
Abstract
Epidermal growth factor receptor (EGFR) inhibitors have been proven as a high-potential therapeutic target for treating advanced non-small-cell lung cancer (NSCLC). However, many patients still suffer from drug-resistant mutations and drug side effects. The current study aims to investigate Osimertinib-based compounds inhibitory activity against double mutant EGFRL858R/T790M by computational methods of 3D-QSAR, molecular docking, and molecular dynamic (MD) simulations. CoMFA and CoMSIA approaches were used to create the 3D-QSAR models. Molecular docking and molecular dynamics simulation was performed to generate the binding mode and stability of the investigated inhibitors. The CoMFA model achieved good predictability with Q2 = 0.663, R2 = 0.978, SEE = 0.115, and an acceptable value for the coefficient of determination Rtest2 = 0.756. In addition, based on the information retained by the CoMFA contour maps, we proposed four new molecules (T1–T4) with significantly higher inhibitory activity. Furthermore, molecular docking and MD simulation analysis were utilized to confirm the 3D-QSAR results, supporting the stability of the proposed molecules in the 3W2O receptor. Finally, the selected molecules showed favorable pharmacokinetic properties and were non-toxic. The results provide significant information about the proposed Osimertinib-based compounds as potential H1975 Cell Line drugs that encourage further innovative experimental and clinical research.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The main target of clinical cures for NSCLCs is the transmembrane receptor tyrosine kinase epidermal growth factor receptor (EGFR), which belongs to the ErbB family. Until now, the third generation of EGFR inhibitors has been developed. Gefitinib [1] and Erlotinib [2] were the first reversible tyrosine kinase inhibitors and were quite effective in treating NSCLC. However, over 50% of first-generation EGFR patients developed a resistant mutation (T790M) in less than a year. To avoid the occurrence of resistance associated with the T790M mutation, a second EGFR inhibitor (Afatinib and Dacomitinib) was designed. However, these inhibitors have many adverse effects that limit their clinical use. The discovery of third-generation inhibitors reduced the toxicity of second-generation EGFR inhibitors such as WZ4002 [3], Rociletinib [4], and Osimertinib [5], as shown in Fig. 1.

The structures of the second line of EGFRL858R/T790M treatment drugs
In March 2017, the first and third generation of EGFR tyrosine kinases (Osimertinib) received complete Food and Drug Administration (FDA) approval to be second-line treatments for NSCLCs with EGFR-T790M [6]. The side effects of Osimertinib clinical use are nausea, diarrhea, hyperglycemia, and pneumonia [7, 8]. The severe side effects prompted the finding of a new practical, potent inhibitor of EGFRL858R/T790M, to exceed the defects of secondary effects and drug resistance that appeared with the use of Osimertinib. Several studies have been performed on the efficacy and the molecular mechanisms of Osimertinib and the influence of EGFR-TKIs as pretreatment on the efficiency of Osimertinib in patients with the EGFR T790M mutation [9, 10]. In addition, recent research examined the impact of different acrylamides on the activity and selectivity of Osimertinib [11], which indicates that Osimertinib analogues were more active and selective for the EGFR T790M mutant compared to Osimertinib. The results encourage the design of new potent Osimertinib analogues targeting the H1975 cell line. Compounds based on heterocyclic rings play a vital role in drug design and the development of potential drugs [12,13,14]. Pyrimidine [15], thiazole [16], quinoline [17], and imidazole derivatives [18] exhibit diverse biological activities, with pyrimidine being particularly significant in the synthesis of antitumor drugs, including EGFR kinase inhibitors [19, 20]. In recent studies, the pyrimidine scaffold has been explored as a novel 1H-pyrazolo[3,4-d] pyrimidine analog, exhibiting remarkable inhibitory action against EGFRL858R/T790M kinase by Gaber et al. [21]. Similarly, Hao et al. conducted research on a series of pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione congeners, which displayed specific EGFR inhibitory action [22]. Novel 2,4-diaryl pyrimidines were synthesized and evaluated by Jianheng et al. as selective EGFRL858R/T790M inhibitors, with the most promising substance exhibiting excellent kinase inhibitory action against EGFR double mutation and inhibiting the proliferation of cancer cells containing the EGFRL858R/T790M mutation [23].
