
Abstract
Purpose of review
IgG4-related disease (IgG4-RD) is an autoimmune fibrosing condition which is being increasingly recognized. This condition has been classically managed with glucocorticoids (GC) with variable dosing and frequent relapses. Multiple observational reports studied different GC doses and steroid-sparing agents. Rituximab (RTX) was reported as an effective induction treatment, with less relapses after 1 year of treatment. In the absence of guidelines, an international management consensus was made in 2015.
Recent findings
In the last few years, significant advances have been made to clarify IgG4-RD pathophysiology. The new classification criteria will enable investigators to design new clinical trials with more homogeneous populations. Furthermore, B and T lymphocytes have a central role in the inflammatory cascade. More solid evidence has been published supporting treatment with GC, RTX, and some other steroid-sparing agents like mofetil mycophenolate. The combination of GC and immunosuppressants has been pointed by a meta-analysis as the most effective treatment for IgG4-RD, although the number and quality of studies remains limited.
Summary
The use of GC and steroid-sparing agents is effective in IgG4-RD, although the choice between rituximab and other immunosuppressants can be influenced by many factors. Novel agents will be trialed in the following years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
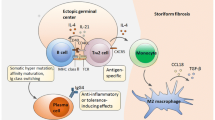
Immunoglobulin-G-4-related disease (IgG4-RD) is a rare fibro-inflammatory condition. There are few epidemiologic studies available, but a Japanese nationwide survey showed a prevalence of 6 cases per 100,000 inhabitants for IgG4-RD [1]. This entity encompasses several classical diseases like type 1 autoimmune pancreatitis, Kuttner’s pseudotumor or Mikulicz’s disease and Riedel’s thyroiditis. The first reports of this common denominator date back to 2003 [2]. They all share pathology findings, including storiform fibrosis, lymphoplasmacytic infiltrates and obliterative phlebitis. IgG4-RD immunohistochemical features include IgG4-positive plasma cells and an IgG4/IgG4-positive plasma cell ratio ≥ 40% [3]. The exact pathological mechanisms which lead to fibrosis are still unclear but recent advances have shed some light on the different pathways involved. IgG4-RD seems an antigen-driven disease. Several antigens including galectin-3 [4], laminin 511 [5], among others, have been identified as potential triggers of the inflammatory response. This inflammation is driven by the activation of B and T lymphocytes, resulting in the oligoclonal production of T cytotoxic lymphocytes and B cell precursors (plasmablasts) [6, 7]. These cells, along with other contributors like Th2 lymphocytes, T follicular helper lymphocytes, or M2 macrophages, enable the production of IgG4-secreting plasma cells and multiple cytokines. Finally, the pro-inflammatory cells migrate to the target tissues, where there is fibroblast activation and secondary fibrosis, leading to tissue damage and organ dysfunction [8, 9]. The role of serum IgG4 is controversial, as it does not have a significant pro-inflammatory function, other mimicker conditions can raise its levels, and 10 to 50% of the patients will have normal levels [10, 11]. Two sets of diagnostic criteria are available: the Japanese comprehensive criteria [12] (based on organ involvement, serum IgG4 levels, and/or IgG4 immunostaining) and the diagnostic pathology consensus [3] (based on pathology and IgG4 immunostaining correlated with clinical findings). Both of them are used in clinical practice for diagnostic purposes. In 2019, the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for IgG4-RD were published [13]. This is the first set of international classification criteria for IgG4-RD. They have a multiple-step approach (entry criteria, exclusion criteria, and inclusion criteria) and they are score-based (inclusion criteria score ≥ 20 points summarizing histopathology, immunostaining, serum IgG4 concentration, and different typical organ involvement). Specificity was 98% and sensitivity 82%. ACR/EULAR classification criteria were designed to provide homogenized cohorts of patients for research but can guide the diagnostic process by ruling out other mimicker conditions.
Since IgG4-RD is still a novel disease, several factors have conditioned the ability to evaluate novel treatments. First, the lack of previous classification criteria prevented from ensuring that patients recruited in different studies had the same standardized characteristics. Hopefully, the new ACR/EULAR criteria will address this problem. Second, there was no validated outcome measurement for IgG4-RD, and outcomes have been reported in heterogeneous ways. The IgG4-responder index (IgG4-RI) is a measurement tool developed for research, based on the preexisting granulomatosis with poliangiitis BVAS scale [14], recently redesigned and validated, which might be used in future trials [15]. In third place, there is no universal biomarker to monitor IgG4-RD activity. Multiple research groups have reported the correlation between serum plasmablast levels [16], cytotoxic T lymphocyte populations [17], and changes in positron emission tomography/computerized tomography uptake (PET-CT) [18] with the level of activity of the disease and response to treatment. The validation and generalization of these techniques will provide more indicators to test new treatments.
