
Abstract
Sclerotinia sclerotiorum, a homothallic plant pathogen, undergoes sexual reproduction via haploid selfing (equivalent to clonal reproduction), and produces long-lasting surviving vegetative structures called sclerotia, enhancing clonal persistence and spread. Thus it is not surprising to detect clones of the species. Whether outcrossing can occur in the homothallic S. sclerotiorum remains unanswered. Early studies showed that S. sclerotiorum has a clonal population structure, consistent with its life history traits. However, recent studies using polymorphic and co-dominant molecular markers showed frequent genetic recombination, suggesting outcrossing. This review focuses on recent developments in population genetics studies related to detecting recombination, random association of alleles and dynamic mating type (MAT) alleles in Sclerotinia. Despite frequent reports of random association of alleles, the mechanisms for outcrossing in a homothallic species remain elusive. Recent intriguing findings are: the MAT genes in Sclerotinia are subject to inversion or deletion in every meiotic generation, the MAT gene deletion is related to ascospore dimorphism and mating type switching in S. trifoliorum, and ascospore dimorphism was also observed in S. sclerotiorum. Determining the nature of the dimorphic ascospores and their prevalence in relation to environmental cues could significantly advance our understanding how S. sclerotiorum populations behave in nature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sclerotinia sclerotiorum is a ubiquitous necrotrophic plant pathogen. It causes various diseases generally called white mold or stem rot on more than 400 plant species including many economically important crops (Boland and Hall 1994). Management of Sclerotinia diseases is difficult because resistance to Sclerotinia is inadequate or unavailable in many crop cultivars (Bolton et al. 2006). The pathogen produces recalcitrant vegetative surviving structure sclerotia, but no other forms of asexual spores (Bolton et al. 2006). Sclerotia can germinate by means of mycelium or are induced to go through sexual processes by means of self-fertilization to produce apothecia with ascospores. However, its behavior in nature is more complicated than what is expected from laboratory observations, as indicated in many population genetics studies. Because of its microscopic nature and scarcity of observable morphological features, a better understanding of its behavior in nature relies on molecular population genetics studies.
Population genetics, a subdiscipline of genetics, is the study of genetic variation of populations and its change over time and space, and molecular population genetics is the study of genetic variations at molecular level (Casillas and Barbadilla 2017). Studying the changes and differences in genetic variation through mathematical modeling allows inference about the behavior of the populations and prediction of their evolutionary potential (Casillas and Barbadilla 2017). Even though uniform crops in monoculture are ideal environments for pathogen proliferation and disease outbreaks, agricultural practices such as crop rotation, deployment of resistant cultivars, and application of pesticides often impose purifying (negative) selection on plant pathogens. According to Fisher’s fundamental theorem of natural selection, pathogen populations with less genetic variation for fitness traits may be vulnerable to extinction (McDonald 1997) and populations with high levels of genetic variation can easily adapt to adverse environments (McDonald and Linde 2002). On the other hand, populations that were highly adapted to a certain environment could have less genetic diversity due to the erosion of genetic variation under selection pressure (Falconer and Mackay 1996). Therefore, studying phenotypic and genetic diversity and population structure provides necessary information for designing and implementing effective disease management strategies. A thorough understanding of a pathogen population may allow prediction of its evolutionary potential. However, microorganisms like Sclerotinia have only a few observable morphological traits that can be used in determining population differentiation. Studies on the limited observable traits of S. sclerotiorum, such as sclerotial formation, mycelial growth and pigmentation, oxalic acid production, virulence and fungicide resistance showed that these quantitative traits often exhibit wide range of variations and usually are not related to origins of host plants or geography (Morrall et al. 1972; Price and Colhoun 1975; Atallah et al. 2004; Durman et al. 2005; Mert-Turk et al. 2007; Irani et al. 2011; Attanayake et al. 2012, 2013). Because of the limited usefulness and the scarcity of the observable features of S. sclerotiorum, molecular population genetics studies are crucial for a better understanding of the behavior of S. sclerotiorum populations in nature.
Life history traits and early population genetics studies of S. sclerotiorum
Although S. sclerotiorum does not produce conidia (asexual spores), it produces numerous recalcitrant surviving vegetative structures called sclerotia and it produces sexual ascospores through self-fertilization (haploid selfing), both enhancing clonal reproduction (Bolton et al. 2006). Sclerotia can survive in soil for many years (Steadman 1983) and may regenerate new sclerotia in the absence of a host plant (Williams and Western 1965), prolonging its survival. Sclerotial density in soil can reach up to 700 sclerotia per square meter of soil (Gilbert 1987) or more than 160 sclerotia in a single diseased cabbage head (Mahalingam et al. 2016). Sclerotia can germinate either myceliogenically or carpogenically under cool (4–20 °C) and moist conditions via self fertilization and produce apothecia with ascospores (Steadman 1983). Ascospores released to the air can travel about 100 m radius under crop canopy (Steadman 1983; Ben-Yephet et al. 1993), but ascospores travelling 3–4 km from the source was also reported (Cubeta et al. 1997). Although S. sclerotiorum can reproduce sexually, homothallic sexual reproduction in a haploid organism is considered equivalent to clonal reproduction (Billiard et al. 2012).