The present research uses computational approaches: 3D-QSAR modeling, molecular docking, and molecular dynamic simulation (MD) to investigate new potent EGFRL858R/T790M kinase inhibitors. The 3D-QSAR study was utilized to develop a structure of 28 acrylamide derivatives compounds to prove the inhibition effect of EGFRL858R/T790M kinase. The stability of the four suggested compounds was determined using ligand-receptor interactions detected using molecular docking and dynamic simulation. Finally, the toxicity of new compounds was evaluated by studying ADMET properties.
2 Materials and Methods
2.1 Data Set
Based on previous studies, a dataset of N-(3-amino-4-methoxyphenyl) acrylamide derivatives (28 compounds) with antiproliferative action against H1975 cell lines was used in this study [24]. The 3D-QSAR models were produced by splitting the data into two sets: a training set of 23 compounds and a test set of 5 compounds. In the CoMFA and CoMSIA models, the activity values (IC50 in nM) were evolved into matching pIC50 values (−log IC50) and utilized as the primary dependent variable (Table 1).
2.2 Minimization and Alignment
Structural alignment is a capital step and the critical parameter in 3D-QSAR modeling [25,26,27]. The twenty-eight molecules were constructed with a sketch module and minimized using the corresponding Gasteiger–Huckel atomic partial charges under the standard Tripos force field [28] and 0.001 kcal/mol as the convergence criterion of the Powell gradient algorithm [29] on the SYBYL-X 2.0 program [28, 30]. The aligned data set is displayed in Fig. 2.

Aligned compounds of data set using compound 17i as a template
2.3 3D-QSAR Modeling
After the alignment step, several CoMFA and CoMSIA models were established to find a reliable model using SYBYLX 2.1. A hybridized sp3 carbon atom with a Van Der Waals radius of 1.52 was used in the CoMFA technique to compute the steric and electrostatic fields, with a default value of 30 kcal/mol defined for the energy cutoff calculations. The same grid was used for CoMFA approach, and the CoMSIA model was utilized to calculate additional fields (hydrophobic, H-bond donor, H-bond acceptor, steric, and electrostatic).
2.4 Y‑Randomization Test
To evaluate the proficiency of the created 3D-QSAR models, the Y-randomization study was put into use. The pIC50 values were arbitrarily shuffled and a novel 3D-QSAR model was produced after each change. Therefore, although we get low values for Q2 and R2, the original model is effective in predicting the activity of new inhibitors. Otherwise, the original model fails due to the overfitting problem of the training set.
2.5 Molecular Docking
Molecular docking is widely employed in drug discovery and understanding of molecular interactions. It allows to prevent the orientation and the optimal position of the ligand in the active site of the protein [31, 32]. In the present investigation, Surflex-Dock implemented in Sybyl 2.0 is used for molecular docking to study the modes of interaction between the ligands and the protein's active site. In practice, the protein was downloaded with a resolution of 2.35 Å from the RCSB protein database (PDB ID: 3W2O) [33]. Then, all of the water molecules in 3W2O were taken out and polar hydrogen atoms were inserted. Finally, the results were viewed with Discovery Studio 2017 [34] and PyMol software [35]. The re-docking protocol was utilized to validate the docking technique by superimposing the native co-crystallized ligand and its docked pose, then calculating the root mean square deviation (RMSD), which must be less than 2 Å [36].
2.6 Molecular Dynamics (MD) Simulations
Molecular dynamics simulations were carried out with the help of GROMACS package (GROMACS 2020.4) [37]. Using the CHARMM36 forcefield [38], the proposed molecules (T1, T2, T3, and T4) were operated in water for 100 ns. The system dissolves in the presence of TIP3P water molecules inside a truncated octahedral box. A quantity of potassium/chlorine ions was introduced to the combination to neutralize the entire system. In order to eliminate any steric conflicts, the convergence was attained within the maximum force 1000 (kJ mol−1 nm−1) for 5000 steps utilizing the Steepest Descent Method. To guarantee a wholly converged system for the production run, all systems were equilibrated at NVT and NPT ensembles for 100 ps (50,000 steps) and 1000 ps (1,000,000 steps), utilizing time steps of 0.2 and 0.1 fs, respectively. The simulations were run using the Parrinello–Rahman and weak coupling velocity-rescaling methods at a constant temperature of 300 K and a constant pressure of 1 atm or bar (NPT). The verlet technique was used to calculate non-bonded interactions. Using the Particle Mesh Ewald (PME) method, the electrostatic interactions and forces were calculated to account for a homogeneous medium outside the long-range limit. The complex's production took 100 ns.
