
Abstract
Introduction
Pulmonary hypertension (PH) is often complicated by chronic lung diseases (CLDs) such as chronic obstructive pulmonary disease (COPD) and interstitial lung disease (ILD). Differentiating between PH associated with CLD (group 3 PH) and pulmonary arterial hypertension (PAH) in CLD is often difficult and reporting on the efficacy of PAH-specific therapies is inconsistent as a result of the lack of understanding of the heterogeneity of patients with PH.
Methods
A retrospective observational cohort study was conducted to understand the baseline characteristics, comorbidities, and treatment profiles of patients with PH in CLD in a real-world setting using a large-scale claims database (Medical Data Vision). Administrative and clinical data for patients admitted to acute-care hospitals in Japan between April 2008 and January 2021 were analyzed.
Results
A total of 115,921 patients with CLD (109,578 with COPD and 6343 with ILD, of whom 569 and 176 had PH, respectively) were analyzed. This study found lower PH diagnosis rates among patients with COPD and patients with ILD than in previous studies. The majority of PH with CLD patients were elderly (mean age 75.7 years) and male (80.81%). Among patients with CLD prescribed PAH-specific therapies (105 patients with COPD; 64 patients with ILD), most received these as monotherapy (COPD, 84.76%; ILD, 75.56%); the most common were phosphodiesterase 5 inhibitors (COPD, 42.70%; ILD, 18.37%), prostacyclins (oral; COPD, 48.31%; ILD, 24.49%), and endothelin receptor antagonists (ERA) (COPD, 8.99%; ILD, 18.37%). Comorbidities (e.g., pulmonary, cardiac, kidney), home oxygen therapy (HOT), and echocardiography (ECHO) were factors associated with the diagnosis of PH.
Conclusion
This is the first study using an administrative database that provides real-world data on patients with PH in CLD in Japan. Our results indicate that PH may be misdiagnosed or underdiagnosed in Japan which may lead to suboptimal treatment for patients, and supports the need for further evidence to guide appropriate treatment.
Plain Language Summary
Pulmonary hypertension is a disorder affecting the arteries in the lungs and the right heart. It can be associated with a variety of heart and lung conditions, including many chronic lung diseases such as chronic obstructive pulmonary disease and interstitial lung disease. Patients with pulmonary hypertension with chronic lung disease and/or hypoxia can be hard to tell apart from patients with pulmonary arterial hypertension coinciding with chronic lung disease. In Japan, there is not enough data on patient demographics and their disease characteristics for patients with pulmonary hypertension and chronic lung disease, including treatment profiles, and disease management. We identified these patients from a large medical claims database in Japan and analyzed their data. Our study focused on the use of therapies for pulmonary arterial hypertension on patients with pulmonary hypertension and chronic lung disease. The diagnosis rates of pulmonary hypertension for patients with chronic obstructive pulmonary disease and interstitial lung disease were low compared to previous reports, meaning patients with pulmonary hypertension may be misdiagnosed or underdiagnosed which may be resulting in suboptimal treatments. Furthermore, the majority of patients with pulmonary hypertension treated with pulmonary arterial hypertension medication received a single drug as treatment, even though the guidelines recommend the use of combination therapies in certain situations. This study emphasizes the need for further evidence generation for improvements in diagnoses and treatment of patients with pulmonary hypertension in Japan.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
There is lack of real-world evidence on the characteristics, factors associated with pulmonary hypertension (PH) diagnosis, and treatment patterns for patients with PH in chronic lung diseases (CLDs) in Japan. |
This study utilizes a large Japanese claims database to understand the characteristics of patients with PH in CLD and their treatment profiles with a focus on pulmonary arterial hypertension (PAH)-specific therapies. |
What was learned from the study? |
PH diagnosis rates among patients with chronic obstructive pulmonary disease (COPD) and patients with interstitial lung disease (ILD) were found to be lower than in previous studies, monotherapy was the most prescribed PAH-specific therapy, and the characteristics of patients receiving continuous PAH-specific therapy were shown to differ between COPD and ILD. |
The results from this study indicate that patients with PH in CLD may be misdiagnosed or underdiagnosed which may be resulting in suboptimal treatments for patients. |
Further evidence on the real-world clinical state of patients with PH in Japan is necessary for the appropriate treatment of patients. |
Introduction
Pulmonary hypertension (PH) is a pathophysiologic disorder with a poor prognosis which is associated with various circulatory and respiratory diseases. The global prevalence of PH is approximately 1%, with a higher prevalence in individuals over 65 years old [1]. PH is clinically classified into five groups, of which group 1 PH in particular now has more treatment options in Japan owing to the launch of pulmonary vasodilators [2]. Furthermore, PAH-specific therapies are now subsidized by the designated intractable disease medical expenses subsidy when patients are diagnosed with a group 1 PH complicated by respiratory disease [3].
Patients with group 3 PH are classified as either non-severe (pulmonary vascular resistance; PVR ≤ 5 Wood units [WU]) or severe PH (PVR > 5 WU), both of which have poor prognoses [2]. With the exception of inhaled treprostinil, there have been no convincing studies demonstrating improved outcomes with PAH-specific therapies such as pulmonary vasodilators (PVs) in patients with group 3 PH [4, 5]. Despite the lack of evidence, extensive use of PAH-specific therapies for patients with group 3 PH has been reported in the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) registry study [6]. However, the European Society of Cardiology and the European Respiratory Society (ESC/ERS) guidelines [2] do not recommend the use of PAH-specific therapies as a result of the limited evidence and potential negative impact of these drugs on gas exchange, hemodynamics, and outcomes. Patients with severe PH are recommended for referral to PH centers for personalized treatments [2]. The variability in treatment recommendations from these guidelines and studies reflects the clinical difficulties of selecting appropriate treatments for patients with PH.