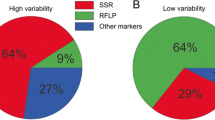
Early studies on population genetics of S. sclerotiorum pioneered by Kohn and associates (Kohli et al. 1992, 1995; Kohn 1995; Cubeta et al. 1997; Kohli and Kohn 1998; Carbone et al. 1999) employed Mycelial Compatibility Groupings (MCGs) and Restriction Fragment Length Polymorphism (RFLP) fingerprinting probed with a repeated dispersed DNA element, and showed that S. sclerotiorum populations have generally low levels of genetic variation and a clonal genetic structure. Mycelial compatibility grouping is based on a self and non-self recognition system, and is likely a multi-genic trait. All the mycelially compatible isolates based on a mycelium-mycelium interaction assay are grouped into a single MCG and the MCG grouping was also highly correlated with DNA fingerprints. Isolates of the same MCG shared the same DNA fingerprint and isolates in different MCGs had different DNA fingerprints. All evidence including association of independent markers showed a clonal genetic structure of S. sclerotiorum, consistent with the life history traits such as sclerotial production and homothallic sexual reproduction. Clonal population structure has been reported from a variety of crops from Australia, Canada, Iran, New Zealand, Turkey, UK and the USA (Kohli et al. 1992, 1995; Kohn 1995; Cubeta et al. 1997; Kohli and Kohn 1998; Carbone et al. 1999; Hambleton et al. 2002; Phillips et al. 2002; Malvárez et al. 2007; Ekins et al. 2011). Many of these populations also exhibited other features of clonal structure such as dominance of a small number of multilocus genotypes at high frequencies from a wider geographic area and repeated recovery of the dominant genotypes over several years in the same area.
Although the MCG grouping is technically easy, occasionally the results could be ambiguous for certain isolates (Kamvar et al. 2017). Moreover, with increasing number of MCGs the number of required pairing tests for every isolate with every MCG increases exponentially and become impractical. Another limitation of MCG grouping is that although isolates of the same MCGs can be identified, their relative relationship with isolates in other MCGs cannot be assessed. Recently the usefulness of MCG in studying genetic diversity was questioned (Kamvar et al. 2017). The RFLP based DNA fingerprinting using Southern hybridization is reliable, but technically demanding, requiring special skills. The hybridizing bands considered as the same size may not be due to descent, creating homoplasy and underestimating genetic diversity. Also the number of fingerprints that can be unambiguously resolved on a polyacriamide gel is conceivably limited. Therefore, techniques with higher resolution are desirable.
Microsatellite (or simple sequence repeat) markers have advantages that can overcome the limitations of the other marker systems discussed above. The locus-specific PCR primers for microsatellite markers developed by Sirjusingh and Kohn (2001) have been broadly used in population genetics studies of S. sclerotiorum. Applications of the microsatellite markers and advanced fragment analysis technologies are found in most of recent studies of S. sclerotiorum and are shown to be the main reason of increased levels of genetic variation detected in S. sclerotiorum populations in recent studies (Lehner et al. 2017; Lehner and Mizubuti 2017).
Genetic variation and population differentiation
Two main components of population genetics are genetic variation within populations and genetic differentiation among populations. Genetic variation can be measured in terms of genic diversity and genotypic diversity. Measures of genic diversity include the proportion of polymorphic loci, number of alleles, average number of alleles per locus and allele frequency, and average expected heterozygosity (He, Nei’s genetic diversity). Measures of genotypic diversity include genotypic richness, the number of genotypes observed in a population (g), and the number of shared genotypes between populations. Stoddart and Taylor’s G (1988), Simpson’s index (1949) and Shannon-Wiener’s index (Shannon and Weaver 1949) are also commonly used indices in estimating genotypic diversities. Caution should be taken when sample sizes differ and a rarefaction approach should be employed to facilitate comparison (Grunwald et al. 2003). A convenient package popper in R incorporating many of the tools is available for most of the population genetics analyses (Kamvar et al. 2014). Genetic variation reported for S. sclerotiourm populations varied widely depending on the markers employed and populations studied, ranging from a few genotypes dominating the populations to almost every isolate being a unique genotype (Malvárez et al. 2007; Clarkson et al. 2013; Aldrich-Wolfe et al. 2015; Clarkson et al. 2017; Kamvar et al. 2017; Lehner et al. 2017).
Besides the levels of genetic variation within populations, another important aspect of population genetics is differentiation among populations (McDonald and Linde 2002). One of the earliest and frequently used methods of quantifying genetic differentiation is based on estimating the degree of inbreeding within a subpopulation relative to the total population and termed as inbreeding coefficient or FST (Wright 1949). FST was first introduced for bi-allelic loci considering two subpopulations and can have theoretical extremes of one to zero. In general, the higher the FST value, the greater the population differentiation (Wright 1978; Hartl and Clark 1997). With the development of molecular tools it was found that many markers have multiple alleles and Nei (1997) redefined the fixation index for multiple alleles and termed as GST. A similar measurement to GST, θ was introduced by Weir and Cockerham (1984) using the analysis of variance frame work taking the sample size differences into consideration. With the rapid development in microsatellite marker technique, a large amount of data became available for many organisms including S. sclerotiorum populations. The classical stepwise mutation model (SMM) is the simplest and most popular model used to explain evolution of microsatellite loci (Kimura and Ohta 1978). To quantify genetic differentiation, Slatkin (1995) incorporated SMM into the pairwise population differentiation index, termed as RST, an analog to FST. Even though it is a highly debated issue whether to use FST, GST, θ, RST or the recently introduced Jost D (Gerlach et al. 2010; Leng and Zhang 2011; Whitlock 2011), many studies have used RST in detecting population differentiation of S. sclerotiorum.
Using RST type analysis, significant population differentiation has been reported among populations from a wide range of geographic scale: from continents, countries, regions, to fields or different seasons of the same fields. For example, significant population differentiation has been observed between canola fields of China and USA (Attanayake et al. 2013) and among several countries in Europe (Clarkson et al. 2017). Populations from North America and South America are differentiated, although they have similar levels of genotypic variability (Lehner et al. 2017). When considering the population differentiation within a country, significant differentiation among canola fields in Australia (Sexton and Howlett 2004), and in Iran (Hemmati et al. 2009), among fields of a variety of crops in different States in the USA (Malvárez et al. 2007; Aldrich-Wolfe et al. 2015) and among dry bean fields in Brazil (Gomes et al. 2011) has been reported. No population differentiation was found between different inoculum sources (ascospores vs mycelium) in the same fields (Sexton et al. 2006; Ekins et al. 2011). However, Atallah et al. (2004) found that ascospore populations differentiated from lesion derived populations, even though samples obtained from different potato fields were not significantly differentiated.
