
Abstract
The term autoallergy denotes autoimmunity accompanying an atopic disease, with antigen-specific IgE as a hallmark. This phenomenon is discussed to contribute to a chronification of the disease and to shape the immune response in chronic atopic dermatitis (AD). In this review, we highlight recent insights into the autoallergic inflammation in AD. Different mechanisms underlying the allergenicity of autoallergens are discussed at the moment: intrinsic functions modulating the immune system as well as molecular mimicry may influence the allergenic potential of these proteins. Finally, the role of specific T cells is discussed.
Cite this as: Hradetzky S, Werfel T, Roesner LM. Autoallergy in atopic dermatitis. Allergo J Int 2015; 24:16–22 DOI: 10.1007/s40629-015-0037-5
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Autoallergy
The formation of a reaction against self has been investigated ever since. The common understanding of the immune system as a guarding system against microbial attacks has been postulated already in the 19th century, but since commensals populating the human body are tolerated, the theory was rethought several times. Nobel laureate Ilya Metchnikoff disagreed already 1892 with the view of a simple fight against non-self and together with theories developed much later, like Polly Matzinger’s danger hypothesis [1], this led to the idea of fighting the (potentially) dangerous instead of fighting the foreign. More and more the question how the immune system can distinguish between friend and foe was set into the focus, and how the system fails in autoimmunity and allergy. In allergic individuals the immune system raises IgE antibodies against per se harmless exogenous proteins like pollen or house dust mite particles. Capture of the allergen by IgE which is bound to Fc-receptors on the cell surface of mast cells or basophils leads to IgE crosslinking and the release of pro-inflammatory mediators, while binding to IgE on dendritic cells leads to facilitated antigen presentation to T cells.
The term autoallergy describes autoimmunity accompanying an atopic disease, with antigen-specific IgE raised against self proteins as a hallmark. This phenomenon has been found in 23 to 91 % with atopic dermatitis (AD) [2], thereof especially severely suffering patients [3, 4, 5] as well as children younger than one year [6, 7]. While the idea of reaction against self in allergic individuals aroused already in the 1920s [8], today several autoallergens have been identified and these can be allocated to three groups (Tab. 1).
Autoantigens, for which cross-reactivity to exogenous allergens is known, e. g., profilin (cross-reactive to birch allergen Bet v 1), have been identified during the 1990s: it was shown that the major epitope of dog albumin shares a homology of more than 80 % between human and other mammals and can be recognized by cross-reactive IgE [9]. This group of autoallergens and the principle of molecular mimicry will be addressed later in this article.
Autoantigens, for which such a cross-reactivity has not been described have been primarily isolated by screening cDNA libraries with sera from atopic patients [10, 11]. The existence of IgE led to the nomenclature according to the International Union of Immunological Societies guidelines Hom s 1 to Hom s 5 (Homo sapiens allergen 1–5). This article highlights molecular aspects of these proteins that may lead to enhanced allergenicity.
Finally, IgE antibodies can be generated as well to autoantigens known from classical autoimmune diseases, such as bullous pemphigoid and Graves‘ disease.
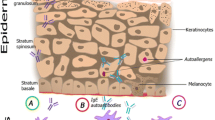
Similar to sensitization to exogenous allergens, autoallergy could be involved in disease exacerbation of AD – with the distinction that allergen avoidance is not possible in the case of autoallergy. By this mechanism also the production of cross-reactive pollen-specific IgE can be boostered [12]. Furthermore it is discussed if autoallergy may lead to chronification of the disease. Since autoallergens are believed to be always accessible, irrespective of e. g. pollen season or dust mite colonization, this may be the missing link to chronicity (Fig. 1).

Autoallergy in atopic dermatitis. Autoallergens like α-NAC or Trx may be released from skin cells by environmental factors, wounding and/or scratching. The mechanism of molecular mimicry may underlie the sensitization against Trx, which shows high sequence homology to the allergen Mala s 13 of the skin-colonizing yeast Malassezia sympodialis. Regarding α-NAC, no such homology has been described up to now.
Along with specific IgE, specific T cells appear to play a pivotal role in autoallergy. T cells shape the clinical picture of AD being the dominant cell type within the skin infiltrate [13] while autoallergy cannot be found in other atopic diseases like asthma or rhinitis [2]. In a healthy individual, allergen-specific T cells are supposed to be tolerogenic while autoantigen-specific T cells are depleted in the thymus by negative selection. However, it is believed that autoreactive T cells that carry T cell receptors with low binding avidity to the autoantigen can bypass negative selection, but being tolerogen [14, 15, 15]. It was shown in a murine system that these cells persist without losing their self-destructive potential and are able to differentiate to memory and effector T cells upon break of tolerance [17]. The characteristics and effector functions of autoallergen-specific T cells, however, are less well understood than those of exoallergen-specific T cells [18]. Yet, there are already some studies investigating T cell responses to autoallergens which point to intrinsic differences between allergens and autoallergens with respect to immunoregulation [19, 20]. Interestingly, whereas exogenous allergens preferentially induce Th2 responses, autoallergens seem to favor Th1 responses [11].
Characteristics of exogenous allergens and autoallergens – what turns a protein to be allergenic?
Why the immune system mixes up self and non-self and why distinct Th-profiles are raised against (auto-)allergens is not completely understood. In the following, several mechanisms that are discussed to play a role are described. Allergic reactions to exogenous allergens and autoallergens both represent IgE-mediated hypersensitivity reactions to antigens that are otherwise harmless [11]. In contrast to the mechanisms enabling Th1 and Th17 immune responses, on which in-depth knowledge has been gained, the question on how Th2 responses are initiated seems much harder to solve. Yet, exogenous allergens obviously do have properties which promote such a response [21]. Among them is a protease activity that many allergens share with parasites [22]. Proteases can facilitate the trespassing of epithelial barriers by allergens, they can impair innate defense mechanisms (e. g., by cleaving surfactant proteins in the lung) and they can act directly immunomodulatory on dendritic cells and T cells [23]. By this, the house dust mite allergen Der p 1 is able to decrease IL-12 secretion by dendritic cells and to reduce the production of Th1 cytokines [24]. Protease effects are also mediated via PAR (protease-activated receptors), which are expressed by skin and lung epithelial cells and which stimulate cytokine release by these cells [23].
Recently, a group of tissue cytokines was identified that is involved in the initiation of Th2 responses, among them TSLP (thymic stromal lymphopoietin), IL-33 and IL-25 [25]. These cytokines have the ability to induce innate Th2-polarized cells (ILCs, innate lymphoid cell 2) [26]. ILC2 are early Th2-like effector cells that produce cytokines such as IL-4, IL-5 and IL-13 and thereby promote T cell polarization towards a Th2 phenotype as well as activate other effector cells, e.g. eosinophils [26].
Some allergens bear adjuvant properties. A prime example is Der p 2 which can colocalize to TLR-4, just AS the endogenous MD-2, and boosts signalling through TLR-4 [27], a signaling pathway well-known from nickel contact hypersensitivity [28]. Such ML(MD-2-related lipid-recognition) domains, which can mediate adjuvant functions, are also known for other major allergens [29]. Plant pollen are always engaged by the immune system together with a large number of small proteins and other molecules, for example lipid-mediators or adenosine, bearing immunomodulatory properties [30]. Microbial components from ambient air, dust or skin flora can also bind to TLRs and other innate immune receptors. By this, they are able to influence the immune response to allergens that are administered simultaneously with the PAMPs (pathogen-associated molecular patterns) [31]. Kücüksezer et al. have shown that such a TLR stimulation is able to break the peripheral allergen-specific T cell tolerance [32]. A similar mechanism is discussed for autoantigens, which either act as DAMPs (danger-associated molecular pattern) or sensitize immune cells towards PAMPs [33]. Interestingly, we described recently that the autoallergen α-subunit of the nascent protein associated complex (α-NAC or Hom s 2) signals via TLR2 itself [34], while many other DAMPs that are released during skin wounding and PAMPs resulting from colonized eczematous skin are shown to trigger different TLRs [35, 36]. Finally, chaperones, heat-shock proteins and stress proteins in general have been linked frequently to autoimmune disorders [37, 38, 39, 40], suggesting an intrinsic mechanism.
Mixing up the proteins – molecular mimicry as a mechanism developing autoallergy
Molecular mimicry is likely to occur in proteins that share at least 50 % sequence homology that displays in similarities on primary and tertiary protein structure [41]. The resulting cross-reactivity has been described for exogenous allergens but can also be found between exogenous allergens and autoantigens [42]. The first description of such a cross-reactivity causing autoallergy was published by Valenta et al. in 1991 [12]. The respective profilins were shortly after identified as a family of pan-allergens that are present in birch and many other pollen and are cross-reactive to human profilin [43].
In addition to pollen allergens, there are also microbial allergens showing cross-reactivity to human autoantigens. The first published allergen was the manganese superoxide dismutase (MnSOD) from Aspergillus fumigatus and its human counterpart, both of which elicit in vitro and in vivo immune responses in individuals sensitized to Aspergillus fumigatus [44]. Later, sensitization to human MnSOD could be demonstrated to correlate with disease activity in AD patients [4]. In this study, cross-reactivity of IgE to fungal and human MnSOD as well as Malassezia sympodialis extract was shown and primary sensitization to M. sympodialis MnSOD was postulated. The impact of this finding was underlined in a subsequent study measuring sensitization to M. sympodialis in as much as 50 % of the AD patients [45]. Ten allergens of M. sympodialis have been described so far [46], one of which is MnSOD (Mala s 11). Another M. sympodialis allergen is Mala s 13, a thioredoxin, for which cross-reactivity to human thioredoxin has been demonstrated at IgE level [47]. We could further demonstrate that T cell clones reactive to Mala s 13 were cross-reactive to human thioredoxin in terms of cell proliferation and cytokine secretion [48]. The role of Malassezia skin colonization for AD pathogenesis has been discussed for a long time and has been corroborated by Clemmensen and Hjorth in 1983 who showed the success of antifungal treatment in patients with head and neck dermatitis and positive skin prick testing against Malassezia [49]. Malassezia species bring with them plenty of immunomodulatory molecules such as indole derivatives and enzymes [46]. Besides, the release of allergens from the yeasts is promoted by elevated skin pH as it is commonly found in AD [50]. Taken together, these findings suggest a primary sensitization to allergens from skin-colonizing Malassezia species with concomitant sensitizations to cross-reactive autoallergens.
However, not every auto-sensitization may be based on molecular mimicry. α-NAC (Hom s 2) is a housekeeping gene and a chaperone which shows no homology to known classical allergens. However, it seems obvious that the amino acid sequence is evolutionary highly conserved among mammals and in part also among dermatophytes and skin-colonizing microorganisms due to its basic function in protein production at the ribosomes. Recently, we identified regions within this autoallergen which are most likely recognized by cytotoxic T cells in sensitized AD patients. Of four putative epitopes, one was found to exhibit high homology with α-NAC from microorganisms, while the remaining three are less or not conserved. So far, it cannot be stated what came first: autoallergy or an allergy against microbes. Clearly, the conserved epitope may represent a driver clone (compare [51]), from which epitope spreading takes place. However, it is also possible that primary sensitization to α-NAC occurs as described above and the homology leads by chance to crossreactivity against microbes.
The occurrence of autoantibodies in small children is also not completely understood. A transient epiphenomenon without particular impact on the atopic disease may be the reason in this case [J. Gutermuth et al., presentation at the 30th Collegum Internationale Allergologicum (CIA) symposium in Petersberg, Germany, 2014]. A causal relationship was assumed due to a significant correlation with sensitization against food allergens [7, 52].
The (cellular cytokine) response to autoallergens
When comparing the exogenous allergen Phl p 1 to the autoallergen α-NAC (Hom s 2) with regard to IFN-γ induction in mononuclear cells of the peripheral blood (PBMCs), cells stimulated with the autoallergen show a distinctly higher IFN-γ release [20]. A comparison of Phl p 1 to the autoallergen Hom s 4 delivers similar results [19]. Looking at human thioredoxin (hTrx) and the crossreactive allergen Mala s 13, we generated T cell lines in the presence of hTrx. After stimulation, these T cell lines released significantly less IL-4 and by trend more IFN-γ than T cell lines generated in the presence of Mala s 13 [Hradetzky et al., unpublished data]. 45 % of blood-derived T cell clones, generated in the presence of Mala s 13 and restimulated with the autoallergen hTrx, belonged to the Th1 subtype [48]. The autoallergen α-NAC also induced a Th1-dominated response in immune cells, which was dependent on IL-12 and mediated through TLR-2 on monocytes [34]. In addition to Th1 cytokines, IL-17 and IL-22 were released by α-NAC-stimulated PBMCs [53]. The induction of T cell cytokines was only efficient when monocytes were present during stimulation, pointing again to the importance of the innate response to autoallergens for the overall immune reaction (beside antigen presentation). We also observed release of the anti-inflammatory cytokine IL-10 by monocytes – however, IL-10 levels were markedly reduced in supernatants from immune cells of AD patients sensitized to α-NAC compared to healthy donors [53]. A similar effect was measured after hTrx stimulation of immune cells from AD patients sensitized to hTrx. In addition, secretion of the Th2 cytokine IL-13 was elevated in PBMCs of AD patients sensitized to the hTrx [Hradetzky et al., in revision].
Cytotoxic T lymphocytes in atopