
Abstract
In this work, nine novel thiazole derivatives of substituted benzenesulfonamide carboxylate were designed, synthesized and characterized (1H NMR, 13C NMR and Mass Spectra) for their possible development as antimalarial agents. All synthesized compounds were subjected to molecular docking, drug-likeness, ADMET properties and molecular dynamics studies by in silico methods using Biovia Discovery Studio (DS) 2020 software. The molecular docking study of all the synthesized compounds was carried out against Plasmodium falciparum cysteine protease falcipain 2 (FP-2, 3BPF) and falcipain 3 (FP-3, 3BPM) enzymes using the CDocker program of DS. Further, the best docked compound was studied by molecular dynamics simulation method followed by MM-PBSA calculation. In molecular docking studies, the synthesized thiazolyl benzenesulfonamides exhibited remarkable binding affinity against FP-2 and FP-3 enzymes. Molecular dynamics studies further confirmed the antimalarial potential of the compounds with the formation of well-defined and stable receptor-ligand interactions against both the falcipain enzymes. One derivative, ethyl 4-methyl-2-(4-methyl-2-(4-methylphenylsulfonamido)pentanamido) thiazole-5-carboxylate possesses promising inhibitory potential against both P. falciparum falcipain 2 and falcipain 3 enzymes. Based upon present findings, the thiazolyl benzenesulfonamide-5-carboxylates can be further evaluated for in vitro and in vivo antimalarial effectiveness towards possible development as antimalarial lead molecules and/ or potent antimalarial drug candidates.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Presently, the arsenal of antimalarial drugs is limited and needs to be fortified. The emergence of Plasmodium falciparum resistance to most quinoline and antifolate based antimalarial drugs threatened the gains in the treatment of malaria [1]. This lead to the development of artemisinin-based combination therapy [2, 3] but unfortunately, there is currently emerging report of artemisinin resistance [4, 5]. In the light of the present state of malaria infection and treatment, there is urgent need for the development of new chemotherapeutic agents to sustain the fight against malaria.
Heterocyclic compounds possessing nitrogen and sulphur heteroatom constitute the structural backbone of a number of biologically active compounds [6]. Among the heterocycles reported as an important class of medicinal compounds are the five-membered rings. Thiazole scaffolds are present in several chemotherapeutic agents such as sulfathiazole, ritonavir, tiazofurin and abafungin used as antimicrobial, antiretroviral, antineoplastic and antifungal agents respectively [7]. Recently, thiazoles have been reported as antimalarial agent. Sharma et al. [8] evaluated the antimalarial potential of two water soluble derivatives of nocathiacin against asexual blood stages of P. falciparum and found that one of the derivatives inhibited parasite growth with mean 50% inhibitory concentration of 51.55 nM. Sahu et al. [9] reported the antimalarial properties of thiazole-1,3,5-triazines using inhibition of P. falciparum dihydrofolate reductase (Pf-DHFR). Kalita et al. [10] also reported novel phenyl thiazolyl clubbed S-triazine derivatives as antifolate based antimalarial agent with 78% inhibition of parasite growth at 50 ug/mL. This report shows that thiazole ring is likely to improve on antimalarial property of compounds or possibly confer on molecules antimalarial potency.
Previous work in our group had shown further that sulphonamide functionality is a privileged antimalarial agent [11,12,13]. We have also observed that incorporating other functionalities with reported antimalarial properties improved the antimalarial potency of sulphonamide derivatives. The literature search has shown clearly that there is need for devoted research towards the discovery and development of new antimalarial agent that will utilize new mode of action so as to avoid easy development of resistance which happens to be the major problem of current antimalarial drugs including artemisinin combination therapy.Though biochemical characterization of four falcipains have been achieved, falcipain inhibitors research have been limited to falcipain-2/3 because of their hemoglobinase activity [14]. Falcipain-2 and falcipain-3 of P. falciparum represent the key enzymes in the life-cycle of the parasite. Both enzymes are involved in hemoglobin hydrolysis, an essential pathway to provide free amino acids for malaria parasite’s metabolic needs. In addition, falcipain-2 is involved in cleaving ankirin and band 4.1 protein, a cytoskeletal element essential for the stability of red cell membrane [15]. Inhibition of either of these enzymes blocks hydrolysis of hemoglobin and completion of the parasite developmental cycle. Other malaria parasites express close homologs of falcipain-2/3 proteases, suggesting that agents that target the falcipains will also be active against other human malaria parasites [16]. In this study, we explored the synergistic antimalarial properties arising from the successful incorporation of thiazoles to substituted benzenesulphonamides.
2 Experimental
2.1 Chemicals and Instruments
1-Hydroxybenzotriazole (HOBT), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), triethylamine (TEA), l-valine, l-leucine, l-alanine, ethyl acetoacetate, N-bromosuccinimide (NBS), were procured from Avra Chemical Ltd. Thiourea, 4-methylbenzene sulphonyl chloride, 4-nitrobenzene sulphonyl chloride, 4-bromobenzenesulphonyl chloride, benzenesulphonyl chloride, were procured from Sigma-Aldrich. 1H NMR and 13C NMR spectra were recorded on Advance 300 MHz, 400 MHz and 500 MHz spectrometers in CDCl3 and DMSO-d6 using TMS as internal standard. FT-IR spectra were recorded on Thermo Nicollet Nexus 670 spectrometer. Mass spectra were obtained on Agilent LCMS instrument. HRMS were measured on Agilent Technologies 6510, Q-TOFLC/MS ESI-Technique. Melting points were determined in open glass capillary tubes on a Stuart melting point apparatus and are uncorrected. All reactions were monitored using thin layer chromatography (TLC) on pre-coated silica gel 60 F254 (mesh); spots were visualized under UV light.
2.2 Synthesis of Compounds
2.2.1 Synthesis of Ethyl-2-Amino-4-Methylthiazole-5-Carboxylate (2a)
To a mixture of ethyl acetoacetate (1a, 1.0 g, 7.68 mmol) in water (7.69 mL) and THF (3.00 mL) below 0 °C was added NBS (1.64 g, 9.22 mmol, 1.20 equiv.). The reaction mixture was stirred at room temperature for 5 h and thin-layer chromatography (TLC, 20% ethyl acetate/hexane, on silica gel plate), showed disappearance of 1a. Thiourea (0.58 g, 7.68 mmol, 1.00 equiv.) was added and the reaction mixture was heated to 80 °C for 2 h. After cooling to r.t., the reaction mixture was filtered to get rid of the insoluble substance, then NH3.H2O (3.0 mL) was added to the filtrate. The resulting yellow floccules were stirred at room temperature for 10 min and filtered. The filter cake was washed with water (50 mL × 3) and recrystallized with ethyl acetate, then dried to give the target compound [17]
Yield (1.25 g, 87.5%), Pale yellow solid, M.p = 180–181 °C, Lit 178–179 °C.
1H-NMR (300 MHz, CDCl3) δ 5.55 (s, 2H, thiazole-2-NH2), 4.27 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.53 (s, 3H, thiazole-4-CH3), 1.33 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ 169.21(thiazole-2-C), 162.55 (C=O), 159.01(thiazole-4-C), 111.51(thiazole-5-C), 60.62 (OCH2CH3), 17.27(OCH2CH3), 14.44(thiazole-4-CH3), ESI–MS: m/z 187 [M + H]+.
