed wetlands for wastewater treatment. Archaea 2018:9319345. https://doi.org/10.1155/2018/9319345
Lapage SP, Shelton JE, Mitchell T (1970) Methods in microbiology. Norris J, Ribbons D (eds), vol 3A, Academic Press, London
Lennette EH, Ballows A, Hausler WJ Jr, HS (1985) Manual of clinical microbiology. American Society for Microbiology, Washington, DC
Levine M (1918) Differentiation of B. coli and B. aerogenes on a simplified eosin-methylene blue agar. J Infect Dis 23(1):43–47
Meerbergen K, Van Geel M, Waud M, Willems KA, Dewil R, Van Impe J et al (2017) Assessing the composition of microbial communities in textile wastewater treatment plants in comparison with municipal wastewater treatment plants. Microbiologyopen 6:e00413
Meerburg FA, Vlaeminck SE, Roume H, Seuntjens D, Pieper DH, Jauregui R et al (2016) High-rate activated sludge communities have a distinctly different structure compared to low-rate sludge communities, and are less sensitive towards environmental and operational variables. Water Res 100:137–145
Mukhtar S, Mirza MS, Awan HA, Maqbool A, Mehnaz S, Malik KA (2016) Microbial diversity and metagenomic analysis of the rhizosphere of Para Grass (Urochloa mutica) growing under saline conditions. Pak J Bot 48:779–791
Mukhtar S, Mirza MS, Mehnaz S, Mirza BS, Malik KA (2018) Diversity of Bacillus-like bacterial community in the rhizospheric and non-rhizospheric soil of halophytes (Salsola stocksii and Atriplex amnicola) and characterization of osmoregulatory genes in halophilic Bacilli. Can J Microbiol 64:567–579
Naether A, Foesel BU, Naegele V, Wüst PK, Weinert J, Bonkowski M, Alt F, Oelmann Y, Polle A, Lohaus G (2012) Environmental factors affect acidobacterial communities below the subgroup level in grassland and forest soils. Appl Environ Microbiol 78:7398–7406
Nascimento AL, Souza AJ, Andrade PAM, Andreote FD, Coscione AR, Oliveira FC, Regitano JB (2018) Sewage sludge microbial structures and relations to their sources, treatments, and chemical attributes. Front Microbiol 9:1462
Nogales B, Lanfranconi MP, Piña-Villalonga JM, Bosch R (2011) Anthropogenic perturbations in marine microbial communities. FEMS Microbiol Rev 35:275–298
Paruch L, Paruch AM, Eiken HG, Sørheim R (2019) Aquatic microbial diversity associated with faecal pollution of Norwegian waterbodies characterized by 16S rRNA gene amplicon deep sequencing. Microb Biotechnol 12(6):1487–1491
Płaza G, Jałowiecki Ł, Głowacka D, Hubeny J, Harnisz M, Korzeniewska E (2021) Insights into the microbial diversity and structure in a full-scale municipal wastewater treatment plant with particular regard to Archaea. PLoS ONE 16(4):e0250514
Schuster SC (2007) Next-generation sequencing transforms today’s biology. Nat Methods 5:16
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett W, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Gen Biol 12:60
Shah F, Ahmad N, Zahid D, Masood K, Ahmad S (2010) The Hudiara Drain wastewater effect on the distribution of surrounding herbaceous vegetation. Pak J Bot 42:1745–1754
Stiborova H, Vrkoslavova J, Pulkrabova J, Poustka J, Hajslova J, Demnerova K (2015) Dynamics of brominated flame retardants removal in contaminated wastewater sewage sludge under anaerobic conditions. Sci Total Environ 533:439–445
Su LS (2016) Isolation and identification of heavy metal-tolerant bacteria from an industrial site as a possible source for bioremediation of cadmium, lead, and nickel. Adv Environ Biol 10:10–15
Tan ZY, Xu XD, Wan ET, Gao JL et al (1997) Phylogenetic and genetic relationships of Mesorhizobium tianshanense and related Rhizobia. Int J Syst Bacteriol 47:874–879
Thomson ISI (2003) Masil Khan, Hammad Naqi Khan and Hania Aslam. PJBS 6:167–175
Tourna M, Stieglmeier M, Spang A, Könneke M, Schintlmeister A, Urich T, Engel M, Schloter M, Wagner M, Richter A (2011) Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc Natl Acad Sci 108:8420–8425
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Wang X, Hu M, Xia Y, Wen X, Ding K (2012) Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in china. Appl Environ Microbiol 78:7042–7047
Winnepenninckx B, Backeljau T, de Wachter R (1993) Extraction of high molecular weight DNA from molluscs. Trends Genet 9:407–412
Yamin MT, Ahmad N (2007) Influence of Hudiara drain water irrigation on trace elements load in soil and uptake by vegetables. J Appl Sci Environ 11(2). https://doi.org/10.4314/jasem.v11i2.55029
Yang C, Zhang W, Liu R, Li Q, Li B, Wang S et al (2011) Phylogenetic diversity and metabolic potential of activated sludge microbial communities in full-scale wastewater treatment plants. Environ Sci Technol 45:7408–7415
Zhao J, Zhao X, Chao L, Zhang W, You T, Zhang J (2014) Diversity change of microbial communities responding to zinc and arsenic pollution in a river of northeastern China. J Zhejiang Univ Sci B 15(7):670–680
Acknowledgements
We are highly thankful to WWF-Pakistan for their help in sampling of wastewater from Hudiara Drain.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflict of interest in the publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Badar, A., Mukhtar, S., Mehnaz, S. et al. Microbial diversity analysis of Hudiara Drain wastewater using culture-independent and culture-dependent approaches. Environmental Sustainability 5, 65–76 (2022). https://doi.org/10.1007/s42398-022-00215-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42398-022-00215-y