The gold standard diagnostic approach for PH is right heart catheterization (RHC); however, other approaches include electrocardiography (ECG), chest radiography, pulmonary function tests, lung scans, echocardiography (ECHO), and the evaluation of biomarkers B-type natriuretic peptides (BNP) and N-terminal-pro hormone BNP (NT-proBNP) [2, 7, 8]. Despite the availability of multiple assessment techniques, it remains difficult to differentiate between patients with group 3 PH and those with group 1 PH with coincident chronic lung disease (CLD) as the severity of pulmonary vascular disease and parenchymal CLD likely overlap with each other [7]. Chronic obstructive pulmonary disease (COPD) and interstitial lung disease (ILD) are among the CLDs that can cause complications in PH and patients demonstrate reduced functional ability and worse outcomes, with COPD being the most prevalent [7, 9, 10].
Although there are previous reports on the diagnosis and treatment of group 3 PH and group 1 PH in Japan, these are mainly limited to specialized centers [11,12,13] and there is no comprehensive analysis of patients with group 3 and group 1 PH including non-specialists to date. There is thus a lack of real-world data on PH particularly from non-specialized centers in Japan. This study aimed to understand the demographics, clinical characteristics, and actual clinical practices used for the treatment of patients with CLDs (COPD and ILD) with PH, including group 1 PH, using electronic health records from Medical Data Vision (MDV)—a large, anonymized administrative claims database of Diagnosis Procedure Combination (DPC) hospitals in Japan. The overall objective of this study was divided into four sub-objectives and included the analysis of (1) demographics and clinical characteristics of patients with PH in CLDs; (2) treatment patterns of PAH-specific therapies; (3) factors associated with diagnosis of PH with CLDs by comparing patients with and without PH; and (4) comparison of patient characteristics of three groups of patients with PH categorized by the length of their respective PAH drug treatments.
Methods
Study Design and Data Sources
This retrospective observational cohort study of Japanese patients with PH in CLD using the MDV database, a hospital-based administrative claims database in Japan, was conducted between April 2008 and January 2021. The database comprised anonymized administrative and laboratory data from approximately 460 acute care hospitals and covers over 40 million patients, including 39% of elderly patients [14]. The MDV dataset includes patient IDs, month and year of birth, diagnoses coded according to the International Classification of Diseases, Tenth Revision (ICD-10) codes, Japanese standard disease codes, medications, laboratory tests, months of diagnoses, dates of procedures, prescriptions, and dates of admission and discharge. The anonymized MDV data are used for epidemiological, and health economics and outcomes research. Informed consent for the analyses in this study was waived because of the anonymous nature of the database.
Study Population
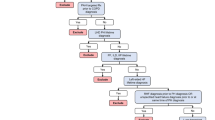
The study population was identified from the database between April 2008 to January 2021 (Fig. 1). All potential incident cases were identified using a systematic algorithm developed by a clinical expert by referring to previous studies [15,16,17,18]. In accordance with previous studies, a multidirectional approach was applied to identify patients with PH in CLD to ensure the accuracy of the diagnosis. Patients with PH were identified as patients who had a confirmed diagnosis of PH based on ICD-10 code I27.0 and had received ECHO and/or RHC within 1 month before/after the first recorded ICD-10 code for PH. Patients meeting the selection criteria were followed up from their initial definitive identification of PH (index date) until the earliest of the end of the observational period, death, or known exit from the data source (follow-up period). Further, a minimum look-back period of 6 months from the first claims record to the index month was included. Patient data were examined for PAH-associated comorbidities and factors associated with diagnosis during the baseline period (6 months prior to the month of first diagnosis of PH, i.e., index month). The study population was identified from the database as per the eligibility criteria (patient selection and attrition can be found in Fig. 2).

Study overview. Study design with patient definitions for objectives 1, 2, 3, and 4 and the definitions of index date, index month, PAH medication date, baseline period, CLD month, and PH onset month. ECHO echocardiography, I270 ICD-10 code for primary pulmonary hypertension, LD lung disease, MDV Medical Data Vision, PAH pulmonary arterial hypertension, PH pulmonary hypertension, PV pulmonary vasodilator, RHC right heart catheterization

Attrition diagram. Flow of patient selection and patient attrition over the observational period. COPD chronic obstructive pulmonary disease, ECHO echocardiography, IPF idiopathic pulmonary fibrosis, I270 ICD-10 code for primary pulmonary hypertension, LD lung disease, PAH pulmonary arterial hypertension, PH pulmonary hypertension, RHC right heart catheterization
In this study, two populations were included on the basis of the study objectives (Fig. S1). Population 1 included patients who met either the COPD or ILD algorithms (Figs. S2 and S3) and those who met the PH algorithms (Fig. S4) during the data period. Patients who did not have any claims records during or prior to the 6 months before the index month were excluded. This population was analyzed for objectives 1, 2, and 4. The study population was divided into two cohorts. Patients in cohort A were defined as patients with PH who also received a PAH-specific treatment on or after the index month. These patients are represented in this study as the “PAH therapy group.” Cohort B comprised patients who were not prescribed PAH-specific therapies on or after the index month in the population, and are referred to as the “no treatment group” in this study. Cohort A was further divided into two subcohorts: cohort A-1, which included patients in cohort A who were prescribed PAH-specific therapies continuously (90% proportion of days covered) for at least 6 months; and cohort A-2, consisting of patients in cohort A who were not included in cohort A-1. Population 2 was analyzed for objective 3 and included patients who met either the COPD or ILD algorithms during the data period. Patients who did not have any claims records for more than 6 months before the CLD month (first definitive diagnosis month of CLD) and within 6 months before the CLD month were excluded.