2.7 Binding Energy Calculations
MM-PBSA binding energy of proposed compounds (T1–T4) was calculated for biomolecular interactions by the g-mmpbsa tool [39]. This tool uses the GROMACS and APBS packages to compute the enthalpy components of the MM-PBSA interaction.
2.8 In Silico Pharmacokinetics ADMET Study
Many candidate drugs do not go through clinical trials for ADMET properties (adsorption, distribution, metabolism, excretion, and toxicity) that are unacceptable for their toxicity or inefficiency [40, 41]. Several online tools that help predict ADMET properties, such as pkCSM [42] and SwissADME online tools [43], are employed to predict the ADMET properties of target-designed compounds.
3 Results and Discussions
3.1 3D-QSAR Models
The optimal CoMFA and CoMSIA models and numerous field combinations that showed cross-validated coefficient values greater than 0.5 are given in Table 2. The CoMFA model was chosen, characterized by the best statistical parameters (a significant value of the cross-validated coefficient Q2 and a correlation coefficient R2). Q2 = 0.663 characterized the CoMFA model, R2 = 0.978, N = 3, SEE = 0.115, and Rtest2 = 0.756, which indicates that this model has good stability and strong predictive ability. The statistical findings also indicate that the steric and electrostatic field contributions are 67% and 33%, respectively, suggesting that the steric field play a more important role in enhancing inhibitory activity. Figure 3 presents the correlation among predicted and observed pIC50 for training and test sets for the CoMFA analysis.

The optimal CoMFA model plots
3.2 3D-QSAR Contour Maps
The CoMFA contour maps have provided information on favorable and unfavorable regions of various fields, such as steric and electrostatic, in 3D space that can increase or decrease activity. Using molecule 17i as a template, the CoMFA steric and electrostatic contour maps were displayed in Fig. 4a, b.

CoMFA contour map of the steric field and electrostatic field with compound 17i
In the steric contour map of CoMFA (Fig. 4a), the yellow contour located on the pyrimidine group suggests that adding voluminous radicals at this site may decline the biological activity of compound 17i. The large green zone around methoxy and meta positions of the phenyl group shows the possibility of appropriate substitution and the addition of bulky radicals to enhance activity. At the same time, we see that the tiny green region surrounding the methyl radical on the indole group indicates the possibility of replacing the methyl group with another group to obtain the desired activity.
Likewise, in CoMFA electrostatic contour map (Fig. 4b), we can see that the contribution of the red contour is more dominant than that of the blue counterpart, suggesting that electropositive groups or atoms can decrease activity. In addition, the blue region near the phenyl fraction associated with piperidine indicates that electron-donation substitutes are favored at those positions that would exhibit good inhibitory activity. Furthermore, in the blue region near the pyrimidine group and the meta position of phenyl group, substituents with electron-donating properties are preferred at positions with good inhibitory activity.
3.3 Y-Randomization Test of Model
To confirm that the 3D-CoMFA model is not due to an accidental correlation of the training set, we used a y-randomization test. Numerous random interferences of the dependent variable (pIC50) were realized. Table 3 shows the low Q2cv (LOO), and R2train values attained, confirming that the CoMFA model is appropriate for predicting new inhibitors.
3.4 Designing of New Potent Inhibitors
Based on CoMFA contour maps on the structural characteristics of compound 17i, new analogues of N-(3-amino-4 methoxyphenyl) acrylamide were designed. The new compounds (T1, T2, T3, and T4) and their predicted activities are exposed in Table 4.
3.5 Molecular Docking Results
Molecular docking was carried out to understand the nature of the interactions between these new candidates (T1, T2, T3, and T4) in the active site of the Protein (PDB ID: 3W2O) and compare them with Osimertinib as EGFR inhibitors. The results are displayed in Table 5.