2.2.2 Synthesis of Substituted Benzenesulfonamoyl Alkanamides (3a-i)
Appropriate substituted benzenesulfonyl chloride (1.82 mmol) was added in portions for 1 h to an aqueous solution of l-amino acid (1.5 mmol) containing sodium carbonate (Na2CO3, 1.82 mmol) at – 5 °C. The slurry formed was stirred at room temperature for 4 h (TLC (MeOH/DCM, 1:9 monitored). The mixture was acidified to pH 2 [13, 18]
2.2.3 Synthesis of Subsituted-4-Methyl-2-(3-Methyl-2-(4-Methylphenylsulfonamido)Butanamido)Thiazole-5-Carboxylate (4a–i)
To a solution of substituted benzenesulphonamoyl alkanamides (3a–l, 0.8 eq) in dichloromethane (DCM) (20 mL) was added triethylamine (TEA) (1.5 eq), EDC.HCl (1.2 eq), HOBT (1.0 eq) at O oC, after stirring for 15 min was added ethyl-2-amino-4-methylthiazole-5-carboxylate (0.8 eq). The resulting mixture was allowed to warm to room temperature and stirred for 18–24 h (monitored with TLC). On The completion of the reaction, the mixture was diluted with DCM, washed with Water (2X 50 mL), then the organic layer was washed with 1N HCl (50 mL), 5% NaHCO3 (50 mL), and brine solution (1 X 50 mL) and was dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was re-crystallized with methanol to obtain the target products [13, 19, 20].
Ethyl 4-methyl-2-(3-methyl-2-(phenylsulfonamido)butanamido)thiazole-5-carboxylate (4a) Yield (0.467 g, 70.8%), white solid, M.p = 244–245 °C.1H NMR (500 MHz, CDCl3) δ 9.73 (s, 1H, NH of amide), 7.85–7.83 (m, 2H, Ar–H), 7.52 (t, J = 7.4 Hz, 1H, Ar–H), 7.46 (t, J = 7.6 Hz, 2H, Ar–H), 5.58 (d, J = 7.9 Hz, 1H, SO2-NH), 4.34–4.30 (m, 2H, OCH2CH3), 3.78 (dd, J = 7.8, 5.6 Hz, 1H, CH-C=O), 2.64 (s, 3H, thiazole-4-CH3), 2.17 (dd, J = 12.6, 6.7 Hz, 1H, CH-(CH3)2), 1.36 (t, J = 7.1 Hz, 3H, OCH2CH3), 0.88 (d, J = 6.8 Hz, 3H, CH3), 0.81 (d, J = 6.8 Hz, 3H, CH3), 13C NMR (126 MHz, CDCl3) δ 169.26 (C=O of amide), 162.53 (thiazole-2-C), 158.08 (C=O), 156.39 (thiazole-4-C), 138.76, 133.32, 129.28, 127.31 (four aromatic carbons), 116.59 (thiazole-5-C), 62.33 (CH-C=O), 61.03 (OCH2CH3), 31.22 (CH-(CH3)2), 18.97 (CH3 of Val), 17.45 (CH3 of Val), 17.08 (thiazole-4-CH3), 14.37 (OCH2CH3), ESI–MS: m/z 426 [M + H]+. ESI–MS: m/z 426 [M + H]+. ESI-HRMS: calcd. For C18H23N3O5S2 [M + H]+ 426.1157; Found 426.1163.
Ethyl 4-methyl-2-(4-methyl-2-(phenylsulfonamido)pentanamido)thiazole-5-carboxylate (4b) Yield (0.52 g, 76.5%), white solid, M.p = 241–242 °C.1H NMR (400 MHz, CDCl3) δ 9.76 (s, 1H, NH of amide), 7.90–7.86 (m, 2H, Ar–H), 7.60–7.55 (m, 1H, Ar–H), 7.53–7.48 (m, 2H, Ar–H), 5.35 (d, J = 7.0 Hz, 1H, SO2-NH), 4.31 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.93 (dt, J = 8.9, 5.9 Hz, 1H, CH-C=O), 2.63 (s, 3H, thiazole-4-CH3), 1.64–1.60 (m, 1H, CH-(CH3)2), 1.50 (ddd, J = 13.1, 11.0, 5.4 Hz, 2H, CH2), 1.35 (t, J = 7.1 Hz, 3H, OCH2CH3), 0.82 (d, J = 6.3 Hz, 3H, CH3), 0.63 (d, J = 6.3 Hz, 3H, CH3)). 13C- NMR (101 MHz, CDCl3) δ 169.80 (C=O of amide), 162.60 (thiazole-2-C), 158.34 (C=O), 156.21 (thiazole-4-C), 138.64, 133.42, 129.28, 127.15 (four aromatic carbons), 118.98 (thiazole-5-C), 60.99(CH-C=O), 55.50 (OCH2CH3), 41.63 (CH2), 24.38(CH-(CH3)2), 22.74(CH3 of Leu), 21.06(CH3 of Leu), 17.07 (thiazole-4-CH3), 14.31(OCH2CH3). ESI–MS: m/z 440 [M + H]+. ESI-HRMS: calcd. For C19H25N3O5S2 [M + H]+ 440.1314; Found 440.1318.
Ethyl 4-methyl-2-(3-methyl-2-(4-methylphenylsulfonamido)butanamido)thiazole-5-carboxylate (4c) Yield (0.37 g, 75%), White solid, M.p = 235–236 °C.1H NMR (500 MHz, CDCl3) δ 10.20 (s, 1H, NH of amide), 7.68 (d, J = 8.2 Hz, 2H, Ar–H), 7.16 (d, J = 8.0 Hz, 2H, Ar–H), 6.18 (d, J = 8.4 Hz, 1H, SO2-NH), 4.32 (tt, J = 10.8, 5.3 Hz, 2H, OCH2CH3), 3.86–3.81 (m, 1H, CH-C=O), 2.67 (s, 3H, thiazole-4-CH3), 2.29 (s, 3H, CH3-Ar), 2.12 (dq, J = 13.4, 6.7 Hz, 1H, CH- (CH3)2), 1.36 (t, J = 7.1 Hz, 3H, OCH2CH3), 0.92 (d, J = 6.8 Hz, 3H, CH3), 0.88 (d, J = 6.7 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 169.95 (C=O of amide), 162.42(thiazole-2-C), 158.29 (C=O), 156.30 (thiazole-4-C), 144.06, 136.09, 129.68, 127.24 (four aromatic carbons), 116.42(thiazole-5-C), 62.41 (CH-C=O), 61.00 (OCH2CH3), 31.27(CH-(CH3)2), 21.42(CH3-Ar), 18.97(CH3 of Val), 17.81 (CH3 of Val), 16.98(thiazole-4-CH3), 14.35(OCH2CH3), ESI–MS: m/z 440 [M + H]+. ESI-HRMS: calcd. For C19H25N3O5S2 [M + H]+ 440.1314; Found 440.1320.