Variables and Outcomes
In this study, the outcomes—including treatment patterns and factors associated with diagnosis—were measured for three patient groups: patients with CLDs (COPD or ILD, i.e., the entire study population), patients with COPD, and patients with ILD. Patients with CLDs who met both the COPD and ILD algorithms were assigned to “patients with ILD” and not “patients with COPD.” These assignments were made when both ILD and COPD are present (ILD generally being the more severe disease), which is referred to as combined pulmonary fibrosis and emphysema (CPFE) and is generally classified as a type of pulmonary fibrosis. With this background in mind, Japanese clinical experts have expressed the opinion that the inclusion of patients with both ILD and COPD into the ILD group is appropriate. Furthermore, a feasibility study prior to this investigation found that there were very few patients with both ILD and COPD in the target population.
The variables and outcomes are described in Table S1 of the electronic supplementary material. In this study, the ECHO for determining PH was performed between the month before and the month after the index month as described above. In the outcomes, however, ECHO was performed from between 12 to 2 months before the index date. Therefore, the timelines do not overlap between the ECHO used for diagnosis and those used for the outcomes. The ECHO variable simply indicates that the tests were performed and are not indicative of abnormalities.
The variables and outcomes (demographic and clinical characteristics) measured in this study were gender, age group, body mass index (BMI), smoking status, New York Heart Association (NYHA) Functional Class (FC), Charlson Comorbidity Index, and the corresponding 1-year mortality risk [19, 20]. These variables were assessed for all patients with CLDs (Tables S2, S3) and compared between patients with COPD and patients with ILD (Table S4). Baseline comorbidities, which are typically concurrent with CLDs, were also reported and considered covariates in the multivariate analysis. Baseline laboratory data, such as BNP and NT-proBNP levels, were also considered as they were generally measured in patients with CLDs and PH.
Treatment patterns of PAH-specific therapy were described in terms of monotherapy (one drug class), double therapy (two different drug classes), and triple therapy (three different drug classes) of PAH-specific drugs. The drug classes of PAH-specific therapy used were also described from baseline (first PAH medication) to 5 years (from the index date) for the overall population with CLDs, patients with COPD, and patients with ILD.
Data Analysis
Patient data were analyzed for baseline characteristics, treatment profiles, and clinical outcomes. The analyses conducted throughout the study were primarily descriptive in nature and were performed using R version 4.1.0 or higher. The only missing data was from the laboratory values which are labeled in the tables and were excluded. No attempts at imputation were made.
Descriptive statistics were summarized for demographics, baseline clinical characteristics, and the outcomes of interest. Frequencies and percentages were reported as categorical variables, while means, standard deviations (SDs), medians, minimum and maximum values, 25th and 75th percentile values, and interquartile ranges (IQRs) were reported as continuous variables.
PAH-specific therapies were described as combination subgroups of PAH-specific therapies (mono/double/triple), their utilization, and concomitant diuretic use for the initial treatment (0–90 days from the index date) and at 1–5 years (the last 90 days for each timepoint). Sankey diagrams were used to visualize treatment patterns over 5 years for PAH-specific therapies. All PAH-specific therapies administered during the initial treatment (or the last 90 days for each timepoint, after the 90-day initial treatment period) were composed of one of the combination therapies. The number of patients who ceased PAH-specific treatment or dropped out from the cohort for each 90-day observation period was reported in the “n of patients ceased PAH-specific treatments or censored by the 90-day observation period” variable.
Inferential analysis was conducted to estimate the factors associated with PH diagnosis using a multivariate Cox proportional hazard model. The starting point for the analysis was identified by the index date (the time of diagnosis of COPD/ILD) and the end point was the diagnosis of PH. The onset of CLD was confirmed retroactively to estimate the factors associated with PH diagnosis. The potential factors (explanatory variables) for each model were determined with assistance from clinical experts in the field. Firth’s penalized likelihood was applied in both analyses to mitigate the bias caused by rare events in the dataset if complete separation was detected [21].
Compliance with Ethics Guidelines
This retrospective database analysis study did not collect, transmit, or use identifiable patient data. Based on the Japanese Ethical Guidelines for Medical and Biological Research Involving Human Subjects, this study did not require any approval from an institutional review board. Permission was obtained from the MDV database for the use of their data for this study.
Results
Demographic and Clinical Characteristics
As the study focused on whether PAH-specific therapies were prescribed at least once (PAH therapy group) or not (no treatment group), the demographics (Table 1) and clinical characteristics (Tables 2 and 3) of these patients were studied and compared. Overall, 109,578 patients with COPD and 6343 patients with ILD were included in the analysis. Patients with COPD in the PAH therapy group had a mean (SD) age of 75.19 (8.80) years and 83.81% were male, while those in the no treatment group had a mean (SD) age of 77.25 (8.55) years and 82.54% were male. Patients with ILD who were in the PAH therapy group had a mean (SD) age of 69.13 (13.01) years and 67.19% were male, while those in the no treatment group had a mean (SD) age of 73.63 (8.27) years and 78.57% were male.