In addition to the RST type analysis, population differentiation or population subdivision could also be detected using Bayesian iterative algorithm of population assignment developed by Pritchard et al. (2000), and implemented in the software STRUCTURE. It uses a Markov Chain Monte Carlo (MCMC) method and assigns each individual into pre-determined clusters regardless of their geographic origin, and then using allele frequencies it assigns individuals into subpopulations based on analysis of likelihood (Porras-Hurtado et al. 2013). STRUCTURE analysis determines whether a population is structured or not, and has been widely used in detecting genetic subdivision in populations of S. sclerotiorum (Hemmati et al. 2009; Attanayake et al. 2012, 2013; Sun et al. 2013; Ali et al. 2014; Engelthaler et al. 2016; Clarkson et al. 2017). Detecting hidden genetic subdivision using STRUCTURE is useful for analyzing genetic recombination, particularly in the multilocus Index of Association (IA) analyses.
Genetic recombination
Whether a pathogen population is clonal or recombining (outcrossing) has significant implications in its evolutionary potential (Billiard et al. 2012). Genetic recombination is the hallmark of outcrossing (Milgroom 1996; Taylor et al. 1999). In heterothallic species, the mechanism of requiring mating with a partner of opposite mating type can prevent selfing. However, there are no known mechanisms that can prevent outcrossing in homothallic species. Thus, can outcrossing occur in the homothallic S. sclerotiorum? This question is best addressed by studying association of alleles among different loci. Random association of alleles among unlinked loci would suggest outcrossing, whereas non-random association of alleles would indicate clonal reproduction. Non-random association of alleles is called linkage disequilibrium which sometimes is referred to as gametic disequilibrium to differentiate it from physical linkage (Hedrick 1987). Because of its common usage in the literature concerning S. sclerotiorum, linkage disequilibrium is used in this article, unless indicated otherwise.
Traditionally linkage equilibrium or random association of alleles is estimated by testing association of alleles of loci in pairwise comparisons. The proportion of the pairwise comparisons that are at equilibrium provides indication of likelihood of either clonality or recombination. Significant proportions of pairwise comparisons were reported to be at linkage equilibrium even in S. sclerotiorum populations considered to be clonal. For example, Kohli and Kohn (1998) reported that 44.5 and 80.5% of pairs of loci were at linkage equilibrium. Linkage equilibrium was also reported in other populations even for pairs of loci that are physically linked (Attanayake et al. 2012, 2014). A more powerful analysis is the Multilocus Index of Association (IA) tests (Maynard-Smith et al. 1993), implemented in the program MultiLocus (Agapow and Burt 2001). In the IA test, the null hypothesis is random association of alleles (random mating). In a recombining population, the distribution of genetic distance between pairs of isolates is normal, whereas in clonal populations it is not (Maynard-Smith et al. 1993; Taylor et al. 1999). This is tested using the variance of genetic distances between pairs of isolates and the variance is lower with a normal distribution in recombining populations than those in clonal populations. To test the statistical significance, the observed dataset is randomly shuffled by re-sampling without replacement of alleles within each locus simulating random mating to generate a recombining dataset for computation (Taylor et al. 1999; Agapow and Burt 2001). The significance is the proportion of the reshuffled datasets that have higher IA values than the observed value. It should be noted that the test is not for a significant difference from zero (Taylor et al. 1999). Thus, how the observed data is shuffled profoundly affect the p value. If the population is subdivided, shuffling the data randomly in the whole population will always be biased toward clonality (Maynard-Smith et al. 1993; Taylor et al. 1999; Agapow and Burt 2001).
Many studies have concluded that S. sclerotiorum populations are clonal mainly because the Multilocus Index of Association tests rejected the null hypothesis of random association of alleles (Atallah et al. 2004; Sexton and Howlett 2004; Malvárez et al. 2007; Hemmati et al. 2009; Clarkson et al. 2013; Aldrich-Wolfe et al. 2015; Clarkson et al. 2017; Lehner et al. 2017). Such a conclusion was reached even for populations that had high levels of genotypic diversity, such as in cases where majority (>90%) of the isolates each have a unique genotype and/or where high proportions of pairwise loci were at linkage equilibrium. The statistic association of alleles could be caused by a number of biological processes beside clonal reproduction. Those biological processes should be carefully examined before rejecting the null hypothesis of random mating (Maynard-Smith et al. 1993; Taylor et al. 1999). One of the biological processes that can cause association of alleles is population subdivision (Milgroom 1996). In some of the studies on S. sclerotiorum, population subdivision was not assessed and not known, and in some other cases population subdivision was detected in STRUCTURE analysis but this information was not taken into account in IA analysis.
The statistic significance of the IA value should be interpreted with caution, especially when the null hypothesis of random association of alleles is rejected. That is because other biological processes may lead to an appearance of association of alleles even when the population may actually be recombining. Such biological processes include linkage, isolation by distance, natural selection, epidemical populations, and population subdivision (Maynard-Smith et al. 1993; Taylor et al. 1999). Clone correction can remove the bias presented by over-presentation of certain clonal genotypes such as in epidemic populations. Many examples are available that clone correction increased sensitivity in detecting recombination (Atallah et al. 2004; Sexton and Howlett 2004; Attanayake et al. 2012, 2013; Clarkson et al. 2013; Attanayake et al. 2014; Aldrich-Wolfe et al. 2015; Clarkson et al. 2017; Lehner et al. 2017; Weber 2017). Population subdivision can also lead to false significance of an IA value. When the population is subdivided and the subdivision information is not taken into account, IA analyses of the whole population will skew toward clonality (Milgroom 1996; Taylor et al. 1999) as pointed out in the program manual of MultiLocus and demonstrated in other fungi; Coccidioides immitis (Koufopanou et al. 1997); Cryptococcus gattii (Campbell et al. 2005); Phytophthora infestans (Montarry et al. 2010)\ and Alternaria alternata (Stewart et al. 2013). The MultiLocus program provides an option to specify population subdivision, but this option is seldom used in analyzing S. sclerotiorum populations. Specifying population subdivisions does not affect the statistics of genotype diversity or linkage disequilibrium, but does affect randomization process (the way the alleles are shuffled) and consequently the p values associated with the statistics (Taylor et al. 1999). Specifying the population subdivision will maintain linkage disequilibrium caused by population subdivision in the reshuffled datasets used in calculating statistic significance levels (Agapow and Burt 2001).
