
Abstract
Purpose of Review
Hematopoietic stem cells (HSCs) reside in a specialized microenvironment called the HSC niche. While key components of the niche have been known for several years, recent advances have identified several additional cell types that regulate HSC in the bone marrow (BM). Here, we review our current understanding of the components and dynamics of the HSC niche.
Recent Findings
While the niche has been considered a stable structure, recent advances clearly show that the niche is regulated in a dynamic manner to control HSC traffic and function. Moreover, the niche can rapidly remodel in response to insults to the BM in a process controlled by positive and negative regulators.
Summary
Multiple niche cells have been shown to be dynamically regulated by systemic and local signals to influence how the niche controls HSC function. Elucidating how different components of the niche coordinate to orchestrate HSC behavior is essential to understand how the hematopoietic system adjusts blood cell production to the demands of the body.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The bone marrow (BM) provides diverse microenvironments that support and regulate the hematopoietic stem and progenitor cells responsible for blood cell production. The hematopoietic stem cell (HSC) niche is the most studied and characterized microenvironment, but several in vivo studies have identified niches that regulate the differentiation of other progenitors (e.g., common lymphoid progenitors [1, 2•]) and even mature cells (e.g., macrophages that support late erythropoiesis [3] and dendritic cells that support B cell differentiation [4]). These niches are not static, and different mechanisms regulate their function and structure to ensure normal blood cell production. The objective of this manuscript is to review our current understanding of the HSC niche and highlight how the dynamics of the niche impact HSC function and behavior.
Structure and Components of the HSC Niche
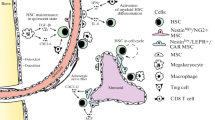
Early studies suggested that the HSC niche was mainly composed of osteoblasts and other cells present in the endosteal surface of the bone [5,6,7,8]. However, it is now clear that sinusoids, arteries, and associated perivascular cells form “vascular niches” that are the key regulators of HSC function [9, 10]. Here, we briefly discuss the main components of BM vascular niches during steady-state hematopoiesis as well as the evidence supporting a role for endosteal cells as niche components. A scheme showing key components of the niche is shown in Fig. 1.

Schematic representation of key cells and regulatory factors in the bone marrow hematopoietic stem cell niche
Vascular Niche Components
Endothelial Cells
The endothelial cells (EC) that line the blood vessels of adult mammals are crucial for proper nutrient and oxygen supply throughout the body, but EC are more than passive conduits; they provide essential signals that direct tissue maintenance and function [11, 12]. This fundamental role is especially important in the highly vascularized environment of the BM, where arteries and arterioles enter the BM and preferentially populate the endosteal region before connecting to a tridimensional venous sinusoidal network drained by a central vein [13, 14•]. During development, HSCs arise from a hemogenic endothelium [11]; they continue to remain closely associated with blood vessels throughout fetal development and adult life [11]. In the BM, most HSCs localize within < 5 μm of a vessel at any given time [13, 15]. EC are major sources of cytokines that regulate HSC function like CXCL12 and stem cell factor (SCF) [11, 12]. Conditional Cxcl12 and Scf deletion in the vasculature leads to a dramatic loss of HSC demonstrating that endothelial cells are critical HSC niche components [1, 16, 17].
Perivascular Stromal Cells
Broadly distributed throughout the BM, an abundant number of perivascular stromal cells can be found connected to one another and to adjacent arterioles or sinusoids [13, 14•]. These mesenchymal-derived cells have been identified using distinct genetic models and flow cytometry methods. For instance, CXCL12-abundant reticular cells (CAR cells) were identified using Cxcl12-gfp reporter mice, Nestin-gfpdim cells using Nestin-gfp mice [13, 18], LepR+ cells using LepR-cre mice [1], Prx1+ cells using Prx1-cre mice [17], and CD45−CD31−Ter119+CD51+CD140a+ cells from WT mice [19]. These methods largely overlap and likely identify the same cell population [20]. Perivascular cell ablation in vivo led to loss of BM HSC and mobilization to the periphery indicating a role for these cells as components of the niche [18]. Similar to endothelial cells, perivascular stromal cells are also major sources of cytokines known to regulate HSC including CXCL12 and SCF [1, 16, 17]. Conditional deletion of either molecule led to loss of HSC in the bone marrow [1, 16, 17]. However, a recent study suggested that Cxcl12 deletion in LepR+ cells does not impact HSC numbers or functions [21••]. As such, additional studies are necessary to clarify this discrepancy but it is clear that perivascular stromal cells are a critical component of the niche.