Patients with CLD also received different tests for the diagnosis of PH. Patients with COPD in the PAH therapy group had a higher rate of RHC when compared with the no treatment group (37.14% vs 19.18%, respectively). This trend remained true for the ILD group, where patients in the PAH therapy group also had a higher rate of RHC compared to the no treatment group (37.50% vs 16.07%, respectively). The usage rate of ECG was high across all groups (> 90%). The results related to comorbidities can be found in Table 2.
The demographic and clinical characteristics of patients who underwent RHC were compared with patients who did not undergo RHC (PAH therapy group, Table S5; no treatment group, Table S6). Although there were some differences in specific categories, the overall characteristics were comparable between the two patient groups.
Treatment Patterns of PAH-Specific Therapies
Treatment patterns were analyzed both independently and together for patients with COPD and patients with ILD (Table 4 and Table S7, respectively), and for patients with CLD as a whole (Figs. S5 and S6). Monotherapy was the most used treatment among patients with COPD (58.33–84.76%, depending on data collection period), compared to those patients receiving double (4.17–33.33%) and triple therapies (0.00–30.00%), as shown in Fig. 3 and Table 4. During the follow-up period, most patients who were initially treated with PAH-specific monotherapy stayed on monotherapy, and a few patients were de-escalated (i.e., triple to double, double to mono, and triple to monotherapy). During the initial treatment, oral prostaglandin (PG, 40.96%) was the most common PAH-specific therapy, followed by phosphodiesterase 5 inhibitor (PDE5i) monotherapy (36.19%) (Fig. 4). Throughout the follow-up period, PDE5i was the most frequently used PAH-specific therapy (31.82–50.00%), followed by oral PG (20.00–40.96%). Most patients who stopped PAH-specific treatment did not restart it throughout the follow-up period.

Sankey diagram representing PAH-specific combination treatment pattern of population with COPD. This figure represents patient flows of PAH-specific combination patterns for the overall population with COPD from the baseline (first PAH medication) to the 90-day period after 5 years from the index date. Each node (Mono/Double/Triple/Not treated) represents the PAH-specific treatment combinations during baseline (90 days) and the last 90-day period after 1–5 years from the index date. The size of the nodes represents the proportion of patients in each 90-day period. In the parentheses, the first number is the number of patients and the second is the percentage of patients out of the total. COPD chronic obstructive pulmonary disease, PAH pulmonary arterial hypertension

Sankey diagram representing drug classes for treatment of population with COPD. This figure represents patient flows of PAH-specific therapeutic categories for the COPD population from the baseline (first PAH medication) to the 90-day period after 5 years from the index date. Each node represents the PAH-specific therapeutic categories during baseline (90 days) and the last 90-day period after 1–5 years from the index date. The size of the nodes represents the proportion of patients in each 90-day period. In the parentheses, the first number is the number of patients and the second is the percentage of patients out of the total. ERA endothelin receptor antagonist, NT not treated, PAH pulmonary arterial hypertension, PDE5i phosphodiesterase 5 inhibitor, PG oral prostaglandin
Monotherapy was the most commonly used PAH-specific treatment (33.33–76.56%) among patients with ILD throughout the observational period (Table 4, Fig. 5). The most common PAH-specific therapy used during the initial treatment was PDE5i monotherapy (43.75% of the overall population), followed by oral PG (18.75%) and ERA monotherapy (14.06%) (Fig. 6). During the follow-up period, PDE5i monotherapy was the most common PAH-specific therapeutic category at 1 and 2 years after the initial treatment (33.33% and 18.75% of patients, respectively), while ERA + oral PG double combination therapy was more common after 3 years from initial treatment (22.22–50.00%).

Sankey diagram representing PAH-specific combination treatment pattern of population with ILD. This figure represents patient flows of PAH-specific combination patterns for the overall population with ILD from the baseline (first PAH medication) to the 90-day period after 5 years from the index date. Each node (Mono/Double/Triple/Not treated) represents the PAH-specific combinations during baseline (90 days) and the last 90-day period after 1–5 years from the index date, respectively. The size of the nodes represents the proportion of patients in each 90-day period. In the parentheses, the first number is the number of patients and the second is the percentage of patients out of the total. ILD interstitial lung disease, NT not treated, PAH pulmonary arterial hypertension

Sankey diagram representing drug classes for treatment of population with ILD. This Sankey diagram represents patient flows of PAH-specific therapeutic categories for the ILD population from the baseline (first PAH medication) to the 90-day period after 5 years from the index date. Each node represents the PAH-specific therapeutic categories during baseline (90 days) and the last 90-day period after 1–5 years from the index date, respectively. The size of the nodes represents the proportion of patients in each 90-day period. In the parentheses, the first number is the number of patients and the second is the percentage of patients out of the total. ERA endothelin receptor antagonist, NT not treated, PAH pulmonary arterial hypertension, PDE5i phosphodiesterase 5 inhibitor, PG oral prostaglandin
For the COPD and ILD populations, 22.22–66.67% of the patients with group 1 PH received concomitant diuretics, with loop diuretics being commonly administered throughout the follow-up period (Table S8).