Lehner and Mizubuti (2017) pointed out that one form of genetic subdivision in S. sclerotiorum is MCG. Isolates within MCGs are more likely to exchange genetic materials resulting in recombination than isolates belonging to different MCGs. In some cases genetic subdivision could be hidden, but detectable using STRUCTURE analysis, as discussed above. For example, Attanayake et al. (2012) found seemingly conflicting results that random association of alleles was found in a significant portion of pairwise loci comparisons, but IA test rejected random association of alleles. However, when the genetic subdivision detected by STRUCTURE was specified in the IA analysis the null hypothesis of random association of alleles could not be rejected (Attanayake et al. 2012). Similarly, Weber (2017) in studying two populations (N > 90 isolates for each population) of S. sclerotiorum from sunflower fields found significant association of alleles in IA tests for both field populations either before or after clone correction, and also detected two genetic subpopulations in each field population using STRUCTURE analysis. When the genetic subdivision detected in STRUCTURE analysis was specified in the IA analysis, the null hypothesis of random association of alleles could not be rejected. These results suggest that the isolates may outcross only with other isolates of the same subpopulation detectable in STRUCTURE analysis. This resembles the situation depicted by Fig. 1b of Maynard-Smith et al. (1993) where frequent recombination occurs between isolates within a subpopulation, but does not occur between isolates from different subpopulations.
In a comprehensive study of population structure of S. sclerotiorum in the UK, Clarkson et al. (2013) studied six populations from five agricultural crops and six populations from wild meadow buttercup with 32 isolates representing each of the 12 populations. Clarkson et al. (2013) analyzed the populations using microsatellite markers and partial DNA sequences of IGS, and found generally high levels of genetic variation and populations are generally not well differentiated. STRUCTURE analysis showed population subdivision of three subpopulations in each population except the EV1 meadow buttercup population in which all isolates belonged to one subpopulation. In IA analysis, all populations showed significant IA values rejecting random association of alleles in all the populations except the EV1 population (Clarkson et al. 2013). A possible explanation is the clonality bias caused by population subdivision in the 11 populations in which random association of alleles was rejected (Clarkson et al. 2013).
Likewise, failure to reject random association of alleles by an insignificant IA value may be due to either outcrossing or mutation, particularly for microsatellite loci that have high mutation rates. Attanayake et al. (2014) developed a novel method to differentiate whether random association of alleles was due to outcrossing or mutation. Attanayake et al. (2012) have observed in pairwise linkage disequilibrium analyses that loci located on the same chromosomes showed linkage equilibrium. It is uncertain that such linkage equilibrium exhibited by physically linked loci was due to outcrossing followed by crossover in meiosis or due to mutation of the microsatellite loci, since IA test could not differentiate the two causes. Measuring linkage disequilibrium decay with increasing distance between loci could help differentiate the two causes because recombination rate due to crossover is related to physical distances between the linked loci, whereas mutation would be independent of the physical distance. This concept, although has been used in genetic mapping in controlled populations, has not been previously applied to fungal population genetics. In a proof-of-the concept study, Attanayake et al. (2014) using 12 microsatellite markers located on three chromosomes showed that linkage disequilibrium decayed with increasing physical distances in eight populations comprising of 268 isolates. The results demonstrated not only that the observed recombination was due to outcrossing, not mutation of the microsatellite markers, but also that outcrossing occurs frequently in populations of S. sclerotiorum. This study provided collaborating evidence of previous observations that different genotypes were detected among sibling ascospores from single apothecia (Atallah et al. 2004).
However, despite the overwhelming evidence of genetic recombination implicating outcrossing, the mechanisms of outcrossing in the homothallic S. sclerotiorum remain elusive. Two mechanisms have been suggested. Mycelia of different genotypes may together form sclerotia on co-infected plants that provide proximity for outcrossing (Sexton and Howlett 2004; Lehner et al. 2015). Heterokaryons were detected in some isolates, providing another possibility of outcrossing (Chitrampalam et al. 2015). There is another possible mechanism of outcrossing in S. sclerotiorum. Because MAT genes control mating behavior (Coppin et al. 1997), it is very intriguing to note the dynamic nature of MAT alleles in relation to mating type switching in Sclerotinia (Chitrampalam et al. 2013; Xu et al. 2016) and the observation of ascospore dimorphism in S. sclerotiorum (Ekins et al. 2006), which could be potentially related to outcrossing.
Dynamic mating type (MAT) alleles and dimorphic Ascospores in Sclerotinia species
The genus Sclerotinia includes species with different reproductive modes: homothallic and heterothallic species (Kohn 1995). Isolates of heterothallic species require a partner of opposite mating type for sexual reproduction, whereas isolates of homothallic species can complete sexual reproduction by itself without a partner. Similar to other fungi, this process is controlled by the mating type (MAT) locus with two alleles (idiomorphs) (Coppin et al. 1997). One idiomorph (MAT1-1) contains a transcription factor with an alpha-1 domain (the alpha box) and the other idiomorph (MAT1-2) encodes a protein with a high mobility group (HMG) DNA-binding domain (Coppin et al. 1997). In the homothallic S. sclerotiorum, the two idiomorphs are arranged in tandem at the same location (Amselem et al. 2011). There are four mating type genes: MAT1-1-1 and MAT1-1-5 of the alpha box and MAT1-2-1 and MAT1-2-4 of the HMG domain (Amselem et al. 2011). However, the arrangement of the genes at the MAT locus was later found to be dynamic and changes in every meiotic generation (Chitrampalam et al. 2013).