Ethyl 4-methyl-2-(4-methyl-2-(4-methylphenylsulfonamido)pentanamido)thiazole-5-carboxylate (4d) Yield (0.48 g, 94%), white solid, M.p = 230–231 °C.1H NMR (400 MHz, CDCl3) δ 9.67 (s, 1H, NH of amide), 7.75 (d, J = 8.3 Hz, 2H, Ar–H), 7.28 (d, J = 8.2 Hz, 2H, Ar–H), 5.23 (d, J = 7.0 Hz, 1H, SO2-NH), 4.32 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.90 (dt, J = 8.8, 5.9 Hz, 1H, CH-C=O), 2.63 (s, 3H, thiazole-4-CH3), 2.36 (s, 3H, CH3), 1.63–1.59 (m, 1H, CH-(CH3)2), 1.58–1.44 (m, 2H, CH2), 1.36 (t, J = 7.1 Hz, 3H, OCH2CH3), 0.83 (d, J = 6.3 Hz, 3H, CH3), 0.65 (d, J = 6.2 Hz, 3H, CH3). 13C-NMR (126 MHz, CDCl3) δ 169.83 (C=O of amide), 162.59 (thiazole-2-C), 158.42 (C=O), 156.40 (thiazole-4-C), 144.56, 135.60, 130.00, 127.43 (four aromatic carbons), 116.49 (thiazole-5-C), 60.98 (CH-C=O), 55.52 (OCH2CH3), 41.62(CH2), 24.44(CH3-Ar), 22.77 (CH-(CH3)2), 21.54(CH3 of Leu), 21.15(CH3 of Leu), 17.13(thiazole-4-CH3), 14.37 (OCH2CH3), ESI–MS: m/z 454 [M + H]+. ESI-HRMS: calcd. For C20H27N3O5S2 [M + H]+ 454.1470; Found 454.1468.
Ethyl 4-methyl-2-(2-(4-methylphenylsulfonamido) propanamido)thiazole-5-carboxylate (4e) Yield (0.38 g, 90.5%), white solid, M.p = 239–240 °C. 1H NMR (400 MHz, DMSO) δ 12.43 (s, 1H, NH of amide), 8.23 (d, J = 8.2 Hz, 1H, SO2-NH), 7.70–7.56 (m, 2H, Ar–H), 7.28 (d, J = 8.0 Hz, 2H, Ar–H), 4.31- 4.19 (m, 2H, CH2), 4.07 (p, J = 7.1 Hz, 1H, CH-C=O), 2.55 (s, 3H, thiazole-4-CH3), 2.28 (s, 3H, CH3-Ar), 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.19 (d, J = 7.1 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ 170.88(C=O of amide), 161.93 (thiazole-2-C), 158.94 (C=O), 155.97 (thiazole-4-C), 142.63, 137.54, 129.30, 126.45(four aromatic carbons), 114.26 (thiazole-5-C), 60.45 (OCH2CH3), 51.40 (CH-CH3), 20.74 (CH3-Ar), 18.36 (CH3), 16.89(thiazole-4-CH3), 14.13(OCH2CH3), ESI–MS: m/z 412 [M + H]. HRMS: calcd. For C17H21N3O5S2 [M + H]+ 412.1001; Found 412.0996.
Ethyl 4-methyl-2-(3-methyl-2-(4-Bromophenylsulfonamido)pentanamido)thiazole-5-carboxylate (4f) Yield (0.45 g, 75.6%), white solid, M.p = 233–234 °C.1H NMR (400 MHz, DMSO) δ 12.43 (s, 1H, NH of amide), 8.39 (d, J = 9.4 Hz, 1H, SO2-NH), 7.62 (d, J = 0.9 Hz, 4H, Ar–H), 4.24 (q, J = 7.0 Hz, 2H, OCH2CH3), 3.77 (dd, J = 9.2, 8.1 Hz, 1H, CH-C=O), 2.55 (s, 3H, thiazole-4-CH3), 1.89 (dd, J = 14.3, 6.8 Hz, 1H, CH- (CH3)2), 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 0.84 (d, J = 6.7 Hz, 3H, CH3), 0.79 (d, J = 6.7 Hz, 3H, CH3). 13C NMR (101 MHz, DMSO) δ 170.28 (C=O of amide), 162.48 (thiazole-2-C), 158.97 (C=O), 156.55 (thiazole-4-C), 140.08, 132.33, 129.06, 126.71 (four aromatic carbons), 114.93(thiazole-5-C), 62.01(CH-C=O), 60.97(OCH2CH3), 31.07(CH-(CH3)2), 19.16 (CH3 of Val), 18.86 (CH3 of Val), 17.49 (thiazole-4-CH3), 14.73 (OCH2CH3), ESI–MS: m/z 504 [M + H]+. ESI-HRMS: calcd. For C18H22N3O5S2Br [M + H]+ 504.0263; Found 504.0258.
Ethyl 2-(2-((4-bromophenyl)sulfonamido)-4-methylpentanamido)-4-methylthiazole-5-carboxylate (4g) Yield (0.31 g, 83.8%), white solid, M.p = 242–243 °C.1H NMR (300 MHz, DMSO) δ 12.57 (s, 1H, NH of amide), 8.43 (d, J = 8.8 Hz, 1H, SO2-NH), 7.64 (s, 4H, Ar–H), 4.25 (q, J = 6.9 Hz, 2H, OCH2CH3), 4.06 (dd, J = 14.3, 8.8 Hz, 1H, CH-C=O), 2.56 (s, 3H, thiazole-4-CH3), 1.61–1.36 (m, 3H, CH2 + CH-(CH3)2), 1.29 (t, J = 7.1 Hz, 3H, OCH2CH3), 0.85 (d, J = 6.4 Hz, 3H, CH3), 0.74 (d, J = 6.3 Hz, 3H, CH3). 13C-NMR (101 MHz, DMSO) δ 170.43 (C=O of amide), 161.89 (thiazole-2-C), 158.68(C=O), 155.94 (thiazole-4-C), 139.52, 131.79, 128.45, 126.16 (four aromatic carbons), 114.38 (thiazole-5-C), 60.38 (CH-C=O), 54.19 (OCH2CH3), 40.59 (CH2), 23.86 (CH-(CH3)2), 22.55 (CH3), 20.97(CH3), 16.90 (thiazole-4-CH3), 14.14 (OCH2CH3), ESI–MS: m/z 518 [M + H]+. ESI-HRMS: calcd. For C19H24N3O5S2Br [M + H]+ 518.0419; Found 518.0419.
Ethyl 4-methyl-2-(3-methyl-2-(4-nitrophenylsulfonamido)pentanamido)thiazole-5-carboxylate (4h) Yield (0.5 g, 79%), Off-white solid, M.p = 240–243 °C.1H NMR (400 MHz, DMSO) δ 12.49 (s, 1H, NH of amide), 8.71 (d, J = 9.3 Hz, 1H, SO2-NH), 8.31–8.16 (m, 2H, Ar–H), 8.01–7.89 (m, 2H, Ar–H), 4.28–4.16 (m, 2H, OCH2CH3), 3.82 (t, J = 8.5 Hz, 1H, CH-C=O), 2.51 (s, 3H, thiazole-4-CH3), 1.93 (dq, J = 13.6, 6.7 Hz, 1H, CH-(CH3)2), 1.32–1.22 (m, 3H, OCH2CH3), 0.87 (d, J = 6.7 Hz, 3H, CH3), 0.80 (d, J = 6.7 Hz, 3H, CH3). 13C-NMR (101 MHz, DMSO) δ 170.02(C=O of amide), 162.30 (thiazole-2-C), 158.74 (C=O), 156.53 (thiazole-4-C), 149.63, 146.37, 128.73, 124.63 (four aromatic carbons), 114.80 (thiazole-5-C), 62.14 (CH-C=O), 60.99 (OCH2CH3), 30.99(CH-(CH3)2), 19.14 (CH3 of Val), 18.86 (CH3 of Val), 17.34(thiazole-4-CH3), 14.63(OCH2CH3).ESI–MS: m/z 471 [M + H]+. ESI-HRMS: calcd. For C18H22N4O7S2 [M + H]+ 471.1008; Found 471.1005.