Factors Associated with Diagnosis of PH
In this study, simple and multiple Cox regression analyses were performed to understand the factors associated with the diagnosis of PH (Tables 5 and 6). The simple Cox regression observed differences in hazard ratios based on differences in diagnostic methods and disease background. The multiple Cox regression model also showed factors associated with the diagnosis of COPD with higher hazard ratios (HRs) for HOT and ECHO (HR 3.501 [p < 0.001] and 3.955 [p < 0.001], respectively) compared with ILD (HR 2.898 [p = 0.010] and 2.384 [p = 0.002], respectively), indicating a statistical significance of these factors in the diagnosis of COPD (p < 0.001). All values from the COPD simple Cox regression model shown in Table 5 were significant; however, only malignancy, HOT, pulmonary gas distribution test, and ECHO were significant for the multiple Cox model (Table 6).
Comparison of Demographic Characteristics of Cohort Subgroups
The patients in the study were categorized as patients receiving a PAH-specific therapy (cohort A) and the no treatment group (cohort B); their demographics were compared to analyze the characteristics of the patients receiving PAH-specific therapy (for at least 6 months) versus those who were not (Table 7). Among the patients with COPD, cohort B consisted of patients older than those in cohorts A-1 and A-2 (mean age of 77.25 [8.55], 73.76 [8.79], and 76.85 [8.01] years, respectively). Cohort B also had a higher percentage of patients with smoking history (64.56%) than cohorts A-1 (55.17%) and A-2 (36.36%). There was also a statistically significant difference found in HOT between the three cohorts.
Among the patients with ILD, those in cohort B were older than those in cohorts A-1 and A-2 (mean ages of 73.63 [8.27], 68.37 [14.49], and 70.00 [12.05] years, respectively). Patients in both cohorts A and B with COPD and ILD were predominantly male (57.14–100%). There were differences in the complication rates among cohorts A-1, A-2, and B for CTD (45.71%, 25.00%, and 11.61%, respectively), and arrhythmia (2.86%, 0.00%, and 20.54%, respectively). The laboratory data comparison of these cohorts is presented in Table S4.
Discussion
Demographic and Clinical Characteristics of Patients with PH in CLDs
As the first nationwide database study on patients with PH in CLD in Japan, our study allows for the comparison of clinical characteristics with studies from Western countries. The estimated nationwide prevalence of idiopathic pulmonary fibrosis, a common type of ILD, as per the MDV database (2008–2019) was 27 per 100,000 population of Japan, which was similar for men in the USA. On the other hand, the prevalence was lower in Japanese women aged < 79 years [22] compared with earlier studies (10 per 100,000 population) [23]. Japan had previously reported around 16–60% of patients with COPD-related PH [24]. However, this study reports much lower PH diagnosis rates among patients with COPD and ILD (0.87% and 1.79%, respectively) compared with previous reports [6, 14, 25], implying that patients may be underdiagnosed and/or misdiagnosed with other diseases. The difference in diagnosis rate may also stem from this being the first study to investigate the prevalence of PH based on COPD and ILD in Japan using a nationwide administrative database that reflects the clinical practices in both referral centers and non-referral hospitals, while previous reports mainly focused on referral centers. It is also suggested that proper diagnosis of PH including group 1 PH, a rare disease, requires expertise such as from referral centers.
Our results demonstrated that even in the PAH therapy group (patients receiving PAH-specific medication), RHC was only performed approximately 37% of the time at diagnosis. Proper training about rare diseases such as group 1 PH and the use of the appropriate diagnostic procedures are required among health professionals, especially general practitioners, so that patients can be appropriately referred to a specialist/cardiologist for RHC [26]. Poor general awareness regarding these rare diseases, delays in referrals, misdiagnoses, and poor compliance to the treatment guidelines may lead to worse prognoses [27]. Our analysis suggests that in the clinical practice of respiratory medicine, which mainly treats CLDs, there are still many barriers to the diagnosis and management of patients with PH in CLD, and there is a need to improve the time to referral, diagnosis, and initiation of treatment.
With regards to demographics of patients with PH, our study population was characterized by higher proportions of elderly patients and male patients. For example, a registry study by Tanabe et al. [11] on patients with PH associated with respiratory disease had a population with a mean age of 67 ± 11 years and was 70% male. Other studies using data extracted from the MDV database in Japan on patients with idiopathic pulmonary fibrosis and COPD also estimated a high percentage of elderly patients and a predominantly male population which is in line with our study [22, 28]. Our findings are consistent with studies using the COMPERA registry which involved elderly patients [6, 29, 30] as well as another recently published study using data from both the COMPERA and ASPIRE registries [30] which partially observed patients (patients with idiopathic pulmonary artery hypertension (IPAH) with a lung phenotype and patients with group 3 PH) with similar backgrounds to our study. The trends in our study population are aligned with these previous reports, suggesting that our study has properly identified patients with COPD and patients with ILD in Japan and may describe the real-world clinical practices for PH in these populations.
Treatment Patterns of PAH-Specific Therapies
The proportion of patients on PAH monotherapy in this study (84.76% for COPD and 76.56% for ILD during the initial treatment period) was markedly higher than that in a recent Japanese registry study, where the proportion of patients on oral/inhaled monotherapy was 23.1% and 20.3% between 2008–2015 and 2016–2020, respectively [13]. In contrast, most patients with IPAH with CLD received monotherapy [31], thus showing a similar trend to our study. The higher proportion of the patients in this study receiving monotherapy may indicate a preference for conservative treatments to avoid side effects in high-risk patients. Moreover, this study demonstrated that few patients were de-escalated in their treatments, which was also reported in previous studies [32, 33]. This suggests the underutilization of combination therapies, which have shown better adherence and lower discontinuation rates compared with monotherapy treatments [32]. The latest guidelines recommend initial monotherapy for patients with group 1 PH with cardiopulmonary comorbidities, followed by regular follow-up assessments and individualized therapies. In group 3 PH, if the patient has severe PH, an individualized treatment approach is recommended, but there is no consensus treatment strategy. In order to facilitate the development of new treatment options such as inhaled treprostinil [4, 5], further research is required to understand the patient backgrounds and physician perspectives regarding treatment strategies for PH.