It has been recently found that S. sclerotiorum has peculiar MAT alleles for a homothallic species (Chitrampalam et al. 2013). A 3.6-kb sequence region of the MAT locus inverts itself in half of its ascospores during meiosis. This inversion affects orientation of three of the four MAT genes: a truncation at the 3′-end of MAT1-1-1 gene, and inversion of MAT1-2-1 and MAT1-2-4 genes (Chitrampalam et al. 2013). The inversion creates two MAT alleles: inversion positive (Inv+) and inversion negative (Inv-). Although MAT1-1-1 is truncated in the Inv+ isolates, the Inv+ isolates are capable of selfing (Chitrampalam et al. 2013). Therefore, the Inv+ and Inv- isolates are capable of self-fertilization (homothallic) and are indistinguishable phenotypically. The inverted region is flanked by a repeating motif of 250 bps in opposite orientation (inverted repeats) (Chitrampalam et al. 2013). Similarly such inversions of the MAT locus also occur in the sister species S. minor and the inverted DNA region is flanked by a 256-bp inverted repeat motif (Chitrampalam and Pryor 2015).
Sclerotinia trifoliorum, on the other hand, is a heterothallic species and its definitive identifying character is ascospore dimorphism (4 large and 4 small ascospores in every ascus) (Kohn 1979; Uhm and Fujii 1983). Large ascospore-derived strains are homothallic and produce dimorphic ascospores again, but small ascospore-derived strains are heterothallic, which is termed mating type switching (Uhm and Fujii 1983). The mating type switching is due to DNA deletion of a 2891-bp region, causing the loss of the entire two MAT1-2 genes (Xu et al. 2016). The deleted DNA region is flanked by a 146-bp DNA repeating motif arranged in the same direction (direct repeats). The repeating motifs in the three Sclerotinia species can be unambiguously aligned, indicating a common origin (Xu et al. 2016). Through tetrad analysis, Xu et al. (2016) found that strains derived from the small ascospores have the MAT1-2 genes missing and the strains derived from the large ascospores have the intact and complete MAT1-1 and MAT1-2 genes. Because of the lack of MAT1-2 genes, the small ascospore strains are heterothallic, requiring mating with microconidia from strains derived from large ascospores (Uhm and Fujii 1983). The DNA deletion is consistently associated with the small size of ascospores and heterothallism in S. trifoliorum (Xu et al. 2016). Direct repeat-mediated mating type switching is also reported in other fungi, although the repeating sequence motifs are of different origins from that of the Sclerotinia species (Wilken et al. 2014; Yun et al. 2017).
It is of particular interest to note that ascospore dimorphism was also observed in some isolates of S. sclerotiorum and S. minor (Ekins et al. 2006). Both species were described to produce only monomorphic ascospores (Kohn 1979). Ekins et al. (2006) carefully examined ascospores in asci and discovered that one out of three S. sclerotiorum isolates had 4.9% of the asci showing ascospore dimorphism and three out of five S. minor isolates with 1 to 3.5% of the asci showing ascospore dimorphism. Whether those small ascospores are hetherothallic or not was not assessed (Ekins et al. 2006; Ekins, personal communication), although this observation was incorrectly cited as “both large and small ascospores were found to be homothallic” (Malvárez et al. 2007). If what we know in S. trifoliorum also occurs in S. sclerotiorum, the small-sized ascospores could also be due to deletion or loss-of-function of certain MAT genes affected during the MAT gene inversion process either by chance or potentially in response to yet unknown environmental cues. Further research is needed to determine whether the small-sized ascospores are capable of self-fertilization and whether the ascospore dimorphisms observed in S. sclerotiorum and S. minor are due to deletion or non-function of certain MAT genes. That would possibly explain why genetic recombination is so common in some populations of S. sclerotiorum.
Summary
S. sclerotiorum is homothallic in sexual reproduction via self-fertilization (haploid selfing), not requiring mating with another partner. Strict homothallism in haploid organisms is equivalent to clonal reproduction. Even though S. sclerotiorum does not produce any asexual spores, it produces long-lasting recalcitrant vegetative surviving structures called sclerotia, which are an efficient means of clonal reproduction, further enhancing clonal persistence and spread. Thus it is not surprising that clones of the species are detected in many populations. However, although the mechanisms of heterothallism can prevent self-fertilization, homothallism has no known mechanisms preventing outcrossing. Whether and how outcrossing can occur in the homothallic S. sclerotiorum is still unknown. Early studies showed that S. sclerotiorum has a clonal population structure evidenced by association of independent markers such as MCG and DNA RFLP fingerprints, and prevalence and persistence of a few dominant genotypes, consistent with the life history traits of S. sclerotiorum. However, recent studies using highly polymorphic and co-dominant molecular markers such as microsatellites showed frequent genetic recombination, suggesting outcrossing. This review focused on recent developments in detecting random association of alleles and discovery of the dynamic nature of mating type (MAT) alleles in Sclerotinia species. Random association of alleles is frequently reported in S. sclerotiorum populations. Hidden population subdivisions were sometimes overlooked in Index of Association analyses, creating bias toward clonality, suggesting genetic recombination is more prevalent in S. sclerotiorum populations than recognized. Yet, the mechanisms for such widespread genetic recombination in a homothallic species remain elusive. Suggested mechanisms include formation of sclerotia by mycelia of different genotypes that co-infect host plants, providing proximity for outcrossing and formation of heterokaryon. The revelation of the relationship among MAT gene deletion, ascospore dimorphism, and mating type switching in S. trifoliorum, in addition to the observation of ascospore dimorphisms in S. sclerotiorum is intriguing. The recent intriguing findings include: the genes in the mating type (MAT) locus in Sclerotinia species are subject to inversion or deletion in every meiotic generation mediated by repeating DNA sequence motifs, the MAT gene deletion is related to ascospore dimorphism and mating type switching in S. trifoliorum, and ascospore dimorphism was also observed in isolates of S. sclerotiorum and S. minor. These findings support a speculation that the ascospore dimorphism in S. sclerotiorum and S. minor could also be due to deletion or otherwise loss-of-function of certain MAT genes. Determining the nature of the dimorphic ascospores and their occurrence and prevalence in relation to environmental cues could significantly advance our understanding how S. sclerotiorum populations behave in nature.