Ethyl 4-methyl-2-(4-methyl-2-((4-nitrophenyl)sulfonamido)pentanamido)thiazole-5-carboxylate (4i) Yield (0.35 g, 76.1%), off-white solid, M.p = 237–238 °C.1H-NMR (400 MHz, DMSO) δ 12.62 (s, 1H, NH of amide), 8.74 (d, J = 8.7 Hz, 1H, SO2-NH), 8.43–8.13 (m, 2H, Ar–H), 8.10–7.80 (m, 2H, Ar–H), 4.29–4.18 (m, 2H, OCH2CH3), 4.17–4.08 (m, 1H, CH-C=O), 2.52 (s, 3H, thiazole-4-CH3), 1.63–1.46 (m, 2H, CH2), 1.41 (ddd, J = 13.7, 8.3, 5.6 Hz, 1H, CH-(CH3)2), 1.28 (t, J = 7.1 Hz, 3H, CH3), 0.86 (d, J = 6.5 Hz, 3H, CH3), 0.77 (d, J = 6.4 Hz, 3H, CH3). 13C-NMR (101 MHz, DMSO) δ 170.67(C=O of amide), 162.31 (thiazole-2-C), 159.02 (C=O), 156.52 (thiazole-4-C), 149.68, 146.43, 128.69, 124.67 (four aromatic carbons), 114.85 (thiazole-5-C), 60.98 (OCH2CH3), 54.89 (CH-C=O), 41.02 (CH2), 24.47(CH-(CH3)2), 23.10 (CH3), 21.56 (CH3), 17.37 (thiazole-4-CH3), 14.63 (OCH2CH3), ESI–MS: m/z 485 [M + H]. HRMS: calcd. For C19H24N4O7S2 [M + H]+ 485.1165; Found 485.1165.
2.3 Molecular Docking
Molecular docking studies were performed on Biovia Discovery Studio (DS) 2020 software (Dassault Systèmes BIOVIA, San Diego, USA). Prior to docking, ligands and proteins were prepared according to the standard protocol of DS software. The three dimensional structure of compounds were generated and their energy minimization was carried out using the CHARMM (Chemistry at Harvard Macromolecular Mechanics)-based smart minimiser. It performs 2000 steps of Steepest Descent followed by Conjugate Gradient algorithm with an RMSD gradient of 0.01 kcal/mol [21]. The x-ray crystal structures of FP-2 (PDB id: 3BPF) and FP-3 (PDB id: 3BPM) proteins were retrieved from the RCSB Protein Data Bank (http://www.rcsb.org/). The chain A of 3BPFand 3BPM determined at a resolution of 3.2 and 3.2 Å, respectively were used in the study [22, 23]. The protein molecules were prepared by necessary cleaning, removal water molecules and energy minimization following the standard protocol. The energy minimization was performed using CHARMM-based smart minimiser method at maximum steps of 200 with RMSD gradient of 0.1 kcal/mol [24].
A receptor grid was generated around the binding cavity (active sites) of the energy minimized protein molecule by specifying the key amino acid residues. The co-crystal ligand (active inhibitor) of the respective protein molecule was selected to define the amino acid residues for predicting the binding sites. For the generated receptor grid boxes, the binding site spheres were set with radius of 2.90 and 2.50 Å and x, y, z dimensions – 57.253734, – 0.877123, – 15.038906 and 6.754358, 13.667812, – 16.835423 for 3BPF and 3BPM, respectively [25, 26].
The validation of the docking protocol was done by re-docking the co-crystal ligand into the predicted binding sites of the target receptor protein followed by analyzing the binding modes of the test compound in the docked complex with comparison to the binding modes of the co-crystal inhibitor. The co-crystallized ligand was re-docked using flexible docking simulations into the original structure of the receptor molecule using all docking parameters to the software’s default values [25].
The docking was performed using simulation-based docking protocol CDocker of the DS 2020. CDocker uses a CHARMm-based molecular dynamics (MD) algorithm to dock compounds into the active site(s) of a receptor molecule. All docking and consequent scoring parameters used were kept at their default settings [27, 28]. CDocker energy (–) and CDocker interaction energy (–) of docked compounds were calculated. All docked poses were scored and ranked, and the best pose for each compound was selected. The binding affinities of docked compounds were predicted by analyzing interactions of receptor-ligand complexes. The binding modes of the best docked pose were analyzed with the help of 3D receptor-ligand complex. Different non-bonding interactions (hydrogen bonding, hydrophobic etc.) were also analyzed with the help of 2D diagram of receptor-ligand complexes. The docking results of test compounds were compared with that of the co-crystal ligands. The docking energies of the co-crystal ligands were used as reference ligands to evaluate the binding affinity of test compounds.
2.4 Drug-Likeness Studies
The molecular properties and drug-likeness parameters of the compounds, 4a–i were calculated based on theoretical approaches using DS 2020. Molecular properties such as log of octanol/water partition coefficient (LogP), molecular weight (MW), number of hydrogen bond acceptors (nHBAs), number of hydrogen bond donors (nHBDs) and molecular polar surface area (PSA) incorporated in Lipinski’s rule of five [29], and other physicochemical parameters like number of aromatic rings (nARs), number of rings (nRs) and number of rotable bonds (nRotBs) were also calculated [30]. The relatively higher lipophilicity of the target compounds could be accounted for the good antimicrobial activities due to the increased intracellular concentration.
2.5 ADMET Prediction
The ADME-Toxicity (ADMET) parameters were calculated using the ADMET descriptor protocol of DS 2020 software. Six mathematical models (aqueous solubility, blood–brain barrier penetration, cytochrome P450 (CYP) 2D6 inhibition, hepatotoxicity, intestinal absorption and plasma protein binding) were used for quantitative prediction of properties related to ADMET characteristics or pharmacokinetics (PKs) of drug molecules [31, 32].
