
Abstract
Purpose of review
Osteoarthritis (OA) is the most common form of arthritis, and pain is the primary symptom of the disease, yet analgesic options for treating OA pain remain limited. In this review, we aimed to give an update on the current clinical and preclinical studies targeting two pathways that are being investigated for treating OA pain: the nerve growth factor (NGF) pathway and the transient receptor potential vanilloid-1 (TRPV1) pathway.
Recent findings
Antibodies against NGF, small molecule inhibitors of TrkA, TRPV1 agonists, and TRPV1 antagonists are all in different stages of clinical and pre-clinical testing for the treatment of OA pain. NGF antibodies have shown efficacy in the primary endpoints tested compared with placebo; however, rapidly progressive OA has been consistently observed in a subset of patients and the cause remains unclear. TRPV1 agonists have also demonstrated reduced pain with no serious adverse events—the most common adverse events include a burning or warming sensation upon administration.
Summary
Targeting the NGF and TRPV1 pathways appears effective for reducing OA pain, but further work is needed to better understand which patients may benefit most from these treatments. The anti-NGF antibody tanezumab and the TRPV1 agonist CNTX-4975 have both received fast-track designation from the FDA for the treatment of OA pain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) is the most common form of arthritis, affecting an estimated 303 million people globally in 2017 [1], and pain is the predominant symptom associated with this disease. OA is a degenerative disease of the synovial joints including the knee, hip, facet joints of the spine, and hand. Risk factors for OA include aging, prior joint injury, obesity, female sex, and genetics [2]. These risk factors are related to the underlying pathogenesis of OA, which is a complex process impacted by altered biomechanics, chronic low-level inflammation, and aging. All of these processes can promote degradation and remodeling of the joint tissues, which ultimately results in failure of the structural integrity of the joint.
OA is a slowly developing, chronic disease with no known cure or strategies for reducing or stopping the progression of joint damage [3]. Management of symptoms is also limited—NSAIDs are one of the few strongly recommended pharmacological therapies for OA [4], yet the efficacy of oral NSAIDs declines with time and these drugs are associated with adverse gastrointestinal and cardiac effects that may not make them a suitable choice for all patients [5]. Therefore, chronic pain associated with OA has a large individual and societal impact. The most recent estimate of global years lived with disability due to OA reached 9,604,000 in 2017, an increase of 31.4% from 2007 [1].
As OA develops, the pain experience also changes [6]. Early OA pain is often associated with specific activities, which results in individuals limiting these activities in an attempt to prevent pain. Chronic OA pain is associated with more constant pain as well as by intense episodes of unpredictable pain. Sensitization of the nervous system also occurs in OA and is correlated with OA knee pain severity [7]. In addition, a recent study demonstrated that quantitative sensory testing, which tests for nervous system sensitization, can help to predict those individuals who are likely to develop persistent knee pain [8]. Overall, the individual perception of chronic OA pain is shaped by a combination of biological, psychological, and social factors, but there is evidence that ongoing peripheral input from the OA joint remains a strong contributor toward the maintenance of this pain [6, 9]. For example, total joint replacement reduces both pain and nervous system sensitization for the majority of patients [10,11,12], suggesting that targeting the peripheral sources of pain may be effective in OA. In addition, peripherally restricted drugs targeting sensory neurons may reduce the addictive adverse effects associated with centrally acting drugs such as opioids [13].
In this narrative review, we are focusing on two peripheral analgesic targets for which drugs have recently received FDA fast-track designation status for OA pain: the nerve growth factor (NGF) pathway and the transient receptor potential vanilloid-1 (TRPV1) pathway. We searched PubMed using different combinations of the following search criteria: “Osteoarthritis” “mice” “rat” “transgenic” “NGF” “TrkA” “pain” “sensitization” “tanezumab” “fulranumab” “fasinumab” “capsaicin” “CNTX-4975” “resiniferatoxin” “zucapsaicin” “civamide” “NEO6860” “mavatrep” “JNJ39439335” “TRPV1”. In addition, we searched www.clinicaltrials.gov and rheumatology conference abstracts for active and recently completed clinical trials associated with these pathways.
Targeting NGF signaling
NGF levels are elevated in different disease states, particularly those associated with inflammation [14•], and NGF plays an important role in nociception and pain mediation through binding to its high affinity receptor, tropomyosin receptor kinase A (TrkA) [15]. In OA joints, NGF is elevated in synovial fibroblasts [16, 17], synovial macrophages [16], chondrocytes [18, 19] and within osteochondral channels [19]. In addition, NGF levels were higher in the osteochondral channels and synovium of OA patients with symptomatic chondropathy compared with the asymptomatic group [16, 20]. NGF and its receptor TrkA have received much attention as possible targets for treating OA pain, especially after reports of significant pain reduction in patients with moderate to severe knee OA [21]. However, a hold was placed by the US Food and Drug Administration (FDA) on all trials in 2010 due to adverse events including rapidly progressive OA (RPOA) in subjects that received the treatment [22]. The clinical hold was lifted in 2015, and the clinical trials of anti-NGF antibodies were resumed [23]. Therefore, we focused on clinical trials performed between years 2015 and 2019 (Table 1). Two humanized monoclonal antibodies are currently in phase 3 clinical trials, tanezumab (Pfizer and Eli Lilly) and fasinumab (Regeneron and Teva). Fulranumab is another monoclonal antibody against NGF that was discontinued by Janssen Research & Development (https://www.jnj.com/media-center/press-releases/janssen-announces-discontinuation-of-fulranumab-phase-3-development-program-in-osteoarthritis-pain).
Tanezumab
Tanezumab is the anti-NGF antibody furthest along in development. Indeed, the FDA has granted tanezumab a fast-track designation for the treatment of chronic pain in patients with OA and chronic low back pain (https://www.pfizer.com/news/press-release/press-release-detail/pfizer_and_lilly_receive_fda_fast_track_designation_for_tanezumab).
This past year, a pooled analysis was performed on the placebo-controlled, phase III OA trial data from 2008 to 2010, in order to evaluate the efficacy (4 trials that were completed before the FDA hold) and safety (in terms of common adverse effects not related to joint function and adverse effects related to neurologic function) (9 trials) of intravenous injections of tanezumab in specific subgroup of OA patients including patients with diabetes and severe OA symptoms, and those aged ≥ 65 years [24]. Patients received tanezumab, tanezumab with oral NSAID, active comparator (naproxen, celecoxib, diclofenac, or oxycodone), or placebo. Tanezumab (2.5, 5, and 10 mg) and naproxen (500 mg) showed improved WOMAC pain, WOMAC physical function, and Patient’s Global Assessment (PGA) compared with placebo-treated group at week 16. These conclusions are similar to two other recent reviews [25, 26]. However, only higher doses of tanezumab (5 and 10 mg) showed improved outcomes when compared with naproxen 500 mg. Tanezumab 5 mg and 10 mg also showed similar efficacy in at risk OA patients compared with nondiabetic patients, patients with less severe OA symptoms, and patients aged < 65 years, respectively. Safety analysis was performed in 7491 patients (pooled from 9 trials): 15.6% had diabetes, 22.3% had severe OA symptoms, and 36.0% were aged ≥ 65 years at baseline. The incidence of common adverse effects (not including rapidly progressive OA) was similar in tanezumab and active comparator group, but both groups showed higher adverse effects when compared with the placebo group. In addition, a greater number of adverse effects were reported when tanezumab was combined with NSAID compared with tanezumab alone. Adverse effects included arthralgia, headache, pain in extremity, paresthesia, peripheral edema, nasopharyngitis, and hypoesthesia [24]. In addition, adverse effects of abnormal peripheral sensation were more frequently reported in tanezumab-treated patients than in placebo or active comparator patients, but tanezumab was not associated with an increase in adverse effects related to decreased sympathetic nervous system function. Separately, a blinded Adjudication Committee reviewed and adjudicated the joint-related adverse effects and determined that higher doses of tanezumab and tanezumab combined with NSAIDs were associated with an increase in rapidly progressive OA [27].
More recently, the efficacy and joint safety outcomes for subcutaneous injection of tanezumab were evaluated for several treatment regimens after the serious adverse events associated with intravenous route [22, 27, 28]. In a randomized, double-blind, multicenter trial of subcutaneous injection of tanezumab, different strategies were implemented in order to reduce the incidence of adverse events seen in previous studies, including preventing the concomitant use of NSAIDs, excluding patients at risk of RPOA and patients who are not suitable candidates for total joint replacement, in addition to administering the lowest efficacious dose of the antibody [29••] (Trial ID NCT02697773). Inclusion criteria focused on patients with moderate to severe hip or knee OA who had not responded to or were unable or unwilling to take acetaminophen, NSAIDs, and tramadol or opioids. Three treatment regimens were tested over 16 weeks: tanezumab 2.5 mg at baseline and at week 8 (2.5 mg/2.5 mg: 231 patients); tanezumab 2.5 mg at baseline and 5 mg at week 8 (2.5 mg/5 mg: 233 patients); or placebo at baseline and at week 8 (232 patients). The two tanezumab treatment groups (2.5 mg/2.5 mg and 2.5 mg/5 mg) showed statistically significant improvements in WOMAC Pain, WOMAC Physical Function, and patient’s global assessment of OA (PGA-OA) scores compared with placebo at week 16, although all 3 groups improved compared with baseline. However, tanezumab-treated patients showed a higher risk of rapidly progressive OA with 5 patients developing it in the 2.5/2.5 mg group (2.2%) and one patient in the 2.5/5 mg group (0.4%). Eight patients in the 2.5/2.5 mg group (3.5%), 16 patients in the 2.5/5 mg group (6.9%), and 4 in the placebo (1.7%) required total joint replacements. This suggests a dose-dependent increase in total joint replacement in tanezumab-treated patients. No osteonecrosis or any pathological fractures were reported [29••].
In another randomized, double-blind study, the efficacy and joint safety events of subcutaneous tanezumab versus NSAID were evaluated for hip and knee OA for 80 weeks, and results have been presented in abstract format thus far [30, 31]. Subcutaneous tanezumab (2.5 mg or 5 mg every 8 weeks), or NSAIDs (naproxen 500 mg, celecoxib 100 mg, or diclofenac ER 75 mg orally bid) were given to participants over the 56-week treatment period. Primary efficacy endpoints were changed from baseline to week 16 in WOMAC pain, WOMAC function, and PGA-0A. At week 16, treatment with tanezumab 5 mg showed significantly improved WOMAC pain and function scores compared with the NSAID group. Tanezumab 2.5 mg did not show any advantage over NSAID in any of the endpoints. PGA-OA was not improved in any of the treatment groups [30]. Joint safety outcomes including rapidly progressive OA type 1 or 2 (RPOA1 or 2) (RPOA 1 and 2 are described in [27, 32]), primary osteonecrosis, subchondral insufficiency fracture (SIF), and pathologic fractures were assessed for the two tanezumab treatment regimens over the 80-week observation period. RPOA1, RPOA2, and total joint replacement were significantly higher with tanezumab treatment (both groups) compared with NSAID group. Rates of RPOA1 were higher with tanezumab 2.5 mg (2.9%) and tanezumab 5 mg (4.9%) than NSAID (1.1%). Rates of RPOA2 were also higher in tanezumab 2.5 mg (0.3%) and tanezumab 5 mg (1.4%) compared with NSAID (0.1%). Finally, total joint replacement rates were higher with tanezumab 2.5 mg (5.3%) and 5 mg (8.0%) than NSAID (2.6%) [31]. SIF rates were similar in the tanezumab groups (5.8%, 6.9% in tanezumab 2.5 and 5 mg groups, respectively) compared with NSAID (3.9%). Of 2000 patients receiving tanezumab, only one patient in the tanezumab 5-mg group had primary osteonecrosis. No pathological fractures were observed. In summary, patients with 5 mg tanezumab treatment showed improved pain and function compared with patients on a lower dose of tanezumab or NSAID therapy. However, adverse events were more common with tanezumab compared with NSAID.
Fasinumab
The efficacy, tolerability, and joint safety of fasinumab treatment were assessed for 342 patients in a phase IIb/III double-blind, placebo-controlled, randomized clinical trial [33••]. Subcutaneous fasinumab (1 mg, 3 mg, 6 mg, or 9 mg) was given every 4 weeks for a total of four doses. Endpoint analysis was evaluated at 16 weeks. Fasinumab treatment in moderate to severe knee and/or hip OA pain proved to be efficient in improving WOMAC pain and physical function sub-scale scores for all doses compared with the placebo group. However, PGA scores showed statistically significant improvement only with the 1-mg and 9-mg doses over placebo. Similar to what has been observed in tanezumab trials, there was an apparent dose-dependent increase in RPOA with fasinumab treatment (1 mg = 2.4%; 3 mg = 2.4%; 6 mg = 5.9%; 9 mg = 8.4%), with no RPOA reports in the placebo group. Total joint replacement occurred in 3–4 patients per treatment group, including the placebo group, with no evidence of a dose-dependent effect [33••]. Development of this drug continues with five active phase 3 clinical trials underway.
Fulranumab
The efficacy and safety of fulranumab were evaluated in a phase 2 trial in patients with moderate to severe knee osteoarthritis compared with placebo and active comparator (oxycodone). Fulranumab (3 mg or 9 mg) was given subcutaneously every 4 weeks. The primary endpoint was the percent improvement in average osteoarthritis-related pain intensity (OAPI) score up to week 12 or to 28 December 2010 (date of clinical hold), whichever was earlier. No difference was observed between fulranumab (both doses) and placebo (3 mg: p = 0.739; 9 mg: p = 0.843) responder rates, while oxycodone had a lower responder rate compared with both doses of fulranumab (3 mg: p = 0.008; 9 mg: p = 0.012) and placebo (p = 0.0021). Adverse events were similar in all treatment groups except for the fulranumab 3 mg group, which showed the lowest adverse events. Four joint replacements were reported in two of the 9 mg fulranumab patients and one oxycodone patient (none of these replacement was attributed to RPOA or osteonecrosis) [34].