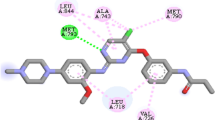
The docking result (Fig. 5) of compound 17i shows that the stability of this molecule is due to the diversity of types of interactions, such as the interaction carbon-hydrogen bond with Leu-788; Ala-743; Met-:793; Gln-791 residues, pi-alkyl interactions with Ala763; Ile759; Val726; Leu792; Lys745 residues, Pi-anion Glu762; Pi-sulfur Met766; Met790; and Pi-sigma with Leu718 residue.

Docking results of compounds 17i with EGFR tyrosine kinase protein (PDB ID: 3W2O)
In addition, docking of the Osimertinib compound shows a hydrogen bond with Leu-788, and Leu-718 residues, a Pi-alkyl with lys745, Ala743, Phe723 residues, and a single Pi-Sigma bond with Val726 residue. Moreover, molecular docking results show the importance of the residues Leu788; Leu718; Lys745; Val726; Ala743; Phe723; Met790; Met766, and Glu762 in the active site of EGFR kinase protein (3W2O).
Finally, all proposed molecular structures (T1–T4) show more exciting interactions and affinity (Table 6). Compared to third-generation EGFR Osimertinib, the newly suggested compounds have better inhibitory activity, making them suitable inhibitors.
3.6 Molecular Docking Validation
The co-crystallized ligand was removed from the protein (PDB ID: 3W2O) and re-docked in the same position to validate the reliability of the molecular docking technique. Figure 6 shows the superimposed view between the original ligand (red color) and the re-docked ligand (blue color) with root mean square deviation (RMSD) of 1.765 Å within reliable range of 2 Å. The docked ligand is connected at a comparable location to the co-crystallized ligand. Consequently, the ligands could interact with the same amino acid residues reported in the co-crystallized ligand. As a result, the docking validation outcomes demonstrate the efficiency and validity of the molecular docking technique.

Re-docking pose of the co-crystalized ligand and RMSD value of 1.765 Å (Red = Original, Bleu = Docked)
3.7 Molecular Dynamic Results
3.7.1 Root Mean Square Deviations RMSD
Using the GROMACS algorithm, RMSD was calculated for the complex (CT1, CT2, CT3, and CT4) based on “Backbone” atoms. RMSD graph (Fig. 7, Row 1) for the protein complex shows that the structure remained stable throughout the simulation time with some fluctuation within the range of ~ 1 Å, typical behavior of the globular Protein. The average value of backbone RMSD is around 2 Å for all four complexes. RMSD was calculated for the Ligand (T1, T2, T3, and T4) based on the Ligand's atoms using GROMACS program, which is displayed in (Fig. 7, Row 1). All the ligands (T1, T2, T3, and T4) remain bound throughout the simulation and have a stable RMSD.

From right to left: a RMSD, b RMSF, and c Radius of gyration of the complexes during 100 ns MD simulation. Compounds CT1 (Row 1), CT2 (Row 2), CT3 (Row 3) and CT4 (Row 4)
3.7.2 Root Mean Square Fluctuations RMSF
Except for specific residues that define a loop or turn in the 3W2O Protein, the RMSF was calculated using the “C-alpha” atoms of the GROMACS, where the fluctuation intensity remained below 3.0 (Fig. 7, Row 2).
3.7.3 The Radius of Gyration (ROG)
Using the GROMACS, Rog was determined for the complex (T1, T2, T3, and T4) based on "C-alpha" atoms the complex based on "C-alpha" atoms was determined. Indicating the stability and compactness of the structures, all four complexes (T1, T2, T3, and T4) display a very stable radius of gyration with a fluctuation of less than 1 Å. The minor opening and shutting of the N and C terminal domains during the MD simulation duration are indicated by the slight variation in the 1 Rog value (Fig. 7, Row 3).
3.7.4 Hydrogen Bonds (Protein–ligand)
Over 100 ns of the simulation time, the total number of hydrogen bonds formed between T1, T2, T3, T4, and 3W2O is shown in (Fig. 8, Row 1). All the ligands (T1, T2, T3, and T4) show a stable network of hydrogen bonding with the 3W2O, with an average number of hydrogen bonds during the simulation.