2.6 Molecular Dynamics Simulation
The compound 4d showed the best binding affinity against the target proteins, 3BPF and 3BPM were further considered for molecular dynamics (MD) simulation study using DS 2020. The protein–ligand complexes generated from the binding affinity prediction analysis were subjected to MD simulation study along with the original crystal structure of the target proteins complexed with the co-crystal inhibitors. The protein–ligand complexes were initially cleaned and prepared using macromolecule tool of DS 2020. The complexes were solvated using explicit periodical boundary condition in a cubic box of water having a distance of 5 Å from the boundary. The system was neutralized by adding 0.15 M NaCl during the solvation process. The solvated systems were energy minimized (5000 steps steepest descent and 5000 steps conjugate gradient with energy RMSD gradient 0.01 kcal/mol), heated (20 ps) and equilibrated (500 ps) using ‘Standard Dynamic Cascade’ protocol of DS 2020. After that, 30 ns production was run in NVT ensemble at 300 K for the whole protein–ligand complexes, where snapshots were saved in every 2 ps. For the electrostatics calculations, the Particle Mesh Ewald (PME) method and to constrain bonds containing hydrogen the SHAKE algorithm were used with the time step of 2 fs. After completing the simulation, RMSD (root-mean-square deviation), RMSF (root mean square fluctuation), ROG (radius of gyration) were computed by taking the starting structure as a reference to evaluate the conformational changes of the protein–ligand complexes. Throughout the simulation period, the distances of different hydrogen bonds formed were also monitored and analysed. Finally, different non-bond interactions were also analysed from the average interaction of the protein–ligand complexes and compared with the interactions obtained from the starting structures [33,34,35].
2.7 Molecular Mechanics-Poisson Boltzmann Surface Area (MM-PBSA)-based Binding Free Energy Calculation
The MM-PBSA based calculation of binding free energy (ΔG) is one of the important parameters to estimate the binding affinity of a compound to a biological macromolecule or target as well as thermodynamic stability of the protein–ligand complex [36]. This technique provides a fast and accurate prediction of absolute binding affinity of a compound within the active binding site of a target protein in the form of binding free energy which is very important for stability and particular potency of the compound [37]. Hence after MD simulation, the binding free energies for each protein–ligand complex were calculated by using ‘Binding Free Energy—Single Trajectory’ protocol of DS 2020 with the MM-PBSA method. In the analysis, the binding free energies of all the generated conformations were calculated and finally, the average binding free energy (ΔG) was determined for each protein–ligand complex.
3 Results and Discussion
3.1 Synthesis
The thiazole hybrids reported in this work were synthesized as outlined in scheme 1 and 2. The compounds (2a) and (3a–i) were synthesized as reported by Meng et al. [17], Ugwu et al. [18] and Ezugwu et al. [13] as shown in the supplementary document. The compounds (4a-i) were synthesized through the condensation reaction of the substituted benzenesulphonamide (3a–i) and ethyl 2-amino-4-methylthiazole-5-carboxylate (2a) upon the addition of peptide coupling reagents EDC-HOBt and TEA. The elucidations of the structures were carried out using FT-IR, 1H-NMR, 13C-NMR, ESI–MS and ESI-HRMS analyses. In the 1H-NMR spectrum of compound 4a, the diagnostic peak at δ9.73 singlet represents N–H of amide. A multiplet peaks from δ 7.83 to 7.85, triplet peaks at δ 7.52 and another triplet peaks at δ 7.46 indicates aromatic protons while a doublet peaks at δ 5.58 is for N–H of sulfonamide part of the compound. In 13C-NMR spectrum, peak at δ169.26 is for carbonyl of amide, peaks at δ 162.53, 158.08 and 156.59 represent thiazole-2-carbon, carbonyl carbon of carboxylate and thiazole-4-Carbon respectively. Four peaks from δ 127.31 to 138.76 represent the aromatic carbons while peaks at δ 116.59 stands for thiazole-5-carbon and the peaks from 14.37 to 62.33 represent the aliphatic carbons. These were also supported with ESI–MS and ESI-HRMS spectra having peaks at m/z 426 and 426.1163 [M + H]+ respectively confirming the structure of compound 4a.

Ethyl-2-amino-4-methylthiazole-5-carboxylate (2a)

Synthesis of subsituted-4-methyl-2-(3-methyl-2-(4-methylphenylsulfonamido) butanamido)thiazole-5-carboxylate (4a-i)
3.2 Docking Simulation
The protein models (3BPF/E64, and 3BPM/Leu) were validated and used for the docking study. Prior to docking, the receptor grid models were generated and optimized in terms of binding site spheres to achieve predictive interactions between receptor molecules and test compounds (Fig. 1).

Receptor grid models A 3BPF and B 3BPM
The reference co-crystal ligands (E64 and Leu) were successfully re-docked to the predicted active sites of protein molecules (3BPF and 3BPM) with acceptable RMSD values of 3.529 and 3.081 Å, respectively. The validation study was performed in order to reproduce the results of ligand binding modes observed experimentally in protein–ligand complexes. Results of validation study confirmed experimental binding modes/conformations of co-crystal inhibitors, E64 and Leupeptin in the binding pocket of respective protein molecules, 3BPF and 3BPM, respectively with predictable protein–ligand interactions (Fig. 2).

Re-docked conformers of receptors, A 3BPF and B 3BPM
The protein–ligand docking was performed to predict the binding affinity of the test compounds as possible novel antimalarial agents with inhibitory activity against P. falciparum cysteine protease falcipain 2 (FP-2) and falcipain 3 (FP-3) enzymes. Molecular docking is a virtual tool intended to find the best binding orientation of small molecules bound to their target protein molecules. It is used to predict the binding affinity as well as biological efficacy of small molecules [14, 15]. Docking plays an important role in the identification of bioactive molecules based on the target protein structure in rational drug design and discovery program. In protein–ligand docking study, CDocker program successfully docked all the compounds, 4a–i into the binding pocket of protein molecules. Compounds could bind well with the pre-defined active site residues of the predicted receptor spheres. Higher binding affinities in terms of CDocker energy and CDocker interaction energy were observed for all the compounds as compared to the co-crystal inhibitor(s). All the nine compounds showed almost similar binding affinities with some degree of variations. Against P. falciparum FP-2 (3BPF), compound 4d, 4g and 4h exhibited comparatively better inhibitory activity than rest of the analogues. On the other hand, compound 4a, 4b, 4d–g were found to be more potent than the other compounds of the series against P. falciparum FP-2 (3BPF). Results of molecular docking are summarized in Table 1.
Among all nine compounds, the compound 4d was found exhibit highest binding affinity against both FP-2 and FP-3. The 3D poses of protein–ligand complexes revealed predicted binding affinity of compounds relative to the molecular orientation of receptor molecule(s). Further analysis of 2D interaction diagram indicates that polar hydrogen bonding interactions were primarily involved between receptor and ligand molecule along with some secondary interactions such as hydrophobic interactions. It was observed that well defined molecular interactions between binding site residues of receptor molecule(s) and complementary moieties/atoms of ligands. Higher the number of hydrogen bonds, higher is the binding affinity. Other non-bonded interactions like hydrophobic bonding were also observed, but to a lesser extent. The compound 4d exhibited well-defined protein–ligand interactions with very good binding modes as compared to rest of the compounds. The details of 3D binding modes and 2D interaction diagrams of compound 4d and co-crystal inhibitors (E64 and Leu) are displayed in Figs. 3 and 4.