In this review, we aim to provide the current available scientific evidence for treatment of IgG4-RD and to give a practical approach to the management of these patients.
Methods
A literature review was performed using Pubmed.gov searching for the MESH terms “IgG4-related disease” and “treatment.” The studies representing either a greater level of evidence (clinical trial, observational study, case series) or the largest number of patients were selected for each treatment. The search was performed on December 30, 2019.
Evidence level was categorized as per the 2014 EULAR recommendations [19]:
-
1A: From meta-analysis of randomized controlled trials
-
1B: From at least one randomized controlled trial
-
2A: From at least one controlled study without randomization
-
2B: From at least one type of quasi-experimental study
-
3: From descriptive studies, such as comparative studies, correlation studies, or case–control studies
-
4: From expert committee reports or opinions and/or clinical experience of respected authorities
Treatment
All the different drugs are summarized in Table 1 including the most representative studies, N of enrolled patients, and treatment schemes.
Glucocorticoids (GC)
This is the classical treatment, initially reported in patients with type 1 autoimmune pancreatitis (AIP) [20]. GC are characteristically effective for the induction of IgG4-RD. The proposed dosages and treatment duration for AIP (prednisone 0.6 mg/Kg/day for 2–4 weeks and then progressive taper down) have been trialed in IgG4-RD recently (Table 1). Masaki et al. showed that 93% of the individuals had a partial or complete response to prednisolone after 1 year of treatment [21], with an average maintenance dose of 7 mg/day. A small RCT by Wu et al. [22] has recently confronted higher doses of prednisone (0.8–1 mg/Kg/day) with moderate doses (0.6–0.5 mg/Kg/day), showing no statistically significant differences at 24 weeks. Both treatment regimens were able to reduce serum IgG4-RI and IgG4 levels with a similar magnitude of effect. Concerns over the potential long-term side effects (alterations in the glucose and lipid metabolism, osteoporosis, skin frailty) and relapses after induction have prompted the proposal of low doses of GC or adding other drugs for maintenance. In a Japanese study, 459 autoimmune pancreatitis patients were treated with GC [23]. Twenty-three percent of the subjects on GC maintenance treatment (median prednisone 5 mg/day) relapsed, versus 34% in the group who did stop maintenance treatment after GC induction and 92% of the relapses occurred in the first 3 years after induction (Evidence category 1B).
Disease modifying anti-rheumatic drugs (DMARDs)
Multiple corticosteroid-sparing agents have been reported to be useful in IgG4-RD (Table 1). Again, the rationale for the use of these drugs has been based on AIP studies. A prospective British study included 115 patients with type 1 AIP and IgG4-related sclerosing cholangitis (both in the IgG4-RD spectrum). Ninety-seven percent of the patients responded to steroids, but 50% relapsed after a median of 4.6 months. Seventy-one percent of the relapse cases were treated with azathioprine (AZA) plus GC, with a relapse rate of 20% during a median follow-up of 32.5 months. After the first reports using AZA, different drugs have been trialed aiming for less relapses and less long-term side effects from GC.
AZA is an inhibitor of the purine synthesis. The core of the evidence supporting its use in IgG4-RD comes from cohort studies. De Pretis et al. [24] reported the use of AZA in combination with GC for the treatment of relapsing AIP or multiorgan disease. Three patients discontinued AZA due to side effects (anaphylaxis, hepatitis, and nausea/vomiting). After 6 months of treatment, serum IgG4 levels in the AZA group were statistically significantly lower than before starting the treatment. The relapse rate was 30% on AZA, with a mean follow-up of 30 months (Evidence category 3).
A non-randomized trial explored the use of oral cyclophosphamide (CYC) in IgG4-RD. CYC is an alkylating agent used in cancer therapy and in other autoimmune diseases to treat severe manifestations [25]. The main potential side effects are leukopenia, infection, liver alterations, ovarian failure, hemorrhagic cystitis, and long-term use after a certain threshold cumulative dose increases the risk of cancer. The primary outcome of the CYC study was the relapse rate. Thirty eight percent of the patients in the GC only group vs. 12% in the GC + CYC group relapsed after 12 months. The time to the first flare was statistically significantly longer in the GC + CYC group (11 vs. 7 months, p = 0.018). Disease remission was a secondary endpoint, defined as IgG4-RD RI < 3 or decline ≥ 2 points and successfully completed a glucocorticoid taper without relapse. The CYC group achieved remission rates of 88% at 12 months vs. 60% in the GC only group (p not reported). Patients with more than 6 involved organs and high serum IgG4 were more likely to relapse in the GC only group, and direct combined treatment was proposed for this subset of cases. In the CYC group, liver enzyme alterations and gastrointestinal reactions were reported in 2 patients each. Mild infections and diabetes mellitus were reported similarly in both groups (Evidence category 2A). The problem with the study is that it was not randomized so there could have been biases with respect to prescribing CYC.