From right to left: A Hydrogen Bonds (Protein–ligand) and B Average distance between Ligand and the Protein for of the complexes during 100 ns MD simulation. Compounds CT1 (Row 1), CT2 (Row 2), CT3 (Row 3) and CT4 (Row 4)
3.7.5 Average Center-of-Mass Distance
The average Center-of-Mass Distance between (T1, T2, T3, and T4) and 3W2O during 100 ns of the simulation time are exposed in (Fig. 8, Row 2). From the COM-COM distance, none of the ligands (T1, T2, T3, and T4) leaves its binding site, and the distance either decreases or stabilizes after 40 ns to 60 ns of simulation time.
3.7.6 Contact Frequency (CF) Analysis
The contact Freq.TCL module in VMD, with a cutoff of 4 Å, was used to perform contact frequency (FC) analysis. The binding between the 3W2O and the test ligand (T1, T2, T3, and T4), where the residues with higher CF%, are presented Fig. 9. The residues with the highest contacts were Leu718, Phe723, Val726, Ala743, Lys745, Ile759, Met766, Leu788, Met790, Leu792, Met793, Arg841, Leu844, Thr854, Asp855, and Glu762.

Contact frequency (CF) analysis
3.7.7 Potential Energy, Pressure, and Temperature
The system potential energy, pressure, and temperature during 100 ns of MD simulation, as achieved from GROMACS edr file, are shown in Fig. 10. Throughout the 100 ns simulations, the graph shows the convergent potential energy, temperature, and pressure.

From left to right: A Temperature, B pressure, and C potential energy during the 100 ns MD simulations
3.7.8 MM/PBSA Binding Energy
The Molecular Mechanics/Poisson Boltzmann Surface (MM/PBSA) method was selected to re-score the binding free energies of the complexes T1, T2, T3, and T4, because it is the fastest force field-based method for calculating the binding free energy compared to other free energy calculation methods, such as the perturbation free energy (PFE) or thermodynamic integration (TI) methods. The calculated binding free energies are presented in Table 7.
3.8 ADMET Properties
To predict the ADMET properties of new compounds (T1–T4), pKCSM and SwissADME online tools were used, as listed in Table 8.
Molecules with less than 30% intestinal absorption are considered minimally absorbed. However, all new compounds have a better value than 89%, indicating good absorption in the human intestine. The volume of distribution (VDss) is regarded as high when it exceeds 0.45. Furthermore, blood–brain barrier (BBB) and central nervous system (CNS) permeability values are given (0.3 < Log BB < −1 and −2 < LogPS < −3, respectively). For a particular medicine, LogPS larger than −2 suggests CNS invasion, while LogPS < −3 indicates that it is challenging for the drug to enter the CNS. Molecules with logBB greater than −1 are thought to be widely distributed in the brain, but LogBB > 0.3 can cross the BBB. Therefore, the BBB permeability results indicate that the BBB is not penetrating for the four proposed molecules.
Cytochrome P450 (CYP) enzymes are a group of enzymes that have a major role in the metabolism of various compounds, including drugs, environmental chemicals and endogenous substrates. These enzymes are widely distributed in various tissues and organs. CYP3A4 is the most abundant CYP enzyme in the liver and is responsible for the metabolism of more than 50% of all drugs. The results show that all designed compounds are substrates and inhibitors to CYP3A4. A lower clearance index score denotes a drug's ability to persist in the body. The findings demonstrate that each compound under investigation has acceptable clearance index values, indicating its persistence in the body. Finally, the negative Ames toxicity test indicates that all compounds are not mutagenic.