Interactions with different amino acid residues at the active site of target proteins by the ligands after docking. A 4d with 3BPF- 3D, B 4d with 3BPF- 2D, C E64 with 3BPM -3D, D E64with 3BPM -2D. Dotted green lines indicate conventional H-bonding interactions

Interactions with different amino acid residues at the active site of target proteins by the ligands after docking. A 4d with 3BPM- 3D, B 4d with 3BPM- 2D, C Leu with 3BPM -3D, D Leu with 3BPM -2D. Dotted green lines indicate conventional H-bonding interactions
3.3 Drug-Likeness
The results of calculated molecular properties and predicted Lipinski’s parameters are displayed in Table 2. Results revealed that all the compounds, 4a–i showed good drug-like properties based on Lipinski’s rule of five with additional parameters of drug-likeness. All compounds obeyed ‘Lipinski’s rule of five’ and ‘Veber rule’. According to Lipinski’s rule, compounds are more likely to be drug-like and orally bioavailable, if they obey the following criteria: LogPo/w (octanol/water partition coefficient) ≤ 5, MW ≤ 500, nHBAs ≤ 10 and nHBDs ≤ 5 [21]. To further substantiate drug-likeness, Veber et al. stated that compounds with ≤ 10 RotB and molecular PSA of ≤ 140 A2 are more likely to show optimum membrane permeability and good bioavailability [22]. In our study, all the compounds exhibited satisfactory molecular properties and ‘Lipinski’s parameters. The LogP, MW, and molecular PSA indicate good membrane permeability, intestinal absorption and oral bioavailability, whereas, other parameters nHBAs, nHBDs, nR and Rotb bonds facilitate to produce well-defined drug-receptor interactions for optimal drug action [23].
3.4 ADMET Properties
The predicted ADMET data are presented in Table 3. Most of the compounds showed poor aqueous solubility. All the compounds exhibited poor blood–brain barrier (BBB) penetration indicating less probability of producing CNS toxicity. The compounds were found to be non-inhibitors of cytochrome P450 2D6 (CYP 2D6). The CYP 2D6 enzyme is one of the important metabolic enzymes involved in drug metabolism [21]. The predictive hepatotoxicity was observed for all the compounds. The intestinal absorption levels were observed in the range from good to very poor. Plasma protein binding (PPB) data revealed that all the compounds were highly protein bound.
The ADMET plot is displayed in Fig. 5.

ADME plot
3.5 MD Simulation
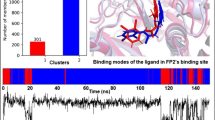
Molecular dynamics simulation was performed to observe the conformational changes and stability of the target proteins in the presence of the test compounds by considering their natural dynamic behaviour. The conformational changes and stability of the proteins were determined in the form of RMSD, RMSF and ROG of the protein–ligand complexes over the simulation period of 30 ns and compared with the control (protein-co-crystal inhibitor complex). After completion of simulation for 3BP F (Fig. 6), it was found that the RMSD values for 3BPF-E64 complex reached the plateau state earlier in comparison to the 3BPF-4d complex. The RMSF values indicated that the amino acid residues were fluctuated more in 3BPF-4d complex (up to 4.5 Å) after residue number 100, but in the case of 3BPF-E64 complex the fluctuations were limited to 2 Å for all the residues. The ROG values indicated that 3BPF-4d complex was not that much compact and tightly bound at the initial state and final state of the simulation in comparison to the 3BPF-E64 complex. All these information indicated that the 3BPF-4d complex was less stable than the 3BPF-E64 complex during the simulation period. Besides, the number of H-bonds formed during the simulation period was also less in 3BPF-4d than 3BPF-E64 complex. In the case of the other target protein i.e., 3BPM (Fig. 7), the 3BPM-4d complex was reached plateau state earlier in comparison to the 3BPM-Leu complex. Till 24 ns the RMSD values for 3BPM-4d was less than 3.5 Å, but after that it was increased and maintained at almost like the deviations of 3BPM-Leu complex. Regarding RMSF values of the amino acid complexes, the 3BPM-4d complex and the 3BPM-Leu complex showed almost similar fluctuations except from 170 to 200 amino acid regions where the 3BPM-4d complex showed significant fluctuations in comparison to the 3BPM-Leu complex. From the ROG values, it was found that the 3BPM-4d complex was less compact and very loosely formed in comparison to the 3BPM-Leu complex. Besides, the number of H-bonds formed within the simulation period was more in 3BPM-Leu than 3BPM-4d complex. All these facts support the formation of less stable complex by 4d in the active site of 3BPM. The deviation of H-bond distances formed during the MD analysis is presented in Fig. 8.

Different outcomes from MD analysis for 3BPF-ligand complexes; A RMSD values, B RMSF values and C ROG values

Different outcomes from from MD analysis for 3BPM-ligand complexes. A RMSD values, B RMSF values, C ROG value

Deviation of H-bond distances formed during the MD analysis. A 3BPF-E64, B 3BPF-4d, C 3BPM-Leu, D 3BPM-4d
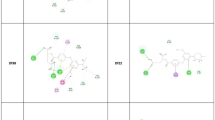
From the interactions generated after MD analysis (Figs. 3 and 4), it was found that E64 formed sixteen conventional hydrogen bond interactions with Gly40, Cys42, Trp43, Lys76, Asn77, Asn81, Gly82, Gly83, Asn87, Asn173 and Asn174; two carbon hydrogen bond interactions with Gly82 and Gly83; two hydrophobic interactions with Cys42 and Ala175 at the active site of 3BPF. On the other hand, the compound 4d formed seven conventional hydrogen bonds with Trp43, Gly82, Gly83, Leu172, Asn173 and His174; two carbon hydrogen bonds with Gly40 and His174; one hydrophobic interaction with Leu172 and one Pi-sulfur interaction with His174.
The leupeptin formed three conventional hydrogen bonds with Cys89 and Asp163; three carbon hydrogen bonds with Cys48, Cys51 and Asp163, and two hydrophobic interactions with Cys51 and His183 in the active site of 3BPM. On the other hand, the compound 4d formed four conventional hydrogen bond interactions with Gly92, Asn182 and Ala184; two carbon hydrogen bonds with Ser55 and Gly91; three hydrophobic interactions with Gly91, His183 and Ala184 and three other types of interactions (Pi-Cation, Pi-Sulfur) with Cys51, Tyr93 and His183. From these analyses, we observed that some interactions are common for both co-crystal inhibitors and compound 4d in the active sites of the target proteins, 3BPF and 3BPM which further indicated the possible inhibitory potential of 4d against FP-2 and FP-3, respectively.
3.6 MM-PBSA Binding Free Energies
The binding free energies (ΔG) of protein–ligand complexes indicate their binding affinity and thermodynamic stability and hence it is directly related to the potency of a compound. In this study, MM-PBSA based approach was used to calculate the ΔG of the all the conformations formed during the 30 ns simulation and finally average value was determined. After calculation, the average ΔG of the 3BPF-4d complex was found to be higher (– 78.477 kcal/mol) than the average ΔG of 3BPF-E64 complex (– 262.928 kcal/mol) which indicated the formation of a less stable complex by the compound 4d than the co-crystal ligand E64 in the active binding pocket of 3BPF. Similarly, the average ΔG for the 3BPM-4d complex was also to be higher (-97.939 kcal/mol) than the average ΔG of3BPM-Leu complex (-170.059 kcal/mol) which also indicated that the compound 4d formed less stable complex than the co-crystal ligand leupeptin in the binding site of 3BPM.