Iguratimod is a molecule with anti-inflammatory effects available in China. Its mechanism of action affects the anti-tumor necrosing factor pathway and multiple chemokines including inteleucin-6 and interleukin-1β. A prospective cohort study has shown some benefits along with intramuscular betamethasone for IgG4-RD induction treatment [26]. The study was limited to 24 weeks, patients had a response rate (total + partial) of 88% at 12 and 24 weeks, with a statistically significant reduction of IgG4-RI (10 vs. 3.6 vs. 3.1), serum IgG4 (12,250 mg/dL vs. 4725 vs. 6020) and CD19+CD24−CD38hi plasmablasts (7.2% vs. 3.8% at 12 weeks). Unfortunately, there was no control group to assess the effect of GC alone or other drugs for comparison. From the safety perspective, iguratimod was found to be very well tolerated as only 5 of 24 had side effects (3 individuals had oral ulcers and 2 had stomach discomfort) (Evidence category 2B).
Leflunomide (LFN) is another pyrimidine synthesis inhibitor. A retrospective cohort study [27] combining LFN with GC for induction obtained a decrease in IgG4-RI (15 to 2.7), while two thirds of the patients were on complete remission and one third on partial remission after 6 months of follow-up. Three patients initially received intravenous GC pulses due to severe manifestations. Only 3 patients relapsed after 6 months while on LFN and maintenance GC, but just 7 individuals were followed for more than 12 months. Serum IgG4 also decreased during follow-up. The main side effects reported were liver enzyme alterations and rash in one patient each (Evidence category 3).
Methotrexate (MTX) acts as a folate antagonist. In a small Italian series [28], ten patients were induced with oral prednisone and subcutaneous or oral MTX mostly as second-line therapy (after six out of ten relapsed during GC treatment and 2 to other DMARDs). All patients responded to GC treatment (IgG4-IR reduction ≥ 2). Six months after adding MTX, two patients were in disease remission (IgG4-RI < 3 and off GC) and 8 were in partial remission (IgG4-RI ≥ 3). No relapses or side effects were reported after a mean follow-up of 21 months (Evidence category 4).
Mofetil mycophenolate (MMF) inhibits de novo purine synthesis. This is the only DMARD with a RCT [29]. In this study, which was non-placebo controlled and open label, Yunyun et al. trialed GC vs. GC + MMF in IgG4-RD treatment-naïve patients where 83% had ≥ 3 organs involved. IgG4-RI scores < 3 points and declining ≥ 2 were recognized as complete remission while IgG4-RD RI scores declining ≥ 2 points but remaining ≥ 3 were considered partial remission. After 3 months, the response rate was significantly higher in the combo group (83 vs. 97%), although statistical differences were neutralized at 6 and 12 months. Clinical relapse was defined as re/appearance of clinical symptoms or imaging findings and serological relapse as isolated serum IgG4 level increase. The differences in total relapse rates (clinical and serological) after 12 months were close to statistical signification (40 vs. 21%, p = 0.056), while clinical relapse rates were statically significantly different (34 vs. 12%, p = 0.034). No differences were found in terms of side effects. The most frequent ones were infection, GC-induced diabetes, and gastrointestinal reactions (Evidence category 1B).
Finally, sporadic use of cyclosporin (N = 1) [30], tacrolimus (N = 5) [31], and 6-mercaptopurine (n = 4) [32], all in combination with GC, have been reported in small numbers of patients for the treatment of relapses after GC induction (Evidence category 4).
Biologics
Rituximab (RTX) is a chimeric monoclonal antibody against CD20, with a B lymphocyte depleting effect. Carruthers et al. reported a single-arm open-label trial using intravenous RTX in 30 IgG4-RD patients, 73% of whom had relapsed on GC monotherapy [33]. These patients had a mean of 3.5 organs involved, mean initial IgG4-RI was 11. All patients received intravenous GC with each of the two RTX infusions, only 4 were on oral prednisone, and none was on GC treatment 2 months after the first infusion. The primary outcome (decline of IgG4-RI ≥ 2 points compared with baseline; and no disease flares before month 6; and no GC use between months 2 and 6) was met in 77% of the participants. Those who did not meet it were still on GC between months 2 and 6. Ninety-seven percent of the patients responded with an IgG4-RI reduction ≥ 2 points after the first 6 months after the RTX infusion. Patients who had elevated baseline serum IgG4 (63%) experienced a statistically significant reduction of this parameter after 6 months (a decrease of CD19+CD27−CD20−CD38hi plasmablast populations after RTX treatment has also been characterized [16]). IgG4-RI decreased between baseline and all subsequent visits (especially in the first 30 days). After 12 months of follow-up, 60% of the participants achieved complete remission (IgG4-RI = 0 and off GC) and 67% if excluding the criterion of isolated serum IgG4 elevation. Only 23% relapsed, within 1 year. The only adverse event attributed to RTX was a urinary infection prompting admission. In 2019, a small retrospective study (N = 14) [34] pointed toward an increased relapse-free rate after a follow-up of 18 months in patients with RTX and either treated with maintenance RTX 1000 mg every 6 months versus patients receiving infusions in the event of new symptoms (100 vs. 29%, p = 0.006) (Evidence category 2A).
A very limited experience based on case reports is available for abatacept (CD80-CD86 co-stimulation blockade, N = 1) [35], dupilumab (IL4-IL13 blocker, N = 1) [36], and infliximab (tumor necrosis factor alpha inhibitor, N = 2) [37] (Evidence category 4).
Others
Bortezomib, a chemotherapeutic agent used in multiple myeloma, has been used in a single IgG4-RD patient [38] (Evidence category 4).
In some clinical scenarios, interventional procedures or surgery might be needed. The mass effect in IgG4-RD affected tissues can cause life-threatening conditions like hydronephrosis and secondary renal failure (IgG4-related retroperitoneal fibrosis) or jaundice and liver/pancreas dysfunction plus cholangitis (IgG4-related pancreatobiliary disease). For hydronephrosis, as in idiopathic retroperitoneal fibrosis, ureteral stenting or surgical ureterolysis and ureteral intraperitonealization have been reported to be useful, generally along with pharmacological treatment [39, 40]. For jaundice due to biliary duct stenosis, endoscopic biliary stenting can be performed. A Japanese prospective cohort [41] with 59 patients with IgG4-pancreatobiliary disease with biliary strictures associated with jaundice or obstructive liver enzyme abnormalities studied the benefits of stenting with or without GC treatment. One month after stenting, patients who were treated with GC vs. no steroids had less incidence of recurrent biliary obstruction (100 vs. 82%, p = 0.0015) and 96% of them had their stents removed (Evidence category 3).
Current treatment approach
After reviewing all the different treatment options for IgG4-RD up to 2019, it is noticeable that the global level of scientific evidence is low. In 2015, an international panel of experts did a systematic review and a set of surveys to develop some treatment recommendations (Table 2) in the absence of formal guidelines [42]. In terms of treating patients, IgG4-RD extension can be minimal and asymptomatic, not warranting treatment. Some involvement can be transient but relapsing, leading to tissue damage on the long term. In the light of this sequence of events, treatment would be desirable. Patients with symptomatic active IgG4-RD should be treated, generally with GC. The use of steroid-sparing agents since the beginning was controversial, with less than half of the experts agreeing on that. The limited experience with DMARDs was acknowledged, and the steroid-sparing agent with a more robust evidence was RTX. In cases of potential risk for irreversible organ damage (aortitis, retroperitoneal fibrosis, proximal biliary strictures, tubulointerstitial nephritis, pachymeningitis, pancreatic enlargement, and/or pericarditis) a combination of GC, RTX, and invasive techniques might be used. The use of drugs for maintenance was supported, especially by prescribing low dose GC in Asian countries. Finally, in the event of relapses, patients should be retreated with GC and steroid-sparing agents could be started, without stating any preference.
In 2020, IgG4-RD treatment landscape has incorporated some new studies to provide us with novel options to treat our patients. New prospective studies on GC dosing have confirmed the utility of GC and helped to narrow the dosage down to 0.5–0.6 mg/Kg/day for induction [22]. The rate of GC tapering is still variable, as is the case in giant cell arteritis [43]. Moreover, evidence favoring the usefulness of DMARDs has provided some new alternatives. Despite the remarkable results of RTX for induction [33], there are relapses if patients are followed for longer than the initial studies. In the French IgG4-RD registry, long-term efficacy of RTX revealed that 42% relapse after 24 months of follow-up (median relapse time 19 months) [44]. In addition, retreatment with RTX might reduce the incidence of flares [34], similar to antineutrophil cytoplasmic antibody-associated vasculitis [45]. With all these treatment options, clinical practice can be variable [46]. Omar et al. [47] published a network meta-analysis in 2019 concluding that RTX maintenance therapy had the lowest relapse rate of all treatments (OR = 0.10, 95% CI (0.01, 1.63)), whereas GC + DMARDs were associated with a lower relapse rate compared with GC alone (OR = 0.39, 95% CI (0.20, 0.80)). Patients treated with GC + DMARDs had a higher remission rate than those given GCs (OR = 3.36, 95% CI (1.44, 7.83)), DMARDs (OR = 55.31, 95% CI (13.73, 222.73)) alone or RTX induction therapy only (OR = 7.38, 95% CI (1.56, 34.94)). Obviously, the quality of the data within the studies analyzed was low and heterogeneous. The rate of adverse events was similar. We also know some predictors of IgG4-RD relapse like baseline elevated serum IgE, IgG4, eosinophilia [48], or a IgG4-RI > 9 [44]. In the light of all these findings, we have created a potential new treatment algorithm for IgG4-RD (Table 3), updating the previous recommendations and exposing our point of view and usual practice. There is still no unique validated treatment approach; thus, other experts might treat patients differently. Furthermore, IgG4-RD management can be variable due to multiple causes including regional differences in drug availability, financial coverage for treatments (individual, insurers or public healthcare systems), disease presentation, comorbidities, and drug tolerance.
Treatment response monitoring
Response to treatment is usually monitored clinically and with imaging techniques according to the individual manifestations of each patient. Computerized tomography is generally available worldwide, while access to magnetic resonance imaging or PET-CT can be limited. Interestingly, PET-CT has prospectively shown changes in 18-fluorodeoxyglucose uptake in response to GC treatment [18] and a correlation with plasmablast quantification [49]. There are no universal biomarkers for IgG4-RD, and general tests as acute phase reactants or complement are unspecific [50]. Serum IgG4 has been reported to diminish in patients with IgG4-RD treated with different drugs [22, 25, 29, 33]. On one hand, serum IgG4 quantification is available. On the other, a significant number of IgG4-RD patients will have normal baseline serum IgG4 levels making this test potentially useful for only some cases [11]. The quantification of different pre-B cell populations using flow cytometry is a technique less accessible, mostly available in reference centers or within IgG4-RD research groups. CD19+CD27−CD20−CD38hi [16] and CD19+CD24−CD38hi plasmablasts [51] have proven to have oligoclonal increased populations pre-treatment which decrease significantly after treatment. Flow cytometry also allows to track CD8α−CD4+SLAMF7+ cytotoxic T effector/memory lymphocyte populations, which have a similar response to GC treatment [17].
Lastly, IgG4-RI was initially designed as a response index based on the Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis [14] and included the serum IgG4 domain [52], with scores from 0 to 4 for each domain. Some flaws were detected once its use was more widespread such as scoring 3 points as a new organ was involved, and during the follow-up, regardless of improvement, the score went down to 2 (persistent or unchanged from last visit) [46]. This IgG4-RI was redesigned and validated in 2018 [15] (the reference includes the form) by simplifying the total scoring system from 0 to 3 (0 = unaffected or resolved; 1 = improved but persistent; 2 = new or recurrence (while off of treatment) or unchanged; 3 = worse or new (despite treatment)). Moreover, the scores were doubled in case of needing urgent treatment due to severe organ dysfunction. The updated IgG4-RI encompasses all the potential organs involved in IgG4-RD and also evaluates the presence of organ damage, excluding serum IgG4 levels. This item will standardize research outcome reports but can also help in treatment response monitoring.
Emerging therapies
With the latest discoveries in terms of IgG4-RD pathophysiology, including the central role of B cells and cytotoxic T lymphocytes, some of the future potential therapeutic targets include CD19+ cells and SLAMF7+ cells [53]. According to Clinicaltrials.gov, a phase two open-label clinical trial was completed studying the effect of XmAb®5871 (NCT02725476), a monoclonal antibody targeting CD19 and the FcγRIIb inhibitory receptor of B cells, resulting in plasmablast inhibition. Final results have not been fully published, but data show a response (IgG4-RI decrease ≥2 points) in 80% of the patients. Another phase I study exploring the combination of RTX and lenalidomide (a thalidomide-like drug) for IgG4-RD (NCT02705638) was completed in April 2019, but results are not available. There is an ongoing phase 2 open-label clinical trial with abatacept (NCT03669861) with an estimated completion date of June 2020. Finally, according to the US Securities and Exchange commission [54], a phase 2b clinical trial might be started in 2020 using inebilizumab, a humanized monoclonal antibody against CD19+ cells.