References
Agapow P-M, Burt A (2001) Indices of multilocus linkage disequilibrium. Molecular Ecology Notes 1:101–102
Aldrich-Wolfe L, Travers S, Nelson BD Jr (2015) Genetic variation of Sclerotinia sclerotiorum from multiple crops in the north Central United States. PLoS One 10:e0139188
Ali S, Gladieux P, Leconte M, Gautier A, Justesen AF, Hovmoller MS, Enjalbert J, de Vallavieille-Pope C (2014) Origin, migration routes and worldwide population genetic structure of the wheat yellow rust pathogen Puccinia striiformis f. sp. tritici. PLoS Pathogens 10:e1003903
Amselem J, Cuomo CA, van Kan JA, Viaud M, Benito EP, Couloux A, Coutinho PM, de Vries RP, Dyer PS, Fillinger S, Fournier E, Gout L, Hahn M, Kohn L, Lapalu N, Plummer KM, Pradier JM, Quevillon E, Sharon A, Simon A, Ten Have A, Tudzynski B, Tudzynski P, Wincker P, Andrew M, Anthouard V, Beever RE, Beffa R, Benoit I, Bouzid O, Brault B, Chen Z, Choquer M, Collemare J, Cotton P, Danchin EG, Da Silva C, Gautier A, Giraud C, Giraud T, Gonzalez C, Grossetete S, Guldener U, Henrissat B, Howlett BJ, Kodira C, Kretschmer M, Lappartient A, Leroch M, Levis C, Mauceli E, Neuveglise C, Oeser B, Pearson M, Poulain J, Poussereau N, Quesneville H, Rascle C, Schumacher J, Segurens B, Sexton A, Silva E, Sirven C, Soanes DM, Talbot NJ, Templeton M, Yandava C, Yarden O, Zeng Q, Rollins JA, Lebrun MH, Dickman M (2011) Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genetics 7:e1002230
Atallah ZK, Larget B, Chen X, Johnson DA (2004) High genetic diversity, phenotypic uniformity, and evidence of outcrossing in Sclerotinia sclerotiorum in the Columbia Basin of Washington state. Phytopathology 94:737–742
Attanayake RN, Porter L, Johnson DA, Chen W (2012) Genetic and phenotypic diversity and random association of DNA markers of isolates of the fungal plant pathogen Sclerotinia sclerotiorum from soil on a fine geographic scale. Soil Biology and Biochemistry 55:28–36
Attanayake RN, Carter PA, Jiang D, Del Rio-Mendoza L, Chen W (2013) Sclerotinia sclerotiorum populations infecting canola from China and the United States are genetically and phenotypically distinct. Phytopathology 103:750–761
Attanayake RN, Tennekoon V, Johnson DA, Porter LD, del Río-Mendoza L, Jiang D, Chen W (2014) Inferring outcrossing in the homothallic fungus Sclerotinia sclerotiorum using linkage disequilibrium decay. Heredity 113:353–363
Ben-Yephet Y, Genizi A, Siti E (1993) Sclerotial survival and apothecial production by Sclerotinia sclerotiorum following outbreaks of lettuce drop. Phytopathology 83:509–513
Billiard S, López-Villavicencio M, Hood ME, Giraud T (2012) Sex, outcrossing and mating types: unsolved questions in fungi and beyond. Journal of Evolutionary Biology 25:1020–1038
Boland GJ, Hall R (1994) Index of plant hosts of Sclerotinia sclerotiorum. Canadian Journal of Plant Pathology 16:93–108
Bolton MD, Thomma BPHJ, Nelson BD (2006) Sclerotinia sclerotiorum (Lib.) de Bary: Biology and molecular traits of a cosmopolitan pathogen. Molecular Plant Pathology 7:1–16
Campbell LT, Currie BJ, Krockenberger M, Malik R, Meyer W, Heitman J, Carter D (2005) Clonality and recombination in genetically differentiated subgroups of Cryptococcus gattii. Eukaryotic Cell 4:1403–1409
Carbone I, Anderson JB, Kohn LM (1999) Patterns of descent in clonal lineages and their multilocus fingerprints are resolved with combined gene genealogies. Evolution 53:11–21
Casillas S, Barbadilla A (2017) Molecular population genetics. Genetics 205:1003–1035
Chitrampalam P, Pryor BM (2015) Characterization of mating type (MAT) alleles differentiated by a natural inversion in Sclerotinia minor. Plant Pathology 64:911–920
Chitrampalam P, Inderbitzin P, Maruthachalam K, Wu B-M, Subbarao KV (2013) The Sclerotinia sclerotiorum mating type locus (MAT) contains a 3.6-kb region that is inverted in every meiotic generation. PLoS One 8:e56895
Chitrampalam P, Qiu C, Aldrich-Wolfe L, Leng Y, Zhong S, Nelson B (2015) Prevalence of inversion positive and inversion negative mating type (MAT) alleles and MAT heterokaryons in Sclerotinia sclerotiorum in the United States. Botany 93:497–505
Clarkson JP, Coventry E, Kitchen J, Carter HE, Whipps JM (2013) Population structure of Sclerotinia sclerotiorum in crop and wild hosts in the UK. Plant Pathology 62:309–324
Clarkson JP, Warmington RJ, Walley PG, Denton-Giles M, Barbetti MJ, Brodal G, Nordskog B (2017) Population structure of Sclerotinia subarctica and Sclerotinia sclerotiorum in England, Scotland and Norway. Frontiers in Microbiology 8:490
Coppin E, Debuchy R, Arnaise S, Picard M (1997) Mating types and sexual development in filamentous ascomycetes. Microbiology and Molecular Biology Reviews 61:411–428
Cubeta MA, Cody BR, Kohli Y, Kohn LM (1997) Clonality of Sclerotinia sclerotiorum on infected cabbage in eastern North Carolina. Phytopathology 87:1000–1004
Durman SB, Menendez AB, Godeas AM (2005) Variation in oxalic acid production and mycelial compatibility within field populations of Sclerotinia sclerotiorum. Soil Biology and Biochemistry 37:2180–2184
Ekins M, Aitken EA, Coulter KC (2006) Homothallism in Sclerotinia minor. Mycological Research 110:1193–1199
Ekins MG, Hayden HL, Aitken EAB, Goulter KC (2011) Population structure of Sclerotinia sclerotiorum on sunflower in Australia. Australasian Plant Pathology 40:99–108
Engelthaler DM, Roe CC, Hepp CM, Teixeira M, Driebe EM, Schupp JM, Gade L, Waddell V, Komatsu K, Arathoon E, Logemann H, Thompson GR 3rd, Chiller T, Barker B, Keim P, Litvintseva AP (2016) Local population structure and patterns of western hemisphere dispersal for Coccidioides spp., the fungal cause of valley fever. MBio 7:e00550–e00516
Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics, 4th edn. Longman, Essx, England
Gerlach G, Jueterbock A, Kraemer P, Deppermann J, Harmand P (2010) Calculation of population differentiation based on Gst and D: forget Gst but not all of statistics! Molecular Ecology 19:3845–3852
Gilbert RG (1987) Crown and stem rot of alfalfa caused by Sclerotinia sclerotiorum. Plant Disease 71:739–742
Gomes EV, Breseguello L, Augusto M, Nasser LCB, Petrofeza S (2011) Microsatellite markers reveal genetic variation within Sclerotinia sclerotiorum populations in irrigated dry bean crops in Brazil. Journal of Phytopathology 159:94–99
Grunwald NJ, Goodwin SB, Milgroom MG, Fry WE (2003) Analysis of genotypic diversity data for populations of microorganisms. Phytopathology 93:738–746
Hambleton S, Walker C, Kohn LM (2002) Clonal lineages of Sclerotinia sclerotiorum previously known from other crops predominate in 1999-2000 samples from Ontario and Quebec soybean. Canadian Journal of Plant Pathology 24:309–315
Hartl DL, Clark AG (1997) Principles of Population Genetics. Third, Edition edn. Sinauer Associate, Inc. Publishers, Sunderland
Hedrick PW (1987) Gametic disequilibrium measures: proceed with caution. Genetics 117:331–341
Hemmati R, Javan-Nikkhah M, Linde CC (2009) Population genetic structure of Sclerotinia sclerotiorum on canola in Iran. European Journal of Plant Pathology 125:617–628
Irani H, Heydari A, Javan-Nikkah M, Ibrahimov AS (2011) Pathogenicity variation and mycelial compatibility groups in Sclerotinia sclerotiorum. Journal of Plant Protection Research 51:329–336
Kamvar ZN, Tabima JF, Grunwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281
Kamvar ZN, Amaradasa BS, Jhala R, McCoy S, Steadman JR, Everhart SE (2017) Population structure and phenotypic variation of Sclerotinia sclerotiorum from dry bean (Phaseolus vulgaris) in the United States. PeerJ 5:e4152
Kimura M, Ohta T (1978) Stepwise mutation model and distribution of allelic frequencies in a finite population. Proceedings of National Academy of Sciences of USA 76:2868–2872
Kohli Y, Kohn LM (1998) Random association among alleles in clonal populations of Sclerotinia sclerotiorum. Fungal Genetics and Biology 23:139–149
Kohli Y, Morrall RAA, Anderson JB, Kohn L (1992) Local and trans-Canadian clonal distribution of Sclerotinia sclerotiorum on canola. Phytopathology 82:875–880
Kohli Y, Brunner LJ, Yoell H, Milgroom MG, Anderson JB, Morrall RAA, Kohn LM (1995) Clonal dispersal and spatial mixing in populations of the plant pathogenic fungus, Sclerotinia sclerotiorum. Molecular Ecology 4:69–77
Kohn LM (1979) Delimitation of the economically important plant pathogenic Sclerotinia species. Phytopathology 69:881–886
Kohn LM (1995) The clonal dynamic in wild and agricultural plant-pathogen populations. Canadian Journal of Botany 73:1231–1240
Koufopanou V, Burt A, Taylor JW (1997) Concordance of gene genealogies reveals reproductive isolation in the pathogenic fungus Coccidioides immitis. Proceedings of the National Academy of Sciences of the United States of America 94:5478–5482
Lehner MS, Mizubuti ESG (2017) Are Sclerotinia sclerotiorum populations from the tropics more variable than those from subtropical and temperate zones? Tropical Plant Pathology 42:61–69
Lehner MS, Júnior TJP, Júnior BTH, Teixeira H, Vieira RF, Carneiro JES, Mizubuti ESG (2015) Low genetic variability in Sclerotinia sclerotiorum populations from common bean fields in Minas Gerais state, Brazil, at regional, local and micro? Scales. Plant Pathology 64:921–931
Lehner MS, De Paula Junior TJ, Del Ponte EM, Mizubuti ESG, Pethybridge SJ (2017) Independently founded populations of Sclerotinia sclerotiorum from a tropical and a temperate region have similar genetic structure. PLoS One 12:1–14
Leng L, Zhang DE (2011) Measuring population differentiation using Gst or D? A simulation study with microsatellite DNA markers under a finite island model and nonequilibrium conditions. Molecular Ecology 20:2494–2509
Mahalingam T, Guruge BMA, Somachandra KP, Rajapakse CS, Attanayake RN (2016) First report of white Mold caused by Sclerotinia sclerotiorum on cabbage in Sri Lanka. Plant Disease 101:249–249
Malvárez G, Carbone I, Grünwald NJ, Subbarao KV, Schafer M, Kohn LM (2007) New populations of Sclerotinia sclerotiorum from lettuce in California and peas and lentils in Washington. Phytopathology 97:470–483
Maynard-Smith J, Smith NH, O'Rourke M, Spratt BG (1993) How clonal are bacteria? Proceedings of National Academy of Sciences of USA 90:4384–4388
McDonald BA (1997) The population genetics of Fungi: tools and techniques. Phytopathology 87:448–453
McDonald BA, Linde C (2002) Pathogen population genetics, evolutionary potential, and durable resistance. Annual Review of Phytopathology 40:349–379