Periarteriolar Ng2+ Cells
Imaging analyses revealed that many HSC associated specifically with arteries and that arteries and arterioles were enriched in quiescent HSCs with reduced levels of reactive oxygen species [13, 21••, 22••]. Arterioles are ensheathed by Ng2+ perivascular cells (identified as Nes-GFPbright cells using Nestin-gfp mice or Ng2+ cells using Ng2-creERT2 mice [13]). Depletion of Ng2-CreERT2-marked cells using an inducible diphtheria toxin receptor (iDTR) line led to a shift in HSC localization away from the arterioles accompanied by a decrease in quiescence [13]. Conditional Cxcl12 deletion in Ng2+ cells recapitulated this HSC phenotype [21••], suggesting a critical role for Ng2+ cell-derived CXCL12 in maintaining HSC quiescence.
Non-myelinating Schwann Cells
Although sympathetic nerves do not regulate HSC directly, they are important regulators of the niche [23]. Sympathetic nerves sit on top of arteries and arterioles and are ensheathed by glial fibrillary acidic protein (GFAP)-positive non-myelinating Schwann cells, which are a major source of active TGFβ in the BM [24]. Although HSC can synthesize TGFβ themselves, they lack the machinery to convert it to active TGFβ, and therefore the GFAP+ cell-derived active TGFβ is critical for TGFβ-mediated HSC quiescence [24]. It has been proposed that active TGFβ promotes HSC quiescence by inhibiting the formation of cytokine-mediated lipid raft clustering, which is thought to augment essential signals for HSC entry into the cell cycle [24]. Surgical sympathectomy of the BM decreased the number of these GFAP+ non-myelinating Schwann cells and reduced the frequency of BM HSC thus suggesting that they are a component of the HSC niche [24].
Sinusoid-Associated Megakaryocytes
Megakaryocytes are large multinucleated cells that reside in sinusoids where they shed new platelets. Imaging analyses showed that approximately 35% of HSC were in direct contact with a megakaryocyte [25,26,27]. In vivo ablation of megakaryocytes led to dramatic increases in HSC numbers due to loss of quiescence and proliferation followed by loss of these cycling HSCs due to exhaustion [25, 26]. Megakaryocytes seem to regulate HSCs by secretion of multiple molecules including CXCL4 [25], TGFβ [26], and CLEC2 [27]. Megakaryocyte ablation or loss of CXCL4 signals led HSCs to relocate away from sinusoids and megakaryocytes suggesting that proximity to megakaryocytes is critical for HSC regulation [25].
Hematopoietic Stem and Progenitor Cells
Most hematopoietic stem and progenitor cells (HSPC) are perivascular and relatively close to each other [28]. This suggested that HSPC might be regulated by their interaction with other progenitors. Indeed, Leiva et al. showed that deletion of the selectin ligand ESL1 in HSPC induced proliferation of wild-type HSC in a non-cell autonomous mechanism [29••]. Endothelial expression of E-selectin regulates HSC quiescence [30], but ESL1 in HSPC functions, instead, by reducing the amount of TGFβ available to neighboring HSPC [29••].