Conclusion
Treatment options in IgG4-RD have evolved during the last 15 years from classic use of GC to immune suppressive medications and RTX. There is some moderate quality evidence favoring the use of GC for induction and maintenance, as well as for RTX in combination with GC. MMF has been shown to have clinical benefit with P values that were not quite significant in small clinical trials. There are no robust guidelines for IgG4-RD management. The use of GC in combination with immunosuppressors (biologics or DMARDs) along with occasional procedures when organ obstruction occurs seems to be supported by the existing literature. Disease severity, acuity, and organ involved as well as drug access and geography can modify the treatment of choice. Since significant advances have been made in the field of IgG4-RD pathophysiology, new drugs against novel therapeutic targets will be trialed in the years to come.
References and Recommended Reading
Uchida K, Masamune A, Shimosegawa T, Okazaki K. Prevalence of IgG4-related disease in Japan based on Nationwide survey in 2009. Int J Rheumatol. 2012;2012:1–5.
Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38:982–4.
Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25:1181–92.
Perugino CA, AlSalem SB, Mattoo H, Della-Torre E, Mahajan V, Ganesh G, et al. Identification of galectin-3 as an autoantigen in patients with IgG4-related disease. J Allergy Clin Immunol. Elsevier Inc. 2019;143:736–745.e6.
Shiokawa M, Kodama Y, Sekiguchi K, Kuwada T, Tomono T, Kuriyama K, et al. Laminin 511 is a target antigen in autoimmune pancreatitis. Sci Transl Med. 2018;10:1–11.
Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol Elsevier Ltd. 2014;134:679–87.
Mattoo H, Stone JH, Pillai S. Clonally expanded cytotoxic CD4 + T cells and the pathogenesis of IgG4-related disease. Autoimmunity. 2017;50:19–24.
Della-Torre E, Lanzillotta M, Doglioni C. Immunology of IgG4-related disease. Clin Exp Immunol. 2015;181:191–206.
Della-Torre E, Rigamonti E, Perugino C, Baghai-Sain S, Sun N, Kaneko N, et al. B lymphocytes directly contribute to tissue fibrosis in patients with IgG4-related disease. J Allergy Clin Immunol: Elsevier Inc.; 2019.
Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2014;74:14–8.
Martínez-Valle F, Fernández-Codina A, Pinal-Fernández I, Orozco-Gálvez O, Vilardell-Tarrés M. IgG4-related disease: evidence from six recent cohorts. Autoimmun Rev. 2017;16:168–72.
Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22:21–30.
Wallace ZS, Naden RP, Chari S, Choi H, Della-Torre E, Dicaire J-F, et al. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease. Arthritis Rheumatol (Hoboken, NJ). 2019;0:1–13.
Stone JH, Hoffman GS, Merkel PA, Min Y-I, Uhlfelder ML, Hellmann DB, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis activity score. Arthritis Rheum. 2001;44:912–20.
Wallace ZS, Khosroshahi A, Carruthers MD, Perugino CA, Choi H, Campochiaro C, et al. An International Multispecialty Validation Study of the IgG4-Related Disease Responder Index. Arthritis Care Res (Hoboken). 2018;70:1671–8.
Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74:190–5.
Della-Torre E, Bozzalla-Cassione E, Sciorati C, Ruggiero E, Lanzillotta M, Bonfiglio S, et al. A CD8α- Subset of CD4+SLAMF7+ Cytotoxic T Cells Is Expanded in Patients With IgG4-Related Disease and Decreases Following Glucocorticoid Treatment. Arthritis Rheumatol (Hoboken, NJ). 2018;70:1133–43.
Zhang J, Chen H, Ma Y, Xiao Y, Niu N, Lin W, et al. Characterizing IgG4-related disease with 18F-FDG PET/CT: a prospective cohort study. Eur J Nucl Med Mol Imaging. 2014;41:1624–34.
van der Heijde D, Aletaha D, Carmona L, Edwards CJ, Kvien TK, Kouloumas M, et al. 2014 update of the EULAR standardised operating procedures for EULAR-endorsed recommendations. Ann Rheum Dis. 2015;74:8–13.
Kamisawa T, Okazaki K, Kawa S, Ito T, Inui K, Irie H, et al. Amendment of the Japanese consensus guidelines for autoimmune pancreatitis, 2013 III. Treatment and prognosis of autoimmune pancreatitis. J Gastroenterol. 2014;49:961–70.
Masaki Y, Matsui S, Saeki T, Tsuboi H, Hirata S, Izumi Y, et al. A multicenter phase II prospective clinical trial of glucocorticoid for patients with untreated IgG4-related disease. Mod Rheumatol Taylor & Francis. 2017;27:849–54.
Wu Q, Chang J, Chen H, Chen Y, Yang H, Fei Y, et al. Efficacy between high and medium doses of glucocorticoid therapy in remission induction of IgG4-related diseases: a preliminary randomized controlled trial. Int J Rheum Dis. 2017;20:639–46.
Kamisawa T, Shimosegawa T, Okazaki K, Nishino T, Watanabe H, Kanno A, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58:1504–7.
de Pretis N, Amodio A, Bernardoni L, Campagnola P, Capuano F, Chari ST, et al. Azathioprine maintenance therapy to prevent relapses in autoimmune pancreatitis. Clin Transl Gastroenterol. 2017;8:e90.
Yunyun F, Yu C, Panpan Z, Hua C, Di W, Lidan Z, et al. Efficacy of cyclophosphamide treatment for immunoglobulin G4-related disease with addition of glucocorticoids. Sci Rep. 2017;7:6195.
Zhang P, Gong Y, Liu Z, Liu Y, Lin W, Li J, et al. Efficacy and safety of iguratimod plus corticosteroid as bridge therapy in treating mild IgG4-related diseases: a prospective clinical trial. Int J Rheum Dis. 2019;22:1479–88.
Wang Y, Li K, Gao D, Luo G, Zhao Y, Wang X, et al. Combination therapy of leflunomide and glucocorticoids for the maintenance of remission in patients with IgG4-related disease: a retrospective study and literature review. Intern Med J. 2017;47:680–9.
Della-torre E, Campochiaro C, Bozzolo EP, Dagna L, Scotti R, Nicoletti R, et al. Methotrexate for maintenance of remission in igg4-related disease. Rheumatol (United Kingdom). 2015;54:1934–6.
Yunyun F, Yu P, Panpan Z, Xia Z, Linyi P, Jiaxin Z, et al. Efficacy and safety of low dose Mycophenolate mofetil treatment for immunoglobulin G4-related disease: a randomized clinical trial. Rheumatology (Oxford). 2019;58:52–60.
Wang L, Zhang P, Wang M, Feng R, Lai Y, Peng L, et al. Failure of remission induction by glucocorticoids alone or in combination with immunosuppressive agents in IgG4-related disease: A prospective study of 215 patients. Arthritis Res Ther. 2018;20:1–12.
Takanashi S, Kaneko Y, Takeuchi T. Effectiveness of tacrolimus on IgG4-related disease. Mod Rheumatol. 2019;29:892–4.
Huggett MT, Culver EL, Kumar M, Hurst JM, Rodriguez-Justo M, Chapman MH, et al. Type 1 autoimmune pancreatitis and IgG4-related sclerosing cholangitis is associated with extrapancreatic organ failure, malignancy, and mortality in a prospective UK cohort. Am J Gastroenterol. 2014;109:1675–83.
Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74:1171–7.
Campochiaro C, Della-Torre E, Lanzillotta M, Bozzolo E, Baldissera E, Milani R, et al. Long-term efficacy of maintenance therapy with rituximab for IgG4-related disease. Eur J Intern Med. 2019.
Yamamoto M, Takahashi H, Takano K, Shimizu Y, Sakurai N, Suzuki C, et al. Efficacy of abatacept for IgG4-related disease over 8 months. Ann Rheum Dis. 2016;75:1576–8.
Simpson RS, Lau SKC, Lee JK. Dupilumab as a novel steroid-sparing treatment for IgG4-related disease. Ann Rheum Dis 2019;0:2–3.
Pasquali T, Schoenfield L, Spalding SJ, Singh AD. Orbital inflammation in IgG4-related sclerosing disease. Orbit. 2011;30:258–60.
Khan ML, Colby TV, Viggiano RW, Fonseca R. Treatment with bortezomib of a patient having hyper IgG4 disease. Clin Lymphoma Myeloma Leuk. Elsevier company. 2010;10:217–9.