Tropomyosin-related kinase inhibition
Tropomyosin-related kinase (TrkA) is a high affinity receptor to NGF, and thus targeting TrkA might provide an alternative way to block NGF signaling. The efficacy of two small molecule TrkA inhibitors has been evaluated for knee OA pain. In a phase II double-blind, placebo-controlled and randomized trial, a single intraarticular injection of GZ389988 (a TrkA inhibitor-formulation in 3-mL i.a. injection vehicle) in the knee of 104 moderate to severe OA patients resulted in reduced WOMAC pain over 12 weeks compared with placebo. Adverse events were associated with injection site inflammatory reactions [35]. Another phase IIa, double-blind clinical trial tested oral administration of ASP7962 (TrkA inhibitor, 100 mg) twice daily for 4 weeks. In this trial, ASP7962 failed to improve WOMAC function and pain scores in 215 knee OA participants over placebo [36]. Why these studies have drawn different conclusions regarding the efficacy of TrkA inhibition remains unclear, but these differences could be due to potential pharmacological differences between the two drugs, different dosing and routes of administration, and different exclusion and inclusion criteria for the study participants (nicely discussed in an editorial published in the November 2019 issue of Osteoarthritis and Cartilage [37•]).
Preclinical OA studies targeting NGF/TrkA
The preclinical testing of NGF antibodies and TrkA inhibitors in models of OA has trailed OA clinical trials, but a number of preclinical studies using different OA models have now demonstrated that anti-NGF can inhibit pain-related behaviors. As clinical trials continue to consistently have cohorts that suffer from rapidly progressive OA, preclinical models provide an important avenue for investigating how exactly blocking the NGF/TrkA pathway provides analgesic relief in OA as well as the causes and risk factors for rapidly progressive OA.
Using the MIA model in both rats and mice, a single treatment with anti-NGF can reverse pain-related behaviors [38,39,40,41]. In addition, one-time injection of a TrkA inhibitor (TrkAd5, 2 mg/kg, subcutaneous (s.c.)) was able to reverse weight-bearing deficits 16 weeks after destabilization of the medial meniscus (DMM) surgery [42].
More extensive studies have examined the effects of longer-term therapy beginning at different time points in the models to better mimic the clinical situation. In one study, the prophylactic and therapeutic potential of long-term anti-NGF therapy was tested in the rat MIA model. Weekly dosing with anti-NGF (muMab911, 10 mg/kg, s.c.) beginning prior to induction of the model was able to prevent the development of weight-bearing asymmetry, but treatment only inhibited MIA-induced hind paw mechanical allodynia on day 28 [43]. Therapeutic treatment, consisting of subcutaneous injection of 10 mg/kg muMab911 or PBS on days 14 and 21 post induction of MIA, was able to inhibit weight-bearing deficits by day 28 and hind paw mechanical allodynia on day 21. Prophylactic anti-NGF treatment in this study did not significantly alter MIA-induced joint damage, but it trended toward more severe cartilage damage compared with the vehicle group; therapeutic treatment had no significant effect on joint damage [43].
A similar study looking at the effects of tanezumab treatment beginning at different time points was conducted using the rat medial meniscal tear (MMT) model [44•]. In this model, pain-related behavior measured by gait deficiency was only observed in MMT rats on day 3 and 7 after surgery, resolving for days 14–28. Tanezumab treatment given weekly beginning on the day of surgery (0.1, 1, or 10 mg/kg, s.c.) was able to prevent this early gait change. However, tanezumab-treated rats exhibited more severe cartilage damage than either vehicle or isotype controls on days 7, 14, and 28. No significant changes in body weight were noted throughout the study; focal areas of alopecia along the mouth/muzzle developed from day 14 in most animals and were present in most of the 1 mg/kg tanezumab group by day 28. In a second set of experiments, tanezumab treatment (0.1 mg/kg, s.c. weekly) was delayed until either day 23 or 57. Tanezumab-treated rats had more severe cartilage degeneration than controls when treatment was started on day 23 and rats were sacrificed on day 37. When treatment was started on day 57, the worsening of joint damage was less apparent by day 71, although rats already had severe cartilage damage by the time treatment was begun in this protocol.
The efficacy of NGF blockade through a vaccine was tested in the partial meniscectomy (PMX) model in male mice [45]. Prophylactic vaccination required boosters to maintain antibody levels. When a booster was given at week 10, weight-bearing deficits were inhibited for 3 weeks before pain behaviors returned again. When vaccination was administered therapeutically beginning at week 10 of the model, with boosters weeks 12 and 15, this therapy resulted in inhibition of weight-bearing deficits from weeks 14 to 18 of the model. Unlike the MMT study [44•], prophylactic NGF vaccination did not impact the development of cartilage damage in this study. One reason for this difference could be due to transient antibody titers resulting in incomplete blockade. Another reason could be due to the difference in model severity—the PMX model induces more slowly progressive cartilage damage compared with the MMT model.
The ability to modulate pain behaviors has also been tested using small molecule inhibitors of TrkA. Prophylactic treatment with a TrkA inhibitor (AR786, 30 mg/kg, orally twice daily) was tested in rats after OA induction by meniscal transection (MNX) surgery [46•]. Treatment with AR786 was withdrawn in one group of rats 2 weeks after arthritis induction and replaced with vehicle treatment. Weight-bearing asymmetry was prevented in MNX-operated rats at all time points when they received AR786 throughout the study compared with vehicle-treated, MNX-operated rats. Likewise, hind paw mechanical allodynia was prevented in arthritic rats that received AR786 throughout the study. Stopping AR786 treatment at day 14 at MNX surgery resulted in the development of weight-bearing asymmetry by day 24, and development of hind paw mechanical allodynia by day 21, demonstrating that ongoing treatment is required for long-term analgesic effects. This study also examined therapeutic administration of AR786 [46•]. The TrkA inhibitor was tested in both the rat MIA and rat MNX models. Treatment beginning on day 14 in both models (AR786, 30 mg/kg, orally twice daily) reduced weight-bearing asymmetry and hind paw mechanical allodynia after 3 days of treatment in the MIA model and following 5 days of treatment in the MNX model.
Finally, a study was recently performed to look at how a gain in function TrkA mutation enhances pain-like behavior in the MIA model [47]. Baseline values were comparable between TrkA knock-in (TrkA KI) and WT mice. A submaximal dose of MIA (0.7 mg/mouse) was associated with slow development of ipsilateral mechanical hypersensitivity starting from day 7 and lasting for up to day 28 in WT mice compared with saline controls [47]. Mechanical hypersensitivity developed more rapidly in TrkA KI and was significantly higher than in WT mice by day 3 post-MIA injection. Area under the curve analysis demonstrated that withdrawal thresholds of TrkA KI mice were lower than WT mice thresholds between days 0 and 7, and between days 21 and 28 after MIA injection [47]. Together this study supports the idea that activation of TrkA contributes to the development of mechanical sensitivity in the MIA model.
Targeting TRPV1
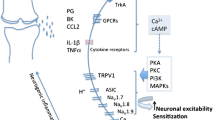
TRPV1 is a ligand-gated, non-selective cation channel expressed predominantly by sensory neurons. TRPV1 responds to a variety of noxious stimuli including capsaicin, intense heat, and acid, and TRPV1-knockout mice are protected against thermal and mechanical hyperalgesia in different types of pain models [48•]. Two pharmacological approaches have been developed to target TRPV1 for treating pain, agonists, and antagonists, although the biological results of these two approaches are quite different [48•]. Agonists of TRPV1 such as capsaicin can cause long-term effects in sensory nerve fibers through a combination of mechanisms including desensitization, nociceptor dysfunction, neuropeptide depletion, and reversible nerve fiber degeneration [49]. Antagonists, on the other hand, block the function of TRPV1 itself. Therefore, agonists to TRPV1 likely have a broader effect than simply targeting this one receptor, although the initial reaction to TRPV1 agonists often causes pain that requires co-administration with an anesthetic [48]. We will discuss all OA clinical trials testing drugs that target TRPV1 (Table 2).