Evidence for Arteriolar and Sinusoidal Niches
Multiple imaging studies using antibodies and transgenic reporter mice have demonstrated that most HSCs are perivascular [13, 14•, 15, 18, 25, 28]. However, the BM contains distinct types of vessels including sinusoids, arterioles, and transitional vessels [13, 31••]. Several lines of evidence support the idea that sinusoids and arteries are spatially distinct niches. HSCs associated with sinusoids are more proliferative than HSCs associated with arterioles. The Frenette group demonstrated that − 35% of all HSCs were located within 20 μm of BM arterioles and regulated by Ng2+ cells [13] while another − 35% of HSCs were in contact with sinusoidal megakaryocytes and away from arterioles [25]. Importantly, megakaryocyte ablation did not affect quiescence or HSC interaction with arterioles suggesting that HSCs in sinusoids and arterioles are independently regulated [25]. This is further supported by a recent study by the same group showing that conditional CXCL12 ablation in periarteriolar Ng2+ cells led to depletion of BM HSCs and reduced localization of HSCs to periarteriolar spaces [21••]. Two additional studies from the Nakauchi and Lapidot laboratories further support the idea that arterioles provide a niche that promotes HSC quiescence. Non-myelinating Schwann cells are critical regulators of HSC quiescence via TGFβ and are also intrinsically associated with arterioles [24]. HSCs associated with arteries have reduced levels of reactive oxygen species (ROS) whereas sinusoids are enriched in ROS+ HSCs [22••]. Additional evidence for a unique role of arterioles in regulation of HSC was provided by Kusumbe et al., wherein they showed that constitutive activation of Notch signaling in the vasculature caused a dramatic expansion in BM arterioles and a large increase in BM HSC numbers indicating that arteriole number controls HSC abundance [31••]. It is important to note, however, that other groups did not find evidence for quiescent HSCs being enriched in periarteriolar areas. Acar et al. using α-catulin-gfp reporter mice to detect HSCs found that only 10% of all HSCs resided close (less than 5 μm) to arterioles; they also did not find any enrichment for quiescent HSC near arterioles [14•]. Additional experiments are necessary to functionally test the concept that sinusoids and arterioles provide unique signals that regulate HSC.
Cells Not Directly Associated with the Vasculature
Macrophages
Another mature hematopoietic cell type that regulates HSC behavior is the macrophage. Unlike megakaryocytes, macrophages do not specifically associate with the vasculature as they are interspersed through the BM and a subset of macrophages “osteomacs” specifically localize to the endosteal surface. Also, and in contrast with megakaryocytes, they do not act directly on HSCs. Instead, macrophages influence HSC release from the niche by acting on perivascular stromal cells to modulate their CXCL12 production [32,33,34]. Depletion of CD169+ macrophages led to a selective downregulation in Cxcl12 expression in perivascular niche cells and resulted in HSC mobilization from the BM [32,33,34]. A recent study also showed that phagocyte depletion increased HSC numbers and that blockade of interferon-γ signaling in macrophage lineage cells regulated HSC quiescence [35]. This study strongly suggests that macrophages not only regulate trafficking but also regulate HSC proliferation.
Neutrophils and Other Myeloid Cells
The first evidence for a role of neutrophils came from the Hidalgo laboratory. This group demonstrated that aged neutrophils that return to the BM from circulation can trigger HSC mobilization by targeting BM macrophages. When aged neutrophils infiltrate the BM, they are homeostatically cleared by BM macrophages via phagocytosis, thereby activating the macrophages and ultimately triggering CXCL12 reduction and HSC egress from the BM [36]. A recent report showed that histamine production is restricted to myeloid cells in the bone marrow (including some myeloid-biased HSCs) and that these myeloid-biased HSCs associated specifically with Gr1+ myeloid cells (likely neutrophils) [37••]. Mice incapable of producing histamine showed reduced HSC quiescence and HSC purified from these mice showed impaired potential to reconstitute myeloid cells [37••]. HSC purified from mice lacking the histamine receptor H2R also showed engraftment defects after transplantation. These results thus strongly suggest that myeloid cells regulate myeloid-biased HSCs directly via histamine-H2R signaling [37••].
Adipocytes
The role of adipocytes in HSC regulation is controversial. A first report suggested that adipocytes were negative regulators of HSC maintenance. This was based on the observations that adipocyte-rich BM contains fewer HSCs and that “fatless” mice with drastically reduced BM adipocytes showed enhanced HSC reconstitution following irradiation and transplantation [38]. However, a subsequent study found no HSC phenotype upon in vivo treatment with the adipocyte-expanding drug troglitazone, suggesting no significant role for adipocytes in homeostatic HSC maintenance [39]. Even more recently, a third study has suggested a role for adipocytes and/or adipocyte progenitors in regulating HSC maintenance in post-irradiation or post-chemotherapy conditions via SCF production but not in the steady state [40••]. Although there appears to be a role for adipocytes, additional studies are required to clarify their role in the regulation of HSC function.
Bone-Associated Cells
Osteoblasts
Early in vitro studies found that osteoblasts are capable of promoting HSC expansion in culture [5, 6], and subsequent studies have found that pharmacological or genetic expansion of the osteoblastic (cells capable of differentiating into osteoblasts) lineage in vivo results in increased BM HSC numbers [7, 8]. However, biglycan-deficient mice, which exhibit a two-fold reduction in osteoblasts, show no significant changes in HSC number or function, and pharmacological osteoblast expansion fails to impact HSC [41]. Furthermore, genetic deletion of key niche factors like CXCL12 or SCF from mature osteoblasts did not cause reductions in BM HSC numbers [1, 17]. Since perivascular stromal cells are also osteoprogenitors [18, 42], the most likely explanation for the results above is that osteoblasts do not regulate HSC directly and, instead, perivascular stromal cells with osteoblastic potential are bona fide niche cells.