4 Conclusion
The results of this research, which aim to investigate the inhibition potency of N-(3-amino-4-methoxyphenyl)-acrylamide derivatives (28 compounds) as EGFRL858R/T790M kinase inhibitors, showed excellent predictive power of the CoMFA model with good statistical results (Q2 = 0.663, R2 = 0.978, Rtest2 = 0.756). Four compounds (T1–T4) with solid inhibitory activity were designed according to CoMFA contour maps. In addition, the docking analysis of the designed compounds (T1–T4) revealed more types and interactions than Osimertinib as an EGFR inhibitor. A 100 ns MD simulation of the four newly designed molecules (T1–T4) showed small fluctuations of RMSD and RMSF, confirming the outcomes of the 3D-QSAR model and molecular docking. The results of ADEMT showed favorable pharmacokinetic properties and non-toxicity. These results will help optimize the discovery of new drugs that can address multidrug resistance. Further research can be carried out to synthesize and evaluate the in vitro activity of the new compounds generated as EGFRL858R/T790M kinase inhibitors.
Referencess
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T (2010) Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 362(25):2380–2388. https://doi.org/10.1056/NEJMoa0909530
Wang Y, Schmid-Bindert G, Zhou C (2012) Erlotinib in the treatment of advanced non-small cell lung cancer: an update for clinicians. Ther Adv Med Oncol 4(1):19–29. https://doi.org/10.1177/1758834011427927
Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Jänne PA (2009) Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462(7276):1070–1074. https://doi.org/10.1038/nature08622
Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, Martin TS, Karp R, Kalken D, Chaturvedi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Allen A (2013) Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M mediated resistance in NSCLC. Cancer Discov 3:1404–1415. https://doi.org/10.1158/2159-8290.CD-13-0314
Finlay MR, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, Brad-bury RH, Brown SJ, Butterworth S, Campbell A, Chorley C, Colclough N, Cross DA, Currie GS, Grist M, Hassall L, Hill GB, James D, James M, Kemmitt PGL (2014) Wrigley Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem 57:8249–8267
Ahmad I, Shaikh M, Surana S, Ghosh A, Patel H (2022) p38α MAP kinase inhibitors to overcome EGFR tertiary C797S point mutation associated with osimertinib in non-small cell lung cancer (NSCLC): emergence of fourth-generation EGFR inhibitor. J Biomol Struct Dyn 40(7):3046–3059
Jänne PA, Yang JCH, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, Ranson M (2015) AZD9291 in EGFR inhibitor–resistant non–small-cell lung cancer. N Engl J Med 372(18):1689–1699. https://doi.org/10.1056/NEJMoa1411817
Patel HM, Shaikh M, Ahmad I, Lokwani D, Surana SJ (2021) BREED based de novo hybridization approach: generating novel T790M/C797S-EGFR tyrosine kinase inhibitors to overcome the problem of mutation and resistance in non-smallcell lung cancer (NSCLC). J Biomol Struct Dyn 39(8):2838–2856. https://doi.org/10.1080/07391102.2020.1754918
Miyashita Y, Ko R, Shimada N, Mitsuishi Y, Miura K, Matsumoto N, Asao T, Shukuya T, Shibayama R, Koyama R, Takahashi K (2021) Impact of the generation of EGFR-TKIs administered as prior therapy on the efficacy of Osimertinib in patients with non-small cell lung cancer harboring EGFR T790M mutation. Thorac Cancer 12(3):329–338. https://doi.org/10.1111/1759-7714.13742
Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, Feeney N, Sholl LM, Dahlberg SE, Redig AJ, Kwiatkowski DJ, Rabin MS, Paweletz CP, Thress KS, Jänne PA (2018) Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol 4(11):1527–1534