Topical capsaicin
A recent systematic review concluded from four older clinical trials that topical capsaicin used at its licensed dose had an increased effect size compared with placebo [50•]. However, these were all relatively small clinical trials and no capsaicin trials adequately blinded their participants due to the warming sensation experienced on its initial application [50•]. A recent randomized phase 2, double-blind clinical trial tested higher doses of topical capsaicin (CGS-200) for treatment of knee OA pain [51]. Vehicle, 1% capsaicin, or 5% capsaicin were applied to both knees of 122 patients for 1 h on 4 consecutive days. Results presented in abstract form so far showed that topical treatment with 5% capsaicin (but not 1% capsaicin) improved WOMAC pain scores on day 35 (primary endpoint) compared with vehicle [51].
CNTX-4975
Another TRPV1 agonist being investigated for the treatment of OA pain by Centrexion Therapeutics is CNTX-4975, a highly potent, highly purified synthetic trans-capsaicin. In 2018, CNTX-4975 received a fast-track designation by the US FDA for treatment of knee OA pain (https://centrexion.com/wp-content/uploads/2018/01/Centrexion-Therapeutics-Announces-Fast-Track-Designation-Granted-by-FDA-to-CNTX-4975-for-Treatment-of-Knee-Osteoarthritis-Pain-.pdf). In a 24-week, randomized, double-blind, placebo-controlled clinical trial, a single intra-articular injection of CNTX-4975 was evaluated at two doses (0.5 mg or 1.0 mg) versus placebo in 175 patients with moderate to severe OA pain and in whom previous treatment was not successful [52••]. The area under the curve for the change from baseline through week 12 in daily WOMAC pain with walking scores (the primary efficacy endpoint) was improved in the 1 mg CNTX-4975 treatment group compared with placebo. Treatment emergent adverse events included arthralgia, upper respiratory tract infection, increased hepatic enzyme, joint effusion, and osteoarthritis, but there were no differences in CNTX-4975 groups compared with placebo.
Civamide
Civamide or zucapsaicin (synthetic cis-isomer of capsaicin) is a TRPV1 receptor agonist. The efficacy and safety of civamide were evaluated in a 12-week randomized, double blind study for 695 knee OA patients with a 52-week open-label extension [53]. Civamide cream (0.075%) or a control cream with a low dose of 0.01% civamide was applied three times daily for 12 weeks. Patients were evaluated on days 21, 42, 63, and 84 (end of the study) on three primary endpoints, namely the time-weighted average (TWA) of change from baseline to end of week 12 (day 84) in the WOMAC pain, WOMAC function scale, and the Subject Global Evaluation (SGE). In the 52-week open-label extension, Osteoarthritis Pain Score and SGE were assessed. Civamide was significantly more efficacious compared with control cream for the TWA of change from baseline to day 84 in WOMAC pain subscale (p = 0.009), WOMAC physical function subscale (p < 0.001), and SGE (p = 0.008). The efficacy was maintained for the 52-week open-label extension. No serious adverse events were reported due to the lack of systemic absorption, but 32 patients withdrew from the study due to an adverse event. The most common adverse event was mild to moderate transient burning sensation on the sites where both creams were applied [53].
Resiniferatoxin
Resiniferatoxin (RTX) is a naturally occurring capsaicin analogue (TRPV1 agonist) derived from the Euphorbia resinifera plant [54]. RTX has been given orphan drug status by the US Food and Drug Administration for the treatment of end-stage diseases, including intractable cancer pain, and intrathecal/epidural RTX is under phase 1 active clinical trials for the treatment of advanced cancer pain by blocking transmission of pain signals to the spinal cord [55].
RTX is also under two active clinical trials for OA pain management. Sorrento Therapeutics announced preliminary results of a small phase 1b double-blinded, placebo-controlled study (NCT03542838). Intraarticular RTX safety and efficacy were evaluated for treatment of moderate to severe OA knee pain. According to a press release, in the best performing RTX dose cohort at day 84, the WOMAC A1 score 10-point scale question “pain at walking on flat surface” showed an average of 5.7-point reduction relative to baseline for RTX, and 3.3-point reduction relative to the saline control (http://investors.sorrentotherapeutics.com/news-releases/news-release-details/sorrento-therapeutics-updates-positive-results-phase-1b). No dose limiting toxicity was found at any dose used, but treatment-emergent adverse events included post-injection pain, tachycardia and hypertension. A phase 3 trial is currently planned but not yet recruiting (NCT04044742).
NEO6860
NEO6860 is a modality selective TRPV1 antagonist, meaning that it specifically antagonizes capsaicin activation of TRPV1 but has little activity against heat or low pH activation of TRPV1 [56]. The analgesic effect of NEO6860 was evaluated in a phase 2 randomized clinical trial after 1 day of oral dosing [56]. NEO6860 (500 mg bid), placebo, or naproxen (500 mg bid) was given to 54 knee OA patients. The primary endpoint was reduction in pain intensity on the Numerical Rating Scale (NRS) after exercise, using the staircase test, 8 h after dosing. NEO6860 showed an analgesic trend (that was not statistically significant) after exercise at 3 and 24 h (not 8 h) versus placebo. The effect was statistically significant only when naproxen was compared with placebo at the 24 h time point. The adverse events that are commonly reported with non-modality-selective TRPV1 antagonist (high body temperature and impairment of heat pain perception) were not seen. Mild adverse effects (but still more than naproxen and placebo) were reported including feeling hot (most common but decreased from 87 to 4% between first and second dose), headache, nausea, dizziness, fatigue, hypoesthesia, and increased blood pressure.
Mavatrep
Mavatrep or JNJ39439335 is a potent, selective, competitive TRPV1 receptor antagonist that was evaluated for painful knee osteoarthritis. In a randomized, placebo- and active-controlled, phase 1b study, 33 knee OA patients were given a single dose of mavatrep (50 mg), naproxen (500 mg TID), or placebo [57]. The primary efficacy endpoint was pain reduction measured by the 4-h postdose sum of pain intensity difference (SPID) based on the 11-point (0–10) Numerical Rating Scale (NRS) for pain after stair-climbing (PASC). Mavatrep showed statistically significant efficacy compared with placebo for 4-h SPID PASC. Patients reported feeling hot as well as changes in heat perception, in addition to dysgeusia and paresthesia. In another double-blind, randomized, placebo-controlled phase 1 study, the pharmacokinetics and pharmacodynamics of mavatrep were evaluated in healthy men (part 1) and in patients with knee osteoarthritis (part 2) [58]. Twenty-four patients with knee OA were given once daily oral mavatrep (JNJ39439335) (10, 25, or 50 mg) or placebo. Efficacy was evaluated using the 11-point NRS score at rest and after stair climbing on days − 1, 8, 15, and 22, 4 h postdose. Both the 25 mg and 50 mg dose groups showed greater mean reduction from baseline in the pain intensity at rest and pain intensity after stair-climbing on day 22 compared with the placebo group (p < 0.05). The 50 mg group also showed significant pain reduction after stair climbing on day 8. All participants reported at least one treatment emergent adverse effect. In the mavatrep-treated groups, the most common adverse effects were thermal burn, headache, paresthesia, dysgeusia, thermohypoesthesia, feeling hot, and hot flush.
Preclinical studies testing targeting of TRPV1
The use of TRPV1 agonists to induce long-term desensitization has also been tested in models of OA, primarily the MIA model, but there is no published pre-clinical data available for the testing of CNTX-4975 in OA models. Pretreatment with intra-articular capsaicin (0.5%) 14 days prior to induction of the rat MIA model inhibited weight-bearing asymmetry from day 14 to 28 after MIA induction, and treatment also protected against bone changes [59]. The authors speculated that the decreased effect of the capsaicin treatment by day 28 may be due to functional recovery of the peripheral capsaicin-sensitive nerve fibers by this time, although this activity was not directly assessed in the study. In a high-dose rat MIA model, intra-articular injection of 0.03% RTX and bupivacaine on day 14 after MIA induction reversed weight-bearing asymmetry over a period of 3 days and inhibited hind paw mechanical allodynia for 10 days [60]. Finally, intra-articular injection of 10 μg RTX with lidocaine in dogs with OA improved locomotion over a period of 4 months or longer, and 2 to 3 years after the injection, there were no signs of accelerated joint degeneration in any of the dogs [61]. However, a randomized, vehicle-controlled, blinded trial must still be performed [61]. In addition to long-term effects presumably due to denervation by TRPV1 agonists, acute analgesic effects of capsaicin have also been tested in the MIA model, and analgesic effects are seen on the order of minutes to hours after injection, particularly in the early phase of the model [62, 63].
Finally, TRPV1 antagonists have also been explored in the MIA pain model and Abbott has shown analgesic efficacy for a number of compounds. A-425619 delivered systemically (i.p.) or A-784168, A-795614, and ABT-102 delivered orally all inhibited weight-bearing asymmetry on day 4 after MIA induction [64,65,66]. Similarly, A-993610, A-995662, or A-889425 given orally all inhibited hind paw grip strength deficits on day 21 after MIA induction [65, 67, 68]. Systemic administration of A-889425 also was shown to reduce the firing of spinal neurons in response to mechanical stimuli directed toward the knee as well as spontaneous firing of these neurons induced by the MIA model, suggesting that blocking TRPV1 reduces painful signaling to the spinal cord [68]. Temperature-neutral TRPV1 antagonists, which are likely to have fewer adverse effects associated with injection, have also been investigated and demonstrate efficacy in this model [69].
Other groups have also tested TRPV1 antagonists in the rat MIA model. In one study, systemic AMG9810 inhibited thermal hypersensitivity but it had no effect on weight-bearing asymmetry or ongoing pain assessed by conditioned place preference [70]. Additionally, in another study, intra-articular or systemic injection of the TRPV1 antagonist JNJ-17203212 inhibited weight-bearing asymmetry on day 14 after MIA induction, but only systemic injection could also inhibit distal mechanical allodynia [71]. Systemic administration of JNJ-17203212 increased core body temperature as previously described, but intra-articular injection had no effect on body temperature [71].
It will be interesting to test whether TRPV1 antagonists are effective in surgical models of OA, since these models may have less inflammation than the MIA model. In addition, another factor to consider in the development of TRPV1 antagonists is the potential interaction between the fatty acid amide hydrolase (FAAH) and TRPV1 pathways [72,73,74], and one study has suggested that inhibition of both FAAH and TRPV1 may be more effective for treating OA pain than one inhibitor alone [75].
Conclusions
While NSAIDs are a heavily relied upon therapy for OA pain, new options are needed for patients for whom NSAIDs are not a safe option and/or do not provide effective relief. Two pathways appear promising as alternative therapeutic targets for the treatment of pain associated with moderate to severe OA, the NGF pathway, and the TRPV1 pathway. Targeting NGF or its high affinity receptor TrkA has shown promising improvements in OA pain and function, with tanezumab receiving a fast-track designation from the US FDA for treatment of OA pain. However, concerns regarding adverse events such as the development of rapidly progressive OA have been raised, particularly associated with high doses and concomitant use of NSAID. TRPV1 modulators have also been attractive targets and are under active clinical investigation for treatment of OA pain. Recently, the US FDA has granted CNTX-4975, a TRPV1 agonist, a fast-track designation for treatment of pain associated with knee OA. Larger trials are required to confirm efficacy and evaluate safety of TRPV1 agonists and antagonists as a treatment of osteoarthritis pain. In addition, for both tanezumab and for CNTX-4975, whether or not these drugs are effective for treating pain associated with earlier stages of OA has yet to be assessed. Preclinical models offer a route for performing mechanistic studies to better understand the precise mechanism of action for these drugs in the context of OA, yet most of the preclinical work performed to date has focused on assessing analgesic efficacy. In conclusion, targeting the NGF and TRPV1 pathways appears effective for reducing OA pain but further work can be done to better understand which patients may benefit most from these treatments.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Disease GBD, Injury I, Prevalence C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1789–858. https://doi.org/10.1016/S0140-6736(18)32279-7.
Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393(10182):1745–59. https://doi.org/10.1016/S0140-6736(19)30417-9.
Osteoarthritis: a serious disease. https://www.oarsi.org/research/oa-serious-disease: Pre Competitive Consortium for Osteoarthritis Osteoarthritis Research Society International 2016.
Kolasinski SL, Neogi T, Hochberg MC, Oatis C, Guyatt G, Block J et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the management of osteoarthritis of the hand, hip, and knee. Arthritis & Rheumatology.n/a(n/a). doi:https://doi.org/10.1002/art.41142.
Osani MC, Vaysbrot EE, Zhou M, McAlindon TE, Bannuru RR. Duration of symptom relief and early trajectory of adverse events for oral NSAIDs in knee osteoarthritis: a systematic review and meta-analysis. Arthritis Care & Research. https://doi.org/10.1002/acr.23884.
Neogi T. The epidemiology and impact of pain in osteoarthritis. Osteoarthr Cartil. 2013;21(9):1145–53. https://doi.org/10.1016/j.joca.2013.03.018.
Neogi T, Frey-Law L, Scholz J, Niu J, Arendt-Nielsen L, Woolf C, et al. Sensitivity and sensitisation in relation to pain severity in knee osteoarthritis: trait or state? Ann Rheum Dis. 2015;74(4):682–8. https://doi.org/10.1136/annrheumdis-2013-204191.
Carlesso LC, Segal NA, Frey-Law L, Zhang Y, Na L, Nevitt M, et al. Pain susceptibility phenotypes in those free of knee pain with or at risk of knee osteoarthritis: the multicenter osteoarthritis study. Arthritis Rheumatol. 2019;71(4):542–9. https://doi.org/10.1002/art.40752.
Malfait AM, Schnitzer TJ. Towards a mechanism-based approach to pain management in osteoarthritis. Nat Rev Rheumatol. 2013;9(11):654–64. https://doi.org/10.1038/nrrheum.2013.138.
Graven-Nielsen T, Wodehouse T, Langford RM, Arendt-Nielsen L, Kidd BL. Normalization of widespread hyperesthesia and facilitated spatial summation of deep-tissue pain in knee osteoarthritis patients after knee replacement. Arthritis Rheum. 2012;64(9):2907–16. https://doi.org/10.1002/art.34466.
Kosek E, Ordeberg G. Lack of pressure pain modulation by heterotopic noxious conditioning stimulation in patients with painful osteoarthritis before, but not following, surgical pain relief. Pain. 2000;88(1):69–78. https://doi.org/10.1016/s0304-3959(00)00310-9.