Osteoclasts
The main function of osteoclasts is to digest the bone during homeostasis. Mice lacking osteoclasts during development have a diminished BM cavity and greatly reduced numbers of hematopoietic cells (including HSCs) [43]. It is less clear whether osteoclasts directly or indirectly regulate the niche during adult steady-state hematopoiesis. Some studies have shown that osteoclast depletion impairs pharmacological HSC mobilization [44] or reduce HSC numbers [45]. However, another study found that osteoclast depletion increases HSC numbers [46]. More studies are needed to formally demonstrate that osteoclasts are a component of the HSC niche during steady-state hematopoiesis.
Other Endosteal Cells
The endosteal regions of the BM are enriched for HSCs under both steady-state and post-transplant conditions [47••]. It has been suggested that the osteolineage cells (OLC) lining the bone surface of these regions might play an important role in HSC regulation [47••, 48•]. Single-cell analysis of OLC located adjacent to HSCs (“proximal” OLC) revealed that these cells have significantly higher levels of mRNA for embigin and angiogenin when compared to OLC not located near HSCs, and subsequent conditional deletion of these molecules in OLC led to loss of HSC quiescence and enhanced proliferation, confirming OLC as bona fide niche components [47••].
Dynamic Regulation of the Niche and Its Effect on HSCs
It is easy to assume that the niche is a stable structure that regulates largely quiescent HSCs. However, multiple reports have shown that BM niches are plastic and dynamically regulated. In this section, we discuss some examples that highlight the dynamic nature of the niche.
Circadian Regulation of CXCL12 Production and HSC Trafficking
HSCs are migratory cells and approximately 1% of all HSC leave their niches to enter the circulation each day [49]. It is thought that this efflux is to refill empty niches in other bones [49] or to recirculate through tissues and become effector cells that contribute to local immunity [50]. Surprisingly, HSC release from the BM into the circulation follows a 24-h circadian rhythm in which maximum egress occurs during the resting phase of the organism (day for mice and night for humans [51, 52]). This circadian release is controlled by regulating CXCL12 production by BM perivascular cells. In turn, these cells are regulated by diverse mechanisms that ensure proper HSC release from their BM niches. The first pathway identified for circadian regulation of HSC trafficking was the sympathetic nervous system (SNS). Light signals received by the eyes are relayed via nerves to the suprachiasmatic nucleus, the major circadian pacemaker, in the brain [52]. Then, these signals are transmitted to the BM via the sympathetic chain to nerve terminals in the bone marrow that produce norepinephrine that acts on perivascular stromal cells via the β3-adrenergic receptor [52]. This receptor activation in turn triggers degradation of the transcription factor SP1, Cxcl12 downregulation, and HSC release [52]. CXCL12 signals via the CXCR4 receptor on HSCs. CXCR4 expression in HSCs is regulated by the molecular clock and follows the same circadian pattern as CXCL12 peaking during the resting phase and reaching a nadir during the activity phase, thus increasing the potency of the CXCL12 signal by regulating receptor abundance [51]. Another pathway that regulates HSC trafficking is a neutrophil-macrophage-perivascular stromal cell axis [36]. Previous studies showed that macrophages crosstalk (through an unknown mediator) with perivascular cells to induce CXCL12 secretion by perivascular cells [32,33,34]. Casanova-Acebes et al. found that neutrophils that have aged in the periphery are recruited back to the bone marrow where they are phagocytosed by BM macrophages; this in turn caused reduced Cxcl12 expression in BM perivascular stromal cells and HSC release [36]. This regulation depends on the expression of the LXR nuclear receptors but it is not known whether these function on macrophages or perivascular stromal cells as both cell types express LXR [36]. A third mediator that regulates circadian CXCL12 production and HSC release from the BM is corticosterone [53]. Corticosterone levels in the circulation also follow circadian rhythms; mice with constant low levels of corticosterone have no oscillations in BM CXCL12 as well and reduced HSC oscillations indicating that this pathway also regulates HSC release from BM HSC niches [53]. Thus, perivascular niches are dynamically regulated by at least three unique pathways to control physiological HSC egress from the BM within a 24-h period.
Regeneration and Reprogramming of the Vascular Niche After Myeloablation
Hematopoietic stem cell transplantation (HSCT) remains the only treatment for many malignant and non-malignant hematopoietic diseases. For the transplant to be successful, the host hematopoietic cells must be eliminated via chemotherapy drugs or radiation to allow the donor cells to engraft. This “myeloablation” not only eliminates the host hematopoietic cells but also dramatically injures the vascular niche leading to the disappearance of most sinusoids and associated perivascular cells whereas arteries and periarteriolar cells are relatively intact [13, 54,55,56]. Regeneration of the vascular niche is the rate-limiting factor for engraftment of donor HSC and restoration of normal hematopoiesis [13, 54, 55]. Niche regeneration is quite fast and, in as little as 2 weeks, the BM can restore an intact vasculature [13, 54, 55]. Niche regeneration is positively and negatively regulated by several mechanisms. A key positive driver of regeneration is vascular endothelial growth factor (VEGF) signaling through its receptor VEGFR2 [54]. Conditional deletion of Vegfr2 in mice did not have any effect in steady-state hematopoiesis but completely abolished vascular regeneration after myeloablation [54]. Vascular regeneration also requires an intact sympathetic nervous system as nerve injury or pharmacological inhibition of β2- and β3-adrenergic receptors prevents regeneration of the sinusoidal network and associated perivascular stromal cells [57]. After myeloablation, BM granulocytes crosstalk with the regenerating vasculature via TNF-α to promote endothelial cell survival and vessel growth [55]; Tnfa−/− mice show delayed niche recovery after vascular injury highlighting the importance of this pathway in regeneration [55]. Vascular regeneration is negatively regulated by NF-κB signaling in the endothelial cells and NF-κB inhibition can be used to accelerate recovery after myeloablation [58•]. In addition, LepR+ cells and hematopoietic progenitors cooperate in inhibiting the regeneration of the sinusoidal vasculature via angiopoietin 1 [59••]. The loss of the sinusoids causes megakaryocytes to relocate to the endosteal surface of the bone where they facilitate HSC engraftment via TGFβ and PDGF-BB [26, 60]; it also causes a dramatic expansion of Adipoq-Cre+ adipocyte progenitors and adipocytes that facilitate hematopoietic regeneration via SCF secretion [40••]. Myeloablation also activates several programs in the vascular niche that support HSC engraftment and restoration of hematopoiesis. Some of these programs include the Notch pathway via Jagged1 and Jagged2 [61, 62, 63•], pleiotrophin [64] and EGF in endothelial cells [65], and Dickkopf-1 activation on perivascular stromal cells and osteoprogenitors [66•]. Together, these pathways regulate regeneration of the niche and engraftment of HSC to restore normal hematopoiesis. Once the niche has regenerated and sufficient numbers of hematopoietic cells have been produced, the BM returns to steady state through incompletely understood mechanisms although TGFβ plays a key role in this process [67].
Conclusions
The BM niche is a complex, multicellular, and heterogeneous structure that maintains hematopoietic stem cells. Different components of the niche are targeted by systemic and local signals in a dynamic way that affects their ability to regulate HSC numbers, trafficking, and function. In addition, the niche is a plastic structure capable of dynamic changes after insults to the bone marrow (e.g., myeloablation, leukemia, aging). Understanding how the different components of the niche regulate each other and cooperate to maintain HSC is one of the frontiers of the HSC niche field.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–5.
• Cordeiro Gomes A, Hara T, Lim VY, Herndler-Brandstetter D, Nevius E, Sugiyama T, et al. Hematopoietic stem cell niches produce lineage-instructive signals to control multipotent progenitor differentiation. Immunity. 2016;45:1219–31. Functional identification of a niche for common lymphoid progenitors.
Chow A, Huggins M, Ahmed J, et al. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med. 2013;19:429–36.
Sapoznikov A, Pewzner-Jung Y, Kalchenko V, Krauthgamer R, Shachar I, Jung S. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nat Immunol. 2008;9:388–95.
Taichman RS. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med. 1994;179:1677–82.
Taichman RS, Reilly MJ, Emerson SG. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood. 1996;87:518–24.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6.
Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41.
Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood. 2015;125:2621–9.
Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–34.
Ramalingam P, Poulos MG, Butler JM. Regulation of the hematopoietic stem cell lifecycle by the endothelial niche. Curr Opin Hematol. 2017;24:289–99.
Doan PL, Chute JP. The vascular niche: home for normal and malignant hematopoietic stem cells. Leukemia. 2012;26:54–62.
Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–43.
• Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526:126–30. Imaging studies that challenge the idea that quiescent HSCs associate with arterioles.
Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–21.
Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–62.
Greenbaum A, Hsu Y-MS, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–30.
Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–34.
Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, et al. PDGFRα and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med. 2013;210:1351–67.
Hanoun M, Frenette PS. This niche is a maze; an amazing niche. Cell Stem Cell. 2013;12:391–2.
•• Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol. 2017;19:214–23. Together with (22), this is the strongest evidence for functionally distinct arteriolar and sinusoidal HSC niches.
•• Itkin T, Gur-Cohen S, Spencer JA, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. 2016;532:323–8. Together with (21), this is the strongest evidence for functionally distinct arteriolar and sinusoidal HSC niches.
Katayama Y, Battista M, Kao W-M, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–21.
Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147:1146–58.
Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20:1315–20.
Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20:1321–6.
Nakamura-Ishizu A, Takubo K, Kobayashi H, Suzuki-Inoue K, Suda T. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. J Exp Med. 2015;212:2133–46.
Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013;15:533–43.
•• Leiva M, Quintana JA, Ligos JM, Hidalgo A. Haematopoietic ESL-1 enables stem cell proliferation in the bone marrow by limiting TGFβ availability. Nat Commun. 2016;7:10222. First demonstration that HSPCs are a component of the HSC niche.
Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. 2012;18:1651–7.
•• Kusumbe AP, Ramasamy SK, Itkin T, Mäe MA, Langen UH, Betsholtz C, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. 2016;532:380–4. Demonstration that expansion of arterioles by modulating Notch signaling also expands HSC.
Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011;208:261–71.
Christopher MJ, Rao M, Liu F, Woloszynek JR, Link DC. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J Exp Med. 2011;208:251–60.
Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116:4815–28.
McCabe A, Zhang Y, Thai V, Jones M, Jordan MB, MacNamara KC. Macrophage-lineage cells negatively regulate the hematopoietic stem cell pool in response to interferon gamma at steady state and during infection. Stem Cells. 2015;33:2294–305.
Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. 2013;153:1025–35.
•• Chen X, Deng H, Churchill MJ, et al. Bone marrow myeloid cells regulate myeloid-biased hematopoietic stem cells via a histamine-dependent feedback loop. Cell Stem Cell. 2017;21:747–760.e7. First report that demonstrates that myeloid cells directly regulates HSC.
Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460:259–63.
Spindler TJ, Tseng AW, Zhou X, Adams GB. Adipocytic cells augment the support of primitive hematopoietic cells in vitro but have no effect in the bone marrow niche under homeostatic conditions. Stem Cells Dev. 2014;23:434–41.
•• Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol. 2017;19:891–903. First description of adipocytes as positive regulators of regeneration. Challenges previous concepts about adipocyte function in the BM.
Lymperi S, Horwood N, Marley S, Gordon MY, Cope AP, Dazzi F. Strontium can increase some osteoblasts without increasing hematopoietic stem cells. Blood. 2008;111:1173–81.
Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33:387–99.
Mansour A, Abou-Ezzi G, Sitnicka E, Jacobsen SEW, Wakkach A, Blin-Wakkach C. Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J Exp Med. 2012;209:537–49.
Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657–64.
Lymperi S, Ersek A, Ferraro F, Dazzi F, Horwood NJ. Inhibition of osteoclast function reduces hematopoietic stem cell numbers in vivo. Blood. 2011;117:1540–9.
Miyamoto K, Yoshida S, Kawasumi M, Hashimoto K, Kimura T, Sato Y, et al. Osteoclasts are dispensable for hematopoietic stem cell maintenance and mobilization. J Exp Med. 2011;208:2175–81.
•• Silberstein L, Goncalves KA, Kharchenko PV, Turcotte R, Kfoury Y, Mercier F, et al. Proximity-based differential single-cell analysis of the niche to identify stem/progenitor cell regulators. Cell Stem Cell. 2016;19:530–43. Identification of embigin and osteolineage cells as a novel niche factor and niche cell, respectively.
• Goncalves KA, Silberstein L, Li S, Severe N, Hu MG, Yang H, et al. Angiogenin promotes hematopoietic regeneration by dichotomously regulating quiescence of stem and progenitor cells. Cell. 2016;166:894–906. Identification of angiogenin as a novel HSPC regulator through a completely novel mechanism.
Wright DE. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294:1933–6.
Massberg S, Schaerli P, Knezevic-Maramica I, Köllnberger M, Tubo N, Moseman EA, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008.
Lucas D, Battista M, Shi PA, Isola L, Frenette PS. Mobilized hematopoietic stem cell yield depends on species-specific circadian timing. Cell Stem Cell. 2008;3:364–6.
Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–7.
Kollet O, Vagima Y, D’Uva G, et al. Physiologic corticosterone oscillations regulate murine hematopoietic stem/progenitor cell proliferation and CXCL12 expression by bone marrow stromal progenitors. Leukemia. 2013;27:2006–15.
Hooper AT, Butler JM, Nolan DJ, et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009;4:263–74.
Bowers E, Slaughter A, Frenette PS, Kuick R, Pello OM, Lucas D. Granulocyte-derived TNFα promotes vascular and hematopoietic regeneration in the bone marrow. Nat Med. 2018;24:95–102.
Doan PL, Russell JL, Himburg HA, Helms K, Harris JR, Lucas J, et al. Tie2(+) bone marrow endothelial cells regulate hematopoietic stem cell regeneration following radiation injury. Stem Cells. 2013;31:327–37.
Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, Barrick C, et al. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med. 2013;19:695–703.
• Poulos MG, Ramalingam P, Gutkin MC, et al. Endothelial-specific inhibition of NF-κB enhances functional haematopoiesis. Nat Commun. 2016;7:13829. Identification of NF-κB signaling as a negative regulator of vascular niche regeneration.
•• Zhou BO, Ding L, Morrison SJ. Hematopoietic stem and progenitor cells regulate the regeneration of their niche by secreting angiopoietin-1. Elife. 2015;4:e05521. First report showing that hematopoietic cells can directly crosstalk with the microenvironment.
Olson TS, Caselli A, Otsuru S, Hofmann TJ, Williams R, Paolucci P, et al. Megakaryocytes promote murine osteoblastic HSC niche expansion and stem cell engraftment after radioablative conditioning. Blood. 2013;121:5238–49.
Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6:251–64.
Poulos MG, Guo P, Kofler NM, Pinho S, Gutkin MC, Tikhonova A, et al. Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep. 2013;4:1022–34.
• Guo P, Poulos MG, Palikuqi B, et al. Endothelial jagged-2 sustains hematopoietic stem and progenitor reconstitution after myelosuppression. J Clin Invest. 2017; https://doi.org/10.1172/JCI92309. Identification of a novel proregenerative molecule in the regenerating endothelium.
Himburg HA, Yan X, Doan PL, Quarmyne M, Micewicz E, McBride W, et al. Pleiotrophin mediates hematopoietic regeneration via activation of RAS. J Clin Invest. 2014;124:4753–8.
Doan PL, Himburg HA, Helms K, Russell JL, Fixsen E, Quarmyne M, et al. Epidermal growth factor regulates hematopoietic regeneration after radiation injury. Nat Med. 2013;19:295–304.
• Himburg HA, Doan PL, Quarmyne M, Yan X, Sasine J, Zhao L, et al. Dickkopf-1 promotes hematopoietic regeneration via direct and niche-mediated mechanisms. Nat Med. 2017;23:91–9. Identification of a novel proregenerative molecule in the vascular niche.
Brenet F, Kermani P, Spektor R, Rafii S, Scandura JM. TGFβ restores hematopoietic homeostasis after myelosuppressive chemotherapy. J Exp Med. 2013;210:623–39.
Acknowledgments
The authors apologize to those whose work was not cited because of space limitations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Margot May, Anastasiya Slaughter, and Daniel Lucas declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Cancer and Stem Cells
Rights and permissions
About this article

Cite this article
May, M., Slaughter, A. & Lucas, D. Dynamic Regulation of Hematopoietic Stem Cells by Bone Marrow Niches. Curr Stem Cell Rep 4, 201–208 (2018). https://doi.org/10.1007/s40778-018-0132-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-018-0132-x