The analysis of docking and MD simulation studies exhibited that the basic molecular scaffold of (4-methyl-2-amidomethylsulfamoyl)thiazole-5-carboxylate was oriented in the binding cavity (active site residues) of receptor molecules. From 2D interaction diagram, the structural moiety could also occupy the binding sites of protein molecule through some hydrophobic interactions like pi-carbon and pi-pi interactions. Such interactions afforded good stability of the complex formed between proteins and ligands, particularly for compound 4d. Basic structural scaffold along with the amido and sulfonamido components from the chain residue interacted significantly with multiple amino acid residues by H-bonding and hydrophobic interactions. Strong H-bonding interactions between several active groups (NH, > CO =) of the amido (NH-CO) and sulfonamido (SO2NH) components and different amino acid residues (Trp43, Cys42, Gly82, Gly83, Leu172, Asn173 and His174) of FP-2 and FP-3 proteins were also observed. The sulfonamido moiety interacted stronger than the amido group with the formation of more number of H-bonds. Substituents such as methyl, pentyl and phenyl substituents also interacted with the different amino acid residues by non-hydrogen bonding interactions for compound 4d. Upon critical analysis protein–ligand-docking complexes, it was assumed that assumed that the basic structural moiety, (4-methyl-2-amido)thiazole-5-carboxylate plays a crucial role in binding affinity of newer thiazole-5-carboxylates. The presence of chain residues and various substituents enhances the binding affinity by increasing the binding strength through the formation of additional H-bonds or other interactions. The sulfonamido and amido components facilitated stronger binding of ligands with their corresponding receptor molecules. Finally, the molecular docking studies reveal that the synthesized compounds could possibly act by the inhibition of cysteine protease FP-2 and FP-3 enzymes in P. falciparum. Since the inhibitory activities against P. falciparumfalcipain enzymes were prominent in MD studies, the present thiazole-5-carboxylate derivatives can be further developed as possible anti-FP antimalarial agents. The compound 4d (Fig. 9) could serve as potent antimalarial lead with promising P. falciparum cysteine protease falcipain 2/3 inhibitory activity.

Compound 4d (4-methyl-2-(4-methyl-2-(4-methylphenylsulfonamido)pentanamido)thiazole-5-carboxylate) with potent anti-FP molecule
4 Conclusion
In this paper, nine novel thiazolyl benzenesulfonamide derivatives were designed, synthesized, characterized and studied for possible antimalarial potential by in silico methods. In molecular docking studies, the synthesized thiazolyl benzenesulfonamides exhibited remarkable binding affinity against P. falciparum cysteine protease falcipain 2 (FP-2) and falcipain 3 (FP-3) enzymes. Molecular dynamics studies confirmed the antimalarial potential of the compounds with the formation of well-defined and stable receptor-ligand interactions against both the falcipain enzymes. The derivative named ethyl 4-methyl-2-(4-methyl-2-(4-methylphenylsulfonamido)pentanamido)thiazole-5-carboxylate possesses promising inhibitory potential against both P. falciparum falcipain 2 and falcipain 3 enzymes. Based upon present findings, thiazolyl benzenesulfonamide-5-carboxylates can be further evaluated for in vitro and in vivo antimalarial effectiveness towards possible development as antimalarial lead molecules and/ or potent antimalarial drug candidates.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
References
Stepniewska K, White NJ (2008) Pharmacokinetics determinants of the window of selection for antimalarial drug resistance. Antimicrob Agents Chemother 52(5):1589–1596. https://doi.org/10.1128/AAC.00903-07
Nosten F, White NJ (2007) Artemisinin based combination treatment of falciparum malaria. Am J Trop Med Hyg 77(6):181–192
Rudrapal M, Chetia D (2021) Malaria and recent developments in antimalarial drugs. In: Neglected tropical diseases and phytochemicals in drug discovery, pp 499–542. https://doi.org/10.1002/9781119617143.ch21
Eastman RT, Fidock DA (2009) Artemisinin-based combination therapies: a virtual tool in efforts to eliminate malaria. Nat Rev Microbiol 7(12):864–874. https://doi.org/10.1038/nrmicro2239
Shukla M, Rathi K, Hassam M, Yadav DK, Karnatak M, Rawat V, Verma VP (2023) An overview on the antimalarial activity of 1, 2, 4-trioxanes, 1, 2, 4-trioxolanes and 1, 2, 4, 5-tetraoxanes. Med Res Rev. https://doi.org/10.1002/med.21979
Ugwu DI, Okoro UC, Ukoha PO, Gupta A, Okafor SN (2018) Novel anti-inflammatory and analgesic agents: synthesis, molecular docking and in vivo studies. J Enzyme Inhib Med Chem 33(1):405–415. https://doi.org/10.1080/14756366.2018.1426573
Ezugwu JA, Okoro UC, Ezeokonkwo MA, Hariprasad KS, Rudrapal M, Ugwu DI, Gogoi N, Chetia D, Celik I, Ekoh OC (2022) Design, synthesis, molecular docking, molecular dynamics and in vivo antimalarial activity of new dipeptide-sulfonamides. ChemistrySelect 7(5):e202103908. https://doi.org/10.1002/slct.202103908
Sharma I, Sullivan M, McCutchan TF (2015) In vitro antimalarial activity of novel semisynthetic nocathiacin I antibiotics. Antimicrob Agents Chemother 59(6):3174–3179. https://doi.org/10.1128/AAC.04294-14
Sahu S, Ghosh SK, Gahtori P, Pratap Singh U, Bhattacharyya DR, Bhat HR (2019) In silico ADMET study, docking, synthesis and antimalarial evaluation of thiazole-1,3,5-triazine derivatives as Pf-DHFR inhibitor. Pharmacol Rep 71(5):762–767. https://doi.org/10.1016/j.pharep.2019.04.006
Kalita JM, Ghosh SK, Sahu S, Dutta M (2017) Rational design and microwave assisted synthesis of some novel phenyl thiazolyl clubbed s-triazine derivatives as antimalarial antifolate. Futur J Pharm Sci 3(1):11–17. https://doi.org/10.1016/j.fjps.2016.09.004
Ugwuja DI, Okoro UC, Soman SS, Soni R, Okafor SN, Ugwu DI (2019) New peptide derived antimalaria and antimicrobial agents bearing sulphonamide moiety. J Enzyme Inhib Med Chem 34(1):1388–1399. https://doi.org/10.1080/14756366.2019
Ugwu DI, Okoro UC, Ukoha PO, Okafor S, Ibezim A, Kumar NM (2017) Synthesis, characterization, molecular docking and in vitro antimalarial properties of new carboxamides bearing sulphonamide. Eur J Med Chem 135:349–369. https://doi.org/10.1016/j.ejmech.2017.04.029