A retrospective study using data from Japanese Respiratory Society (JRS)-approved institutions reported that 80% of patients with severe PH associated with respiratory diseases were treated with a PAH-specific therapy, especially PDE5i [34]; however, beraprost sodium was the most commonly used medication in our study. Moreover, the percentage of ERA usage was higher than expected with the third highest usage rate following PDE5i. The guidelines for the diagnosis and treatment of PH in Japan are prepared by the Japanese Circulation Society (JCS) and are based on ESC/ERS guidelines [35]. The latest guidelines released in 2022 will impact the JCS guidelines, which describe recommendations for PDE5i or ERA as initial monotherapy of group 1 PH with cardiopulmonary comorbidities [2]. Our findings may highlight the need to generate more evidence for PDE5i and ERA in these patients.
It is often difficult to differentiate between patients with group 1 and group 3 PH because of their overlapping characteristics. As a chronic disease, patients with group 1 PH typically require continuous treatment with a PAH-specific therapy. Therefore, it is possible that patients who received PAH-specific therapy for more than 6 months in this present study (cohort A-1) were patients with group 1 PH. On the other hand, patients who discontinued PAH-specific therapy within 6 months (cohort A-2) may have been identified as patients with group 1 PH at the initiation of treatment, but may have in fact been patients with group 3 PH who discontinued treatments because of the negative effects of PAH-specific therapies on patients with group 3 PH [36, 37].
Patients with COPD taking PAH-specific therapies were generally treated with HOT, suggesting that PAH-specific therapies were being used for severe patients with COPD.
A lower proportion of patients with ILD were treated with HOT compared to patients with COPD, suggesting that patients with ILD had mild-to-moderate lung disease compared to those with COPD. When patients with mild-to-moderate ILD were diagnosed with group 1 PH and administered PAH-specific therapies, they tended to receive continuous treatment for over 6 months. Our results imply that the stage of disease at which PAH-specific therapy is prescribed may differ between COPD and ILD. Further research is needed on PH in each type of CLD.
Factors Associated with PH Diagnosis
This study showed that a lung disease diagnosis followed by active ECHO results in higher rates of PH diagnosis in both patients with COPD and patients with ILD. ECHO is readily accessible in Japanese clinical practice, and our results offer real-world data with further confirmation of its importance for PH diagnosis in Japan. Our multivariate analysis also indicated that CTD, including systemic sclerosis, was associated with the diagnosis of PH in patients with ILD. This is in agreement with the JRS guidelines [38]. RHC is still considered the gold standard for the hemodynamic evaluation of pulmonary circulation and should be performed in most patients diagnosed with PH to understand the severity of PH and initiate the appropriate treatment [39]. Even with a mild concomitant lung disease, the prognostic impact on patients with IPAH is significant [40].
In another study conducted by JRS-approved institutions, Tanabe et al. evaluated the status of diagnostic and treatment modalities in patients with PH with respiratory diseases [25]. It is important to note that although there are studies challenging the usage of ECHO for the diagnosis of PH [41,42,43], ECHO is used for both the determination of group 1 and group 3 PH in real-world practices both in Japan [25] and in other countries such as the Netherlands [44]. ECHO was used for diagnosis in 99% of the institutions, while RHC was used in only 36% of institutions with more than half of the pulmonologists considering RHC as only necessary prior to the initiation of a PAH-specific therapy. Despite this, PAH-specific therapies were used in 45% of the institutions, even without confirmation from RHC [25]. This discrepancy, along with the low usage of RHC, indicates the potential for under/misdiagnoses and suboptimal treatments for many patients and emphasizes the need for further studies.
As expected, HOT was shown to be a factor associated with the diagnosis of PH in both patients with COPD and patients with ILD. Since a diagnosis of PH in Japan is medically indicated for the initiation of HOT, our results are consistent with clinical practice.
Study Limitations
There are limitations to be considered in this study. (1) The MDV database predominantly comprises administrative claims data. Therefore, several key variables associated with the diagnosis, severity, and prognosis of PH overall or specifically group 1 PH were not available meaning the clinical classification of PH groups could not be fully assessed. Furthermore, lab-value data were limited to what was available in the database, meaning that data such as lung function testing (e.g., forced expiratory volume in 1 s [FEV1]) was not available as a measure of disease severity. (2) MDV data are limited to a small number of medical institutions in Japan; these institutions are subject to DPC and may not fully reflect the general population, and extrapolation outside of Japan should be approached with caution. In addition, clinical and drug treatment information received at medical institutions other than the target DPC hospitals cannot be obtained or tracked. (3) The use of claims data has limitations in patient identification (e.g., patients with PH or COPD/ILD in the insurance-based name of the disease). (4) This study included a small minority of patients with left-sided heart failure who may have been difficult to categorize between groups 1, 2, and 3. (5) There may have been unmeasured confounding factors that may have influenced the results. (6) This study analyzed patients with group 3 and group 1 PH with CLDs together although there is likely overlap between the two populations as it was not possible to distinguish them on the basis of the available data. Further studies are needed to confirm the robustness of the findings of this study.