Petersen KK, Arendt-Nielsen L, Simonsen O, Wilder-Smith O, Laursen MB. Presurgical assessment of temporal summation of pain predicts the development of chronic postoperative pain 12 months after total knee replacement. Pain. 2015;156(1):55–61. https://doi.org/10.1016/j.pain.0000000000000022.
Berta T, Qadri Y, Tan PH, Ji RR. Targeting dorsal root ganglia and primary sensory neurons for the treatment of chronic pain. Expert Opin Ther Targets. 2017;21(7):695–703. https://doi.org/10.1080/14728222.2017.1328057.
Denk F, Bennett DL, McMahon SB. Nerve growth factor and pain mechanisms. Annu Rev Neurosci. 2017;40:307-25. doi:https://doi.org/10.1146/annurev-neuro-072116-031121.This review provides background on the NGF signaling pathway.
Mantyh PW, Koltzenburg M, Mendell LM, Tive L, Shelton DL. Antagonism of nerve growth factor-TrkA signaling and the relief of pain. Anesthesiology. 2011;115(1):189–204. https://doi.org/10.1097/ALN.0b013e31821b1ac5.
Stoppiello LA, Mapp PI, Wilson D, Hill R, Scammell BE, Walsh DA. Structural associations of symptomatic knee osteoarthritis. Arthritis Rheumatol. 2014;66(11):3018–27. https://doi.org/10.1002/art.38778.
Manni L, Lundeberg T, Fiorito S, Bonini S, Vigneti E, Aloe L. Nerve growth factor release by human synovial fibroblasts prior to and following exposure to tumor necrosis factor-alpha, interleukin-1 beta and cholecystokinin-8: the possible role of NGF in the inflammatory response. Clin Exp Rheumatol. 2003;21(5):617–24.
Iannone F, De Bari C, Dell'Accio F, Covelli M, Patella V, Lo Bianco G, et al. Increased expression of nerve growth factor (NGF) and high affinity NGF receptor (p140 TrkA) in human osteoarthritic chondrocytes. Rheumatology (Oxford). 2002;41(12):1413–8. https://doi.org/10.1093/rheumatology/41.12.1413.
Walsh DA, McWilliams DF, Turley MJ, Dixon MR, Franses RE, Mapp PI, et al. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatology (Oxford). 2010;49(10):1852–61. https://doi.org/10.1093/rheumatology/keq188.
Aso K, Shahtaheri SM, Hill R, Wilson D, McWilliams DF, Walsh DA. Associations of symptomatic knee osteoarthritis with histopathologic features in subchondral bone. Arthritis Rheumatol. 2019;71(6):916–24. https://doi.org/10.1002/art.40820.
Lane NE, Schnitzer TJ, Birbara CA, Mokhtarani M, Shelton DL, Smith MD, et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med. 2010;363(16):1521–31. https://doi.org/10.1056/NEJMoa0901510.
Hochberg MC. Serious joint-related adverse events in randomized controlled trials of anti-nerve growth factor monoclonal antibodies. Osteoarthr Cartil. 2015;23(Suppl 1):S18–21. https://doi.org/10.1016/j.joca.2014.10.005.
Miller RE, Block JA, Malfait AM. What is new in pain modification in osteoarthritis? Rheumatology (Oxford). 2018;57(suppl_4):iv99-iv107. doi:https://doi.org/10.1093/rheumatology/kex522.
Tive L, Bello AE, Radin D, Schnitzer TJ, Nguyen H, Brown MT, et al. Pooled analysis of tanezumab efficacy and safety with subgroup analyses of phase III clinical trials in patients with osteoarthritis pain of the knee or hip. J Pain Res. 2019;12:975–95. https://doi.org/10.2147/JPR.S191297.
Chen J, Li J, Li R, Wang H, Yang J, Xu J et al. Efficacy and safety of tanezumab on osteoarthritis knee and hip pains: a meta-analysis of randomized controlled trials. Pain Medicine. 2016:pnw262. doi:https://doi.org/10.1093/pm/pnw262.
Schnitzer TJ, Marks JA. A systematic review of the efficacy and general safety of antibodies to NGF in the treatment of OA of the hip or knee. Osteoarthr Cartil. 2015;23:S8–S17. https://doi.org/10.1016/j.joca.2014.10.003.
Hochberg MC, Tive LA, Abramson SB, Vignon E, Verburg KM, West CR, et al. When is osteonecrosis not osteonecrosis?: adjudication of reported serious adverse joint events in the tanezumab clinical development program. Arthritis Rheumatol. 2016;68(2):382–91. https://doi.org/10.1002/art.39492.
Schnitzer TJ, Ekman EF, Spierings ELH, Greenberg HS, Smith MD, Brown MT, et al. Efficacy and safety of tanezumab monotherapy or combined with non-steroidal anti-inflammatory drugs in the treatment of knee or hip osteoarthritis pain. Ann Rheum Dis. 2015;74(6):1202–11. https://doi.org/10.1136/annrheumdis-2013-204905.
Schnitzer TJ, Easton R, Pang S, Levinson DJ, Pixton G, Viktrup L et al. Effect of tanezumab on joint pain, physical function, and patient global assessment of osteoarthritis among patients with osteoarthritis of the hip or knee: a randomized clinical trial. JAMA. 2019;322(1):37–48. doi:https://doi.org/10.1001/jama.2019.8044. This is the most recent clinical trial demonstrating efficacy of tanezumab compared with placebo over 16 weeks.
Hochberg M, Carrino J, Schnitzer T, Guermazi A, Vignon E, Walsh D et al. Subcutaneous tanezumab vs NSAID for the treatment of osteoarthritis: efficacy and general safety results from a randomized, double-blind, active-controlled, 80-week, phase-3 study [abstract]. Arthritis Rheum. 2019;71(suppl 10):1302. https://acrabstracts.org/abstract/subcutaneous-tanezumab-vs-nsaid-for-the-treatment-of-osteoarthritis-efficacy-and-general-safety-results-from-a-randomized-double-blind-active-controlled-80-week-phase-3-study/.
Hochberg M, Carrino J, Schnitzer T, Guermazi A, Walsh D, White A et al. Subcutaneous tanezumab versus NSAID for the treatment of osteoarthritis: joint safety events in a randomized, double-blind, active-controlled, 80-week, phase-3 study [abstract]. Arthritis Rheum. 2019;71(suppl 10):2756. https://acrabstracts.org/abstract/subcutaneous-tanezumab-vs-nsaid-for-the-treatment-of-osteoarthritis-efficacy-and-general-safety-results-from-a-randomized-double-blind-active-controlled-80-week-phase-3-study/.
Roemer FW, Hayes CW, Miller CG, Hoover K, Guermazi A. Imaging atlas for eligibility and on-study safety of potential shoulder adverse events in anti-NGF studies (part 3). Osteoarthr Cartil. 2015;23(Suppl 1):S59–68. https://doi.org/10.1016/j.joca.2014.09.018.
Dakin P, DiMartino SJ, Gao H, Maloney J, Kivitz AJ, Schnitzer TJ et al. The efficacy, tolerability, and joint safety of fasinumab in osteoarthritis pain: a phase IIb/III double-blind, placebo-controlled, randomized clinical trial. Arthritis Rheumatol. 2019;71(11):1824–34. https://doi.org/10.1002/art.41012. This is the most recent clinical trial demonstrating efficacy of fasinumab compared with placebo over 16 weeks.