Ezugwu JA, Okoro UC, Ezeokonkwo MA, Bhimapaka C, Okafor SN, Ugwu DI, Ugwuja DI (2020) Synthesis and biological evaluation of Val-Val dipeptide-sulfonamide conjugates. Arch Pharm 353(7):e2000074. https://doi.org/10.1002/ardp.202000074
Marco M, Coterón JM (2012) Falcipain inhibition as a promising antimalarial target. Curr Top Med Chem 12(5):408–444. https://doi.org/10.2174/156802612799362913
Ettari R, Previti S, Di Chio C, Zappalà M (2021) Falcipain-2 and falcipain-3 inhibitors as promising antimalarial agents. Curr Med Chem 28(15):3010–3031. https://doi.org/10.2174/09298673276662007302153
Rosenthal PJ (2020) Falcipain cysteine proteases of malaria parasites: an update. Biochim Biophys Acta Proteins Proteom 1868(3):140362. https://doi.org/10.1016/j.bbapap.2020
Meng G, Wang M, Zheng A, Dou J, Guo Z (2014) Efficient one-pot synthesis of ethyl 2-substitued-4-methylthiazole-5-carboxylates. Green Chem Lett Rev 7(1):46–49. https://doi.org/10.1080/17518253.2014.895858
Ugwu DI, Ezema BE, Eze FU, Ugwuja DI (2014) Synthesis and structural activity relationship study of antitubercular carboxamides. Int J Med Chem 2014:614808. https://doi.org/10.1155/2014/614808
Ezugwu JA, Okoro UC, Ezeokonkwo MA, Bhimapaka CR, Okafor SN, Ugwu DI, Ekoh OC, Attah SI (2020) Novel Leu-Val based dipeptide as antimicrobial and antimalarial agents: synthesis and molecular docking. Front Chem 8:583926. https://doi.org/10.3389/fchem.2020.583926
Ezugwu JA, Okoro UC, Ezeokonkwo MA, Bhimapaka CR (2020) Synthesis of novel valine-based dipeptide carboxamide bearing benzene sulfonamide moiety as antimalarial agent. Commun Phys Sci. 5(2):176–197
Kalita J, Chetia D, Rudrapal M (2020) Design, synthesis, antimalarial activity and docking study of 7-chloro-4-(2-(substituted benzylidene)hydrazineyl)quinolines. Med Chem 16:928–937. https://doi.org/10.2174/1573406415666190806154722
Ghosh S, Chetia D, Gogoi N, Rudrapal M (2021) Design, molecular docking, drug-likeness, and molecular dynamics studies of 1,2,4-trioxane derivatives as novel Plasmodium falciparum falcipain-2 (FP-2) inhibitors. Biotechnologia 102(3):257–275. https://doi.org/10.5114/bta.2021.108722
Rudrapal M, Banu ZW, Chetia D (2018) Newer series of trioxane derivatives as potent antimalarial agents. Med Chem Res 27(2):653–668. https://doi.org/10.1007/s00044-017-2090-8
Ghorab MM, Soliman AM, Alsaid MS, Askar AA (2020) Synthesis, antimicrobial activity and docking study of some novel 4-(4, 4-dimethyl-2, 6-dioxocyclohexylidene) methylamino derivatives carrying biologically active sulfonamide moiety. Arab J Chem 13(1):545–556. https://doi.org/10.1016/j.arabjc.2017.05.022
Wu G, Robertson DH, Brooks CL, Vieth M (2023) Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J Comput Chem 24(13):1549–1562. https://doi.org/10.1002/jcc.10306
Abdel-Hamid MK, McCluskey A (2014) In silico docking, molecular dynamics and binding energy insights into the bolinaquinone-clathrin terminal domain binding site. Molecules 19(5):6609–6622. https://doi.org/10.3390/molecules19056609
Jain AN (2008) Bias, reporting, and sharing: computational evaluations of docking methods. J Comput Aided Mol Des 22(3–4):201–212. https://doi.org/10.1007/s10822-007-9151-x
Ponnan P, Gupta S, Chopra M, Tandon R, Baghel AS, Gupta G, Prasad AK, Rastogi RC, Bose M, Raj HG (2013) 2D-QSAR, docking studies, and in silico ADMET prediction of polyphenolic acetates as substrates for protein acetyltransferase function of glutamine synthetase of Mycobacterium tuberculosis. ISRN Struct Biol. 2013:1–12. https://doi.org/10.1155/2013/373516
Rudrapal M, Chetia D, Singh V (2017) Novel series of 1,2,4-trioxane derivatives as antimalarial agents. J Enzyme Inhib Med Chem 32(1):1159–1173. https://doi.org/10.1080/14756366.2017
Rudrapal M, Mullapudi S (2019) Design, synthesis, drug-likeness studies and bio-evaluation of some novel chalconeimines. Pharm Chem J 53(9):814–821. https://doi.org/10.1007/s11094-019-02084-y
Kashyap A, Chetia D, Rudrapal M (2016) Synthesis, antimalarial activity evaluation and drug-likeness study of some New Quinoline-Lawsone hybrids. Indian J Pharm Sci 78(6):892–911. https://doi.org/10.4172/pharmaceutical-sciences.1000186
Sharma D, Chetia D, Rudrapal M (2016) Design, synthesis and antimalarial activity of new 2-hydroxy-1,4-naphthoquinone-4-hydroxyanilino hybrid Mannich bases. Asian J Chem 28(4):782–788. https://doi.org/10.14233/ajchem.2016.19478
Lin CH, Chang TT, Sun MF, Chen HY, Tsai FJ, Chang KL, Fisher M, Chen CY (2011) Potent inhibitor design against H1N1 swine influenza: structure-based and molecular dynamics analysis for M2 inhibitors from traditional Chinese medicine database. J Biomol Struct Dyn 28(4):471–482. https://doi.org/10.1080/07391102.2011
Noha SM, Schmidhammer H, Spetea M (2017) Molecular docking, molecular dynamics, and structure-activity relationship explorations of 14-oxygenated N-methylmorphinan-6-ones as potent μ-opioid receptor agonists. ACS Chem Neurosci 8(6):1327–1337. https://doi.org/10.1021/acschemneuro.6b00460
Du QS, Huang RB, Wang SQ, Chou KC (2010) Designing inhibitors of M2 proton channel against H1N1 swine influenza virus. PLoS 5(2):e9388. https://doi.org/10.1371/journal.pone.0009388
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10(5):449–461. https://doi.org/10.1517/17460441.2015.1032936
Rastelli G, Del Rio A, Degliesposti G, Sgobba M (2010) Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem 31(4):797–810. https://doi.org/10.1002/jcc.21372 (PMID: 19569205)
Acknowledgements
Dr. James Anayochukwu Ezugwu expresses his sincere gratitude to The World Academy of Sciences (TWAS) and the Council for Science and Industrial Research (CSIR), New Delhi for the joint sponsorship of the experimental work at CSIR-India Institute of Chemical Technology, Hyderabad, Telangana, India under the 2017 CSIR-TWAS Postgraduate Fellowship Award (Grant/Award number: 22/FF/CSIR‐TWAS/2017).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
There is no conflict of interest with other people or organizations that could inappropriately influence or bias the content of the paper.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ezugwu, J.A., Okoro, U.C., Ezeokonkwo, M.A. et al. Design, Synthesis, Molecular Docking, Drug-Likeness/ADMET and Molecular Dynamics Studies of Thiazolyl Benzenesulfonamide Carboxylates as Antimalarial Agents. Chemistry Africa 7, 2353–2368 (2024). https://doi.org/10.1007/s42250-024-00904-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-024-00904-7