Conclusion
To our knowledge, this is the first study to describe the patient and treatment profiles of patients with PH in CLD using a Japanese nationwide administrative database. Considering the low diagnosis rates and high use of monotherapy in our study, it is possible that PH is being under/misdiagnosed in the real-world Japanese clinical setting. Thus, more accurate diagnostic methods for identifying patients with PH can lead to earlier treatment and improved outcomes. We emphasize the need for further evidence for early diagnosis, appropriate evaluation, and treatment of patients with PH in CLD.
Data Availability
The datasets generated and/or analyzed during the current study are not publicly available due to the commercial nature of the data.
Change history
20 December 2023
A Correction to this paper has been published: https://doi.org/10.1007/s41030-023-00247-7
References
Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016;4(4):306–22.
Humbert M, Kovacs G, Hoeper MM, et al. ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG). Eur Heart J. 2022;2022:237.
肺動脈性肺高血圧症 (指定難病86) Japan Intractable Diseases Information Center. https://www.nanbyou.or.jp/entry/253. Accessed 1 Oct 2022.
Waxman A, Restrepo-Jaramillo R, Thenappan T, et al. Long-term inhaled treprostinil for pulmonary hypertension due to interstitial lung disease: INCREASE open-label extension study. Eur Respir J. 2023;61(6):2202414.
Waxman A, Restrepo-Jaramillo R, Thenappan T, et al. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384(4):325–34.
Hoeper MM, Behr J, Held M, et al. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PLoS ONE. 2015;10(12):e0141911.
Nathan SD, Barbera JA, Gaine SP, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. 2019;53(1):1801914.
Singh N, Dorfmüller P, Shlobin OA, Ventetuolo CE. Group 3 pulmonary hypertension: from bench to bedside. Circ Res. 2022;130(9):1404–22.
Medrek SK, Sharafkhaneh A, Spiegelman AM, Kak A, Pandit LM. Admission for COPD exacerbation is associated with the clinical diagnosis of pulmonary hypertension: results from a retrospective longitudinal study of a veteran population. COPD. 2017;14(5):484–9.
Hayes D Jr, Black SM, Tobias JD, Kirkby S, Mansour HM, Whitson BA. Influence of pulmonary hypertension on patients with idiopathic pulmonary fibrosis awaiting lung transplantation. Ann Thorac Surg. 2016;101(1):246–52.
Tanabe N, Kumamaru H, Tamura Y, et al. Multi-institutional prospective cohort study of patients with pulmonary hypertension associated with respiratory diseases. Circ J. 2021;85(4):333–42.
Tamura Y, Kumamaru H, Satoh T, et al. Effectiveness and outcome of pulmonary arterial hypertension-specific therapy in Japanese patients with pulmonary arterial hypertension. Circ J. 2017;82(1):275–82.
Tamura Y, Kumamaru H, Inami T, et al. Changes in the characteristics and initial treatments of pulmonary hypertension between 2008 and 2020 in Japan. JACC Asia. 2022;2(3):273–84.
About MDV database: Medical data vision. https://en.mdv.co.jp/about-mdv-database/. Accessed 1 Oct 2022.
Mueller S, Gottschalk F, Groth A, et al. Primary data, claims data, and linked data in observational research: the case of COPD in Germany. Respir Res. 2018;19(1):161.
Stanford RH, Nag A, Mapel DW, et al. Validation of a new risk measure for chronic obstructive pulmonary disease exacerbation using health insurance claims data. Ann Am Thorac Soc. 2016;13(7):1067–75.
Gershon AS, Wang C, Guan J, Vasilevska-Ristovska J, Cicutto L, To T. Identifying individuals with physician diagnosed COPD in health administrative databases. COPD. 2009;6(5):388–94.
Ley B, Urbania T, Husson G, et al. Code-based diagnostic algorithms for idiopathic pulmonary fibrosis. Case validation and improvement. Ann Am Thorac Soc. 2017;14(6):880–7.
Concept: Charlson Comorbidity Index 2021. http://mchp-appserv.cpe.umanitoba.ca/viewConcept.php?printer=Y&conceptID=1098. Accessed 1 Oct 2022.
Ludvigsson JF, Appelros P, Askling J, et al. Adaptation of the Charlson comorbidity index for register-based research in Sweden. Clin Epidemiol. 2021;13:21–41.
Firth D. Bias reduction of maximum likelihood estimates. Biometrika. 1993;80(1):27–38.
Kondoh Y, Suda T, Hongo Y, et al. Prevalence of idiopathic pulmonary fibrosis in Japan based on a claims database analysis. Respir Res. 2022;23(1):24.
Natsuizaka M, Chiba H, Kuronuma K, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med. 2014;190(7):773–9.
Zhang L, Liu Y, Zhao S, et al. The incidence and prevalence of pulmonary hypertension in the COPD population: a systematic review and meta-analysis. Int J Chron Obstruct Pulmon Dis. 2022;17:1365–79.
Tanabe N, Taniguchi H, Tsujino I, et al. Current trends in the management of pulmonary hypertension associated with respiratory disease in institutions approved by the Japanese Respiratory Society. Respir Investig. 2014;52(3):167–72.
Mandras SA, Ventura HO, Corris PA. Breaking down the barriers: why the delay in referral for pulmonary arterial hypertension? Ochsner J. 2016;16(3):257–62.