Harun P, Iqrar A, Harsha J, Rahul P, Deepak L, Sanjay S (2022) Investigating the impact of different acrylamide (electrophilic warhead) on Osimertinib’s pharmacological spectrum by molecular mechanic and quantum mechanic approach. Comb Chem High Throughput Screen 25:149-166(18). https://doi.org/10.2174/1386207323666201204125524
Al-Mulla A (2017) A review: biological importance of heterocyclic compounds. Der Pharma Chem 9(13):141–147
Reutskaya E, Osipyan A, Sapegin A, Novikov AS, Krasavin M (2018) Rethinking hydrolytic imidazoline ring expansion: a common approach to the preparation of medium-sized rings via side-chain insertion into [1.4] oxa-and [1.4] thiazepinone scaffolds. J Org Chem 84(4):1693–1705
Othman IM, Alamshany ZM, Tashkandi NY, Gad-Elkareem MA, Anwar MM, Nossier ES (2021) New pyrimidine and pyrazole-based compounds as potential EGFR inhibitors: synthesis, anticancer, antimicrobial evaluation and computational studies. Bioorg Chem 114:105078
Sherbiny FF, Bayoumi AH, El-Morsy AM, Sobhy M, Hagras M (2021) Design, synthesis, biological evaluation, and molecular docking studies of novel Pyrazolo [3, 4-d] Pyrimidine derivative scaffolds as potent EGFR inhibitors and cell apoptosis inducers. Bioorg Chem 116:105325
Mikherdov AS, Novikov AS, Kinzhalov MA, Zolotarev AA, Boyarskiy VP (2018) Intra-/intermolecular bifurcated chalcogen bonding in crystal structure of thiazole/thiadiazole derived binuclear (diaminocarbene) PdII complexes. Cryst 8(3):112
Kardile RA, Sarkate AP, Lokwani DK, Tiwari SV, Azad R, Thopate SR (2023) Design, synthesis, and biological evaluation of novel quinoline derivatives as small molecule mutant EGFR inhibitors targeting resistance in NSCLC: In vitro screening and ADME predictions. Eur J Med Chem 245:114889
Osipyan A, Sapegin A, Novikov AS, Krasavin M (2018) Rare medium-sized rings prepared via hydrolytic imidazoline ring expansion (HIRE). J Org Chem 83(17):9707–9717
Joshi G, Nayyar H, Kalra S, Sharma P, Munshi A, Singh S, Kumar R (2017) Pyrimidine containing epidermal growth factor receptor kinase inhibitors: synthesis and biological evaluation. Chem Biol Drug Des 90(5):995–1006
Nasser AA, Eissa IH, Oun MR, El-Zahabi MA, Taghour MS, Belal A, Saleh AM, Mehany ABM, Luesch H, Mostafa AE, Afifi WM, Rocca JR, Mahdy HA (2020) Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFR WT and EGFR T790M. Org Biomol Chem 18(38):7608–7634
Gaber AA, Bayoumi AH, El-Morsy AM, Sherbiny FF, Mehany AB, Eissa IH (2018) Design, synthesis and anticancer evaluation of 1H-pyrazolo [3, 4-d] pyrimidine derivatives as potent EGFRWT and EGFRT790M inhibitors and apoptosis inducers. Bioorg Chem 80:375–395
Hao Y, Lyu J, Qu R, Tong Y, Sun D, Feng F, Tong L, Yang L, Zhao Z, Zhu L, Ding J, Xu Y, Xie H, Li H (2018) Design, Synthesis, and biological evaluation of pyrimido[4, 5-d] pyrimidine-2, 4 (1H, 3H)-diones as potent and selective epidermal growth factor receptor (EGFR) inhibitors against L858R/T790M resistance mutation. J Med Chem 61(13):5609–5622
Li J, An B, Song X, Zhang Q, Chen C, Wei S, Fan R, Li X, Zou Y (2021) Design, synthesis and biological evaluation of novel 2, 4-diaryl pyrimidine derivatives as selective EGFRL858R/T790M inhibitors. Eur J Med Chem 212:113019
Ding S, Dong X, Gao Z, Zheng X, Ji J, Zhang M, Liu F, Wu S, Li M, Song W, Shen J, Duan W, Liu J, Chen Y (2022) Design synthesis and biological evaluation of novel N-(3-amino-4-methoxyphenyl) acrylamide derivatives as selective EGFRL858R/T790M kinase inhibitors. Bioorg Chem 118:105471. https://doi.org/10.1016/j.bioorg.2021.105471
Abdullahi SH, Uzairu A, Shallangwa GA, Uba S, Umar AB (2022) Structure based design of some novel 3-methylquinoxaline derivatives through molecular docking and pharmacokinetics studies as novel VEGFR-2 inhibitors. Chem Afr 5:1967–1978
Zhu J, Wu Y, Xu L, Jin J (2020) Theoretical studies on the selectivity mechanisms of glycogen synthase kinase 3β (GSK3β) with pyrazine ATP-competitive inhibitors by 3D-QSAR, molecular docking, molecular dynamics simulation and free energy calculations. Curr Comput-Aided Drug Des 16(1):17–30