Mayorga AJ, Wang S, Kelly KM, Thipphawong J. Efficacy and safety of fulranumab as monotherapy in patients with moderate to severe, chronic knee pain of primary osteoarthritis: a randomised, placebo- and active-controlled trial. Int J Clin Pract. 2016;70(6):493–505. https://doi.org/10.1111/ijcp.12807.
Krupka E, Jiang GL, Jan C. Efficacy and safety of intra-articular injection of tropomyosin receptor kinase A inhibitor in painful knee osteoarthritis: a randomized, double-blind and placebo-controlled study. Osteoarthr Cartil. 2019;27(11):1599–607. https://doi.org/10.1016/j.joca.2019.05.028.
Watt FE, Blauwet MB, Fakhoury A, Jacobs H, Smulders R, Lane NE. Tropomyosin-related kinase A (TrkA) inhibition for the treatment of painful knee osteoarthritis: results from a randomized controlled phase 2a trial. Osteoarthr Cartil. 2019;27(11):1590–8. https://doi.org/10.1016/j.joca.2019.05.029.
Walsh DA, Neogi T. A tale of two TrkA inhibitor trials: same target, divergent results. Osteoarthritis and cartilage. 2019;27(11):1575-7. https://doi.org/10.1016/j.joca.2019.07.013. This editorial provides discussion on mixed results in two TrkA inhibitor clinical trials.
Ishikawa G, Koya Y, Tanaka H, Nagakura Y. Long-term analgesic effect of a single dose of anti-NGF antibody on pain during motion without notable suppression of joint edema and lesion in a rat model of osteoarthritis. Osteoarthr Cartil. 2015;23(6):925–32. https://doi.org/10.1016/j.joca.2015.02.002.
Bryden LA, Nicholson JR, Doods H, Pekcec A. Deficits in spontaneous burrowing behavior in the rat bilateral monosodium iodoacetate model of osteoarthritis: an objective measure of pain-related behavior and analgesic efficacy. Osteoarthr Cartil. 2015;23(9):1605–12. https://doi.org/10.1016/j.joca.2015.05.001.
Miyagi M, Ishikawa T, Kamoda H, Suzuki M, Inoue G, Sakuma Y, et al. Efficacy of nerve growth factor antibody in a knee osteoarthritis pain model in mice. BMC Musculoskeletal Disorders. 2017;18(1). https://doi.org/10.1186/s12891-017-1792-x.
Sakurai Y, Fujita M, Kawasaki S, Sanaki T, Yoshioka T, Higashino K, et al. Contribution of synovial macrophages to rat advanced osteoarthritis pain resistant to cyclooxygenase inhibitors. PAIN. 2019;160(4):895–907. https://doi.org/10.1097/j.pain.0000000000001466.
McNamee KE, Burleigh A, Gompels LL, Feldmann M, Allen SJ, Williams RO, et al. Treatment of murine osteoarthritis with TrkAd5 reveals a pivotal role for nerve growth factor in non-inflammatory joint pain. Pain. 2010;149(2):386–92. https://doi.org/10.1016/j.pain.2010.03.002.
Xu L, Nwosu LN, Burston JJ, Millns PJ, Sagar DR, Mapp PI, et al. The anti-NGF antibody muMab 911 both prevents and reverses pain behaviour and subchondral osteoclast numbers in a rat model of osteoarthritis pain. Osteoarthr Cartil. 2016;24(9):1587–95. https://doi.org/10.1016/j.joca.2016.05.015.
LaBranche TP, Bendele AM, Omura BC, Gropp KE, Hurst SI, Bagi CM et al. Nerve growth factor inhibition with tanezumab influences weight-bearing and subsequent cartilage damage in the rat medial meniscal tear model. Annals of the rheumatic diseases. 2016;76(1):295-302. https://doi.org/10.1136/annrheumdis-2015-208913. A preclinical study in the rat MMT model saw accelerated cartilage damage with anti-NGF therapy.
von Loga IS, El-Turabi A, Jostins L, Miotla-Zarebska J, Mackay-Alderson J, Zeltins A, et al. Active immunisation targeting nerve growth factor attenuates chronic pain behaviour in murine osteoarthritis. Ann Rheum Dis. 2019;78(5):672–5. https://doi.org/10.1136/annrheumdis-2018-214489.
Nwosu LN, Mapp PI, Chapman V, Walsh DA. Blocking the tropomyosin receptor kinase A (TrkA) receptor inhibits pain behaviour in two rat models of osteoarthritis. Annals of the rheumatic diseases. 2015;75(6):1246-54. https://doi.org/10.1136/annrheumdis-2014-207203. A preclinical study demonstrated analgesic efficacy using a TrkA inhibitor in two models of OA.
Sousa-Valente J, Calvo L, Vacca V, Simeoli R, Arévalo JC, Malcangio M. Role of TrkA signalling and mast cells in the initiation of osteoarthritis pain in the monoiodoacetate model. Osteoarthr Cartil. 2018;26(1):84–94. https://doi.org/10.1016/j.joca.2017.08.006.
Holzer P. The pharmacological challenge to tame the transient receptor potential vanilloid-1 (TRPV1) nocisensor. Br J Pharmacol. 2008;155(8):1145–62. https://doi.org/10.1038/bjp.2008.351. This review provides background on the TRPV1 signaling pathway.
Schumacher MA. Transient receptor potential channels in pain and inflammation: therapeutic opportunities. Pain Pract. 2010;10(3):185–200. https://doi.org/10.1111/j.1533-2500.2010.00358.x.
Persson MSM, Stocks J, Walsh DA, Doherty M, Zhang W. The relative efficacy of topical non-steroidal anti-inflammatory drugs and capsaicin in osteoarthritis: a network meta-analysis of randomised controlled trials. Osteoarthritis and cartilage. 2018;26(12):1575-82. https://doi.org/10.1016/j.joca.2018.08.008. A systematic review describes the efficacy of topical capsaicin clinical trials.
Billard M TJ, Fleming M, Warneke T, Qiu Y, Ly N, Aronstein W, Moore W. A phase 2 double-blind clinical trial to examine the comparative E. Arthritis Rheum. 2019;71(suppl 10).
Stevens RM, Ervin J, Nezzer J, Nieves Y, Guedes K, Burges R et al. Randomized, double-blind, placebo-controlled trial of intraarticular trans-capsaicin for pain associated with osteoarthritis of the knee. Arthritis & Rheumatology. 2019;71(9):1524–33. https://doi.org/10.1002/art.40894. This is the most recent clinical trial demonstrating efficacy of CNTX-4975 compared with placebo.
Schnitzer TJ, Pelletier JP, Haselwood DM, Ellison WT, Ervin JE, Gordon RD, et al. Civamide cream 0.075% in patients with osteoarthritis of the knee: a 12-week randomized controlled clinical trial with a longterm extension. J Rheumatol. 2012;39(3):610–20. https://doi.org/10.3899/jrheum.110192.
Appendino G, Szallasi A. Euphorbium: modern research on its active principle, resiniferatoxin, revives an ancient medicine. Life Sci. 1997;60(10):681–96. https://doi.org/10.1016/s0024-3205(96)00567-x.
Sapio MR, Neubert JK, LaPaglia DM, Maric D, Keller JM, Raithel SJ, et al. Pain control through selective chemo-axotomy of centrally projecting TRPV1+ sensory neurons. J Clin Invest. 2018;128(4):1657–70. https://doi.org/10.1172/JCI94331.