Meiwald A, Gara-Adams R, Rowlandson A, et al. Qualitative validation of COPD evidenced care pathways in Japan, Canada, England, and Germany: common barriers to optimal COPD care. Int J Chron Obstruct Pulmon Dis. 2022;17:1507–21.
Muro S, Suzuki M, Nakamura S, et al. Real-world effectiveness of early intervention with fixed-dose tiotropium/olodaterol vs tiotropium in Japanese patients with COPD: a high-dimensional propensity score-matched cohort analysis. Respir Res. 2021;22(1):180.
Hoeper MM, Pausch C, Grünig E, et al. Idiopathic pulmonary arterial hypertension phenotypes determined by cluster analysis from the COMPERA registry. J Heart Lung Transplant. 2020;39(12):1435–44.
Hoeper MM, Dwivedi K, Pausch C, et al. Phenotyping of idiopathic pulmonary arterial hypertension: a registry analysis. Lancet Respir Med. 2022;10(10):937–48.
Peacock AJ, Ling Y, Johnson MK, et al. Idiopathic pulmonary arterial hypertension and co-existing lung disease: is this a new phenotype? Pulm Circ. 2020;10(1):2045894020914851.
Studer S, Hull M, Pruett J, Elliott C, Tsang Y, Drake W. Retrospective database analysis of treatment patterns among patients with pulmonary arterial hypertension. Pulm Ther. 2020;6(1):79–92.
Ogbomo A, Tsang Y, Kariburyo F, Tsai WL, Panjabi S. Real-world analysis of treatment patterns among hospitalized patients with pulmonary arterial hypertension. Pulm Ther. 2021;7(2):575–90.
Tanabe N, Taniguchi H, Tsujino I, et al. Multi-institutional retrospective cohort study of patients with severe pulmonary hypertension associated with respiratory diseases. Respirology. 2015;20(5):805–12.
Fukuda K, Date H, Doi S, et al. Guidelines for the treatment of pulmonary hypertension (JCS 2017/JPCPHS 2017). Circ J. 2019;83(4):842–945.
Raghu G, Behr J, Brown KK, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–9.
Nathan SD, Behr J, Collard HR, et al. Riociguat for idiopathic interstitial pneumonia-associated pulmonary hypertension (RISE-IIP): a randomised, placebo-controlled phase 2b study. Lancet Respir Med. 2019;7(9):780–90.
膠原病に伴う間質性肺疾患 診断・治療指針 2020: The Japanese Respiratory Society; 2020. https://www.jrs.or.jp/publication/jrs_guidelines/20200630142610.html. Accessed 1 Oct 2022.
Behr J, Ryu JH. Pulmonary hypertension in interstitial lung disease. Eur Respir J. 2008;31(6):1357.
Lewis RA, Thompson AAR, Billings CG, et al. Mild parenchymal lung disease and/or low diffusion capacity impacts survival and treatment response in patients diagnosed with idiopathic pulmonary arterial hypertension. Eur Respir J. 2020;55(6):2000041.
Fisher MR, Forfia PR, Chamera E, et al. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179(7):615–21.
Taleb M, Khuder S, Tinkel J, Khouri SJ. The diagnostic accuracy of Doppler echocardiography in assessment of pulmonary artery systolic pressure: a meta-analysis. Echocardiography. 2013;30(3):258–65.
Janda S, Shahidi N, Gin K, Swiston J. Diagnostic accuracy of echocardiography for pulmonary hypertension: a systematic review and meta-analysis. Heart. 2011;97(8):612–22.
Jansen SMA, Huis In ’t Veld AE, et al. Clinical characteristics of patients undergoing right heart catheterizations in community hospitals. J Am Heart Assoc. 2022;11(17):e025143.
Acknowledgements
Medical Writing and Editorial Assistance
The authors would like to acknowledge Kashika Arora and Rosario Vivek of IQVIA India and Louis Watanabe of IQVIA Japan for their medical writing and editorial assistance. Support for this assistance was funded by Janssen Pharmaceutical K.K.
Funding
This study was sponsored by Janssen Pharmaceutical K.K. who also funded the journal’s Rapid Service Fee.
Author information
Authors and Affiliations
Contributions
Kazuki Kitahara is the corresponding author of this manuscript. Kazuki Kitahara and Junichi Omura contributed to study design, protocol development, data interpretation, manuscript preparation, and manuscript approval. Junichi Omura made medical judgments from the perspective of a physician. Shingo Wada and Seok-Won Kim contributed to data analysis and manuscript preparation. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the entire work, and give their approval for this version to be published.
Corresponding author
Ethics declarations
Conflict of Interest
Kazuki Kitahara and Junichi Omura are employees of Janssen Pharmaceutical K.K. Shingo Wada and Seok-Won Kim have nothing to disclose.
Ethical Approval
This retrospective database analysis study did not collect, transmit, or use identifiable patient data. Based on the Japanese Ethical Guidelines for Medical and Biological Research Involving Human Subjects, this study did not require any approval from an institutional review board. Permission was obtained from the MDV database for the use of their data for this study.
Additional information
The original online version of this article was revised: The alignment of the heading in table 3 was incorrect.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kitahara, K., Omura, J., Wada, S. et al. Patient and Therapeutic Profiles of Pulmonary Hypertension in Chronic Lung Diseases in Japan: A Cohort Study Using a Claims Database. Pulm Ther 10, 21–49 (2024). https://doi.org/10.1007/s41030-023-00243-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41030-023-00243-x