Abdullahi SH, Uzairu A, Shallangwa GA, Uba S, Umar AB (2023) 2D and 3D-QSAR modeling of 1H-pyrazole derivatives as EGFR inhibitors: molecular docking, and pharmacokinetic profiling. Chem Afr 6:1381–1398. https://doi.org/10.1007/s42250-023-00592-9
Dhingra N, Kar A, Sharma R (2020) Towards further understanding the structural requirements of combretastatin-like chalcones as inhibitors of microtubule polymerization. Curr Comput-Aided Drug Des 16(2):155–166
Arakawa M, Hasegawa K, Funatsu K (2007) The recent trend in QSAR modeling-variable selection and 3D-QSAR methods. Curr Comput-Aided Drug Des 3(4):254–262
Clark M, Cramer RD, Opdenbosch NV (1989) Validation of the general purpose tripos 5.2 force field. J Comput Chem 10:982–1012. https://doi.org/10.1002/jcc.540100804
Powell MJD (1977) Restart procedures for the conjugate gradient method. Math Program 12:241–254
Tsai KC, Chen YC, Hsiao NW, Wang CL, Lin CL, Lee YC, Li M, Wang BA (2010) Comparison of different electrostatic potentials on prediction accuracy in CoMFA and CoMSIA studies. Eur J Med Chem 45(4):1544–1551
Sogabe S, Kawakita Y, Igaki S, Iwata H, Miki H, Cary DR, Ishikawa T (2013) Structure-based approach for the discovery of pyrrolo [3, 2-d] pyrimidine-based EGFR T790M/L858R mutant inhibitors. ACS Med Chem Lett 4(2):201–205. https://doi.org/10.1021/ml300327z
BIOVIA DS (2016) Discovery studio modeling environment, release 2017, San Diego: DassaultSystèmes, 2016
DeLano W (2017) The PyMOL molecular graphics system. Delano Scientific, Palo Alto
Tabti K, Elmchichi L, Sbai A, Maghat H, Bouachrine M, Lakhlifi T (2022) HQSAR, CoMFA, CoMSIA docking studies and simulation MD on quinazolines/quinolines derivatives for DENV virus inhibitory activity. Chem Afr 5(6):1937–1958
Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718
Kumari R, Kumar R (2014) Open Source Drug Discovery Consortium, Lynn, A. g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54(7):1951–1962
Safavi A, Ghodousi ES, Ghavamizadeh M, Sabaghan M, Azadbakht O, Babaei H, Zarezade V (2021) Computational investigation of novel farnesyltransferase inhibitors using 3D-QSAR pharmacophore modeling, virtual screening, molecular docking and molecular dynamics simulation studies: a new insight into cancer treatment. J Mol Struct 1241:130667. https://doi.org/10.1016/j.molstruc.2021.130667
Ghaleb A, Aouidate A, Ayouchia HBE, Aarjane M, Anane H, Stiriba SE (2022) In silico molecular investigations of pyridine N-Oxide compounds as potential inhibitors of SARS-CoV-2: 3D QSAR, molecular docking modeling, and ADMET screening. J Biomol Struct Dyn 40(1):143–153. https://doi.org/10.1080/07391102.2020.1808530
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7(1):42717. https://doi.org/10.1038/srep42717
Author information
Authors and Affiliations
Contributions
All contributors aided the study's inception and design. El Bahi S, Boutalaka M, El Alaouy M, Koubi Y, and El Khatabi K: presented idea, draft preparation, data handling, data analysis, and writing; Alaqarbeh M: performed the calculations of molecular dynamics, writing, review, and editing; Choukrad M, Sbai A, and Bouachrine M: data analysis, study justification, reviewing, and supervision; Lakhlifi T: reviewing supervision, and project administration.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
El Bahi, S., Boutalaka, M., Alaqarbeh, M. et al. In-Silico Investigation of Osimertinib Based Compounds as Potential Double Mutant EGFR Kinase Inhibitors Against H1975 Cell Line: Integrating QSAR Modeling, Molecular Docking, MD Simulations, and ADME/Tox Studies. Chemistry Africa 7, 111–129 (2024). https://doi.org/10.1007/s42250-023-00744-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-023-00744-x