Arsenault P, Chiche D, Brown W, Miller J, Treister R, Leff R, et al. NEO6860, modality-selective TRPV1 antagonist: a randomized, controlled, proof-of-concept trial in patients with osteoarthritis knee pain. Pain Rep. 2018;3(6):e696. https://doi.org/10.1097/PR9.0000000000000696.
Mayorga AJ, Flores CM, Trudeau JJ, Moyer JA, Shalayda K, Dale M, et al. A randomized study to evaluate the analgesic efficacy of a single dose of the TRPV1 antagonist mavatrep in patients with osteoarthritis. Scand J Pain. 2017;17:134–43. https://doi.org/10.1016/j.sjpain.2017.07.021.
Manitpisitkul P, Flores CM, Moyer JA, Romano G, Shalayda K, Tatikola K, et al. A multiple-dose double-blind randomized study to evaluate the safety, pharmacokinetics, pharmacodynamics and analgesic efficacy of the TRPV1 antagonist JNJ-39439335 (mavatrep). Scand J Pain. 2018;18(2):151–64. https://doi.org/10.1515/sjpain-2017-0184.
Kalff K-M, El Mouedden M, van Egmond J, Veening J, Joosten L, Scheffer GJ, et al. Pre-treatment with capsaicin in a rat osteoarthritis model reduces the symptoms of pain and bone damage induced by monosodium iodoacetate. Eur J Pharmacol. 2010;641(2–3):108–13. https://doi.org/10.1016/j.ejphar.2010.05.022.
Kim Y, Kim E-h, Lee KS, Lee K, Park SH, Na SH et al. The effects of intra-articular resiniferatoxin on monosodium iodoacetate-induced osteoarthritic pain in rats. The Korean Journal of Physiology & Pharmacology. 2016;20(1):129. https://doi.org/10.4196/kjpp.2016.20.1.129.
Iadarola MJ, Sapio MR, Raithel SJ, Mannes AJ, Brown DC. Long-term pain relief in canine osteoarthritis by a single intra-articular injection of resiniferatoxin, a potent TRPV1 agonist. Pain. 2018;159(10):2105–14. https://doi.org/10.1097/j.pain.0000000000001314.
Abaei M, Sagar DR, Stockley EG, Spicer CH, Prior M, Chapman V, et al. Neural correlates of hyperalgesia in the monosodium iodoacetate model of osteoarthritis pain. Mol Pain. 2016;12:174480691664244. https://doi.org/10.1177/1744806916642445.
Haywood AR, Hathway GJ, Chapman V. Differential contributions of peripheral and central mechanisms to pain in a rodent model of osteoarthritis. Sci Rep. 2018;8(1):7122. https://doi.org/10.1038/s41598-018-25581-8.
Honore P, Wismer CT, Mikusa J, Zhu CZ, Zhong C, Gauvin DM, et al. A-425619 [1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea], a novel transient receptor potential type V1 receptor antagonist, relieves pathophysiological pain associated with inflammation and tissue injury in rats. J Pharmacol Exp Ther. 2005;314(1):410–21. https://doi.org/10.1124/jpet.105.083915.
Honore P, Chandran P, Hernandez G, Gauvin DM, Mikusa JP, Zhong C, et al. Repeated dosing of ABT-102, a potent and selective TRPV1 antagonist, enhances TRPV1-mediated analgesic activity in rodents, but attenuates antagonist-induced hyperthermia. Pain. 2009;142(1–2):27–35. https://doi.org/10.1016/j.pain.2008.11.004.
Cui M, Honore P, Zhong C, Gauvin D, Mikusa J, Hernandez G, et al. TRPV1 receptors in the CNS play a key role in broad-spectrum analgesia of TRPV1 antagonists. J Neurosci. 2006;26(37):9385–93. https://doi.org/10.1523/JNEUROSCI.1246-06.2006.
Puttfarcken PS, Han P, Joshi SK, Neelands TR, Gauvin DM, Baker SJ, et al. A-995662 [(R)-8-(4-methyl-5-(4-(trifluoromethyl)phenyl)oxazol-2-ylamino)-1,2,3,4-tetrahydr onaphthalen-2-ol], a novel, selective TRPV1 receptor antagonist, reduces spinal release of glutamate and CGRP in a rat knee joint pain model. Pain. 2010;150(2):319–26. https://doi.org/10.1016/j.pain.2010.05.015.
Chu KL, Chandran P, Joshi SK, Jarvis MF, Kym PR, McGaraughty S. TRPV1-related modulation of spinal neuronal activity and behavior in a rat model of osteoarthritic pain. Brain Res. 2011;1369:158–66. https://doi.org/10.1016/j.brainres.2010.10.101.
Voight EA, Gomtsyan AR, Daanen JF, Perner RJ, Schmidt RG, Bayburt EK, et al. Discovery of (R)-1-(7-chloro-2,2-bis (fluoromethyl)chroman-4-yl)-3-(3-methylisoquinolin-5-yl) ur ea (A-1165442): a temperature-neutral transient receptor potential vanilloid-1 (TRPV1) antagonist with analgesic efficacy. J Med Chem. 2014;57(17):7412–24. https://doi.org/10.1021/jm500916t.
Okun A, Liu P, Davis P, Ren J, Remeniuk B, Brion T, et al. Afferent drive elicits ongoing pain in a model of advanced osteoarthritis. Pain. 2012;153(4):924–33. https://doi.org/10.1016/j.pain.2012.01.022.
Kelly S, Chapman RJ, Woodhams S, Sagar DR, Turner J, Burston JJ, et al. Increased function of pronociceptive TRPV1 at the level of the joint in a rat model of osteoarthritis pain. Ann Rheum Dis. 2015;74(1):252–9. https://doi.org/10.1136/annrheumdis-2013-203413.
Schuelert N, McDougall JJ. Cannabinoid-mediated antinociception is enhanced in rat osteoarthritic knees. Arthritis Rheum. 2008;58(1):145–53. https://doi.org/10.1002/art.23156.
Schuelert N, Zhang C, Mogg AJ, Broad LM, Hepburn DL, Nisenbaum ES, et al. Paradoxical effects of the cannabinoid CB2 receptor agonist GW405833 on rat osteoarthritic knee joint pain. Osteoarthr Cartil. 2010;18(11):1536–43. https://doi.org/10.1016/j.joca.2010.09.005.
Fowler CJ, Naidu PS, Lichtman A, Onnis V. The case for the development of novel analgesic agents targeting both fatty acid amide hydrolase and either cyclooxygenase or TRPV1. Br J Pharmacol. 2009;156(3):412–9. https://doi.org/10.1111/j.1476-5381.2008.00029.x.
Malek N, Mrugala M, Makuch W, Kolosowska N, Przewlocka B, Binkowski M, et al. A multi-target approach for pain treatment: dual inhibition of fatty acid amide hydrolase and TRPV1 in a rat model of osteoarthritis. Pain. 2015;156(5):890–903. https://doi.org/10.1097/j.pain.0000000000000132.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Osteoarthritis
Rights and permissions
About this article

Cite this article
Obeidat, A.M., Donner, A. & Miller, R.E. An Update on Targets for Treating Osteoarthritis Pain: NGF and TRPV1. Curr Treat Options in Rheum 6, 129–145 (2020). https://doi.org/10.1007/s40674-020-00146-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40674-020-00146-x