Mert-Turk F, Ipek M, Mermer D, Nicholson P (2007) Microsatellite and morphological markers reveal genetic variation within a population of Sclerotinia sclerotiorum from oilseed rape in the Canakkale province of Turkey. Journal of Phytopathology 155:182–187
Milgroom MG (1996) Recombination and the multilocus structure of fungal populations. Annual Review of Phytopathology 34:457–477
Montarry J, Andrivon D, Glais I, Corbiere R, Mialdea G, Delmotte F (2010) Microsatellite markers reveal two admixed genetic groups and an ongoing displacement within the French population of the invasive plant pathogen Phytophthora infestans. Molecular Ecology 19:1965–1977
Morrall RAA, Duczek LJ, Sheard JW (1972) Variations and correlations within and between morphology, pathogenicity and pectolytic enzyme activity in Sclerotinia from Saskatchewan. Canadian Journal of Botany 50:767–786
Nei M (1997) F-statistics and analysis of gene diversity in subdivided populations. Annals of Human Genetics 41:225–233
Phillips DV, Carbone I, Gold SE, Kohn LM (2002) Phylogeography and genotype-symptom associations in early and late season infections of canola by Sclerotinia sclerotiorum. Phytopathology 92:785–793
Porras-Hurtado L, Ruiz Y, Santos C, Phillips C, Carracedo A, Lareu MV (2013) An overview of STRUCTURE: applications, parameter settings, and supporting software. Frontiers in Genetics 4:98
Price K, Colhoun J (1975) Pathogenicity of isolates of Sclerotinia sclerotiorum (Lib.) de Bary to several hosts. Phytopathologische Zeitschrift 83:232–238
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Sexton AC, Howlett BJ (2004) Microsatellite markers reveal genetic differentiation among populations of Sclerotinia sclerotiorum from Australian canola fields. Current Genetics 46:357–365
Sexton AC, Whitten AR, Howlett BJ (2006) Population structure of Sclerotinia sclerotiorum in an Australian canola field at flowering and stem-infection stages of the disease cycle. Genome 49:1408–1415
Shannon CE, Weaver W (1949) The mathematical theory of communication. University of Illinois Press, Urbana
Simpson EH (1949) Measurement of diversity. Nature 163:688
Sirjusingh C, Kohn L (2001) Characterization of microsatellites in the fungal plant pathogen, Sclerotinia sclerotiorum. Molecular Ecology Notes 1:267–269
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Steadman JR (1983) White mold - a serious yied-limiting disease of bean. Plant Disease 67:346–350
Stewart JE, Thomas KA, Lawrence CB, Dang H, Pryor BM, Timmer LW, Peever TL (2013) Signatures of recombination in clonal lineages of the citrus brown spot pathogen Alternaria alternata sensu lato. Phytopathology 103:741–749
Stoddart JA, Taylor JF (1988) Genotypic diversity: estimation and prediction in samples. Genetics 118:705–711
Sun X, Kang S, Zhang Y, Tan X, Yu Y, He H, Zhang X, Liu Y, Wang S, Sun W, Cai L, Li S (2013) Genetic diversity and population structure of rice pathogen Ustilaginoidea virens in China. PLoS One 8:e76879
Taylor JW, Geiser DM, Burt A, Koufopanou V (1999) The evolutionary biology and population genetics underlying fungal strain typing. Clinical Microbiology Reviews 12:126–146
Uhm JY, Fujii H (1983) Heterothallism and mating type mutation in Sclerotinia trifoliorum. Phytopathology 73:569–572
Weber JM (2017) Management of white mold in hybrid sunflower seed crops in the Columbina basin of Central Washington. MS Thesis, Washington State University, Pullman, WA, USA.
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Whitlock MC (2011) G'ST and D do not replace FST. Molecular Ecology 20:1083–1091
Wilken PM, Steenkamp ET, Wingfield MJ, de Beer ZW, Wingfield BD (2014) DNA loss at the Ceratocystis fimbriata mating locus results in self- sterility. PLoS One 9:e92180
Williams GH, Western JH (1965) The biology of Sclerotinia trifoliorum Erikss. And other species of sclerotium-forming fungi II: the survival of sclerotia in soil. Annals of Applied Biology 56:261–268
Wright S (1949) The genetical structure of populations. Annals of Eugenics 15:323–354
Wright S (1978) Evolution and the genetics of populations volume 4 variability within and among populations. University of Chicago Press, Chicago
Xu L, Jardini TM, Chen W (2016) Direct repeat-mediated DNA deletion of the mating type MAT1-2 genes results in unidirectional mating type switching in Sclerotinia trifoliorum. Scientific Reports 6:27083–27083
Yun SH, Kim HK, Lee T, Turgeon BG (2017) Self-fertility in Chromocrea spinulosa is a consequence of direct repeat-mediated loss of MAT1-2, subsequent imbalance of nuclei differing in mating type, and recognition between unlike nuclei in a common cytoplasm. PLoS Genetics 13:e1006981
Acknowledgements
Funding was provided in part by the National Science Foundation, Sri Lanka – Research grant RG/2015/BT/04, the startup funding from Northwest A&F University, and USDA National Sclerotinia Initiative.
Author information
Authors and Affiliations
Corresponding author
Additional information
Section Editor: Sarah J. Pethybridge
Rights and permissions
About this article

Cite this article
Attanayake, R.N., Xu, L. & Chen, W. Sclerotinia sclerotiorum populations: clonal or recombining?. Trop. plant pathol. 44, 23–31 (2019). https://doi.org/10.1007/s40858-018-0248-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40858-018-0248-7