Khosroshahi A, Carruthers MN, Stone JH, Shinagare S, Sainani N, Hasserjian RP, et al. Rethinking Ormond’s disease: “idiopathic” retroperitoneal fibrosis in the era of IgG4-related disease. Medicine (Baltimore). 2013;92:82–91.
Fernández-Codina A, Martínez-Valle F, Castro-Marrero J, Detorres I, Vilardell-Tarrés M, Ordi-Ros J. Idiopathic retroperitoneal fibrosis: a clinicopathological study in 24 Spanish cases. Clin Rheumatol. 2013;32:889–93.
Kuraishi Y, Muraki T, Ashihara N, Ozawa M, Nakamura A, Watanabe T, et al. Validity and safety of endoscopic biliary stenting for biliary stricture associated with IgG4-related pancreatobiliary disease during steroid therapy. Endosc Int Open. 2019;07:E1410–8.
Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol (Hoboken, NJ). 2015;67:1688–99.
Hellmich B, Agueda A, Monti S, Buttgereit F, de Boysson H, Brouwer E, et al. 2018 update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2020;79:19–30.
Ebbo M, Grados A, Samson M, Groh M, Loundou A, Rigolet A, et al. Long-term efficacy and safety of rituximab in IgG4-related disease: data from a French nationwide study of thirty-three patients. PLoS One. 2017;12:e0183844.
Charles P, Terrier B, Perrodeau É, Cohen P, Faguer S, Huart A, et al. Comparison of individually tailored versus fixed-schedule rituximab regimen to maintain ANCA-associated vasculitis remission: results of a multicentre, randomised controlled, phase III trial (MAINRITSAN2). Ann Rheum Dis. 2018;77:1143–9.
Fernández-Codina A, Pinilla B, Pinal-Fernández I, López C, Fraile-Rodríguez G, Fonseca-Aizpuru E, et al. Treatment and outcomes in patients with IgG4-related disease using the IgG4 responder index. Jt Bone Spine. 2018;85:721–6.
Omar D, Chen Y, Cong Y, Dong L. Glucocorticoids and steroid sparing medications monotherapies or in combination for IgG4-RD: a systematic review and network meta-analysis. Rheumatology (Oxford). 2019.
Wallace ZS, Mattoo H, Mahajan VS, Kulikova M, Lu L, Deshpande V, et al. Predictors of disease relapse in IgG4-related disease following rituximab. Rheumatology. 2016;55:1000–8.
Berti A, Della-Torre E, Gallivanone F, Canevari C, Milani R, Lanzillotta M, et al. Quantitative measurement of 18F-FDG PET/CT uptake reflects the expansion of circulating plasmablasts in IgG4-related disease. Rheumatology (Oxford). 2017;56:2084–92.
Martínez-Valle F, Orozco-Gálvez O, Fernández-Codina A. Update in ethiopathogeny, diagnosis and treatment of the IgG4 related disease. Med Clin (Barc). Elsevier España, S.L.U. 2018;151:18–25.
Lin W, Zhang P, Chen H, Chen Y, Yang H, Zheng W, et al. Circulating plasmablasts/plasma cells: a potential biomarker for IgG4-related disease. Arthritis Res Ther. Arthritis Research & Therapy. 2017;19:25.
Carruthers MN, Stone JH, Deshpande V, Khosroshahi A. Development of an IgG4-RD responder index. Int J Rheumatol. 2012;2012:1–7.
Perugino CA, Mattoo H, Mahajan VS, Maehara T, Wallace ZS, Pillai S, et al. Emerging Treatment Models in Rheumatology: IgG4-Related Disease: Insights Into Human Immunology and Targeted Therapies. Arthritis Rheumatol (Hoboken, NJ). 2017;69:1722–32.
Viela Bio Reports Third Quarter 2019 Financial Results and Business Highlights [Internet]. [cited 2020 Apr 8]. Available from: https://www.sec.gov/Archives/edgar/data/1734517/000156459019043518/ck0001734517-ex991_6.htm.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Fernandez Codina reports grants from Scleroderma Society of Ontario, grants from Saint Joseph’s Health Care London Foundation, personal fees from Actelion, grants from Medicina, other from Catalan-Balearic Society of Internal Medicine, outside the submitted work. The other authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and compiled with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Vasculitis
Rights and permissions
About this article

Cite this article
Fernández-Codina, A., Orozco-Gálvez, O. & Martínez-Valle, F. Therapeutic Options in IgG4-Related Disease. Curr Treat Options in Rheum 6, 191–204 (2020). https://doi.org/10.1007/s40674-020-00147-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40674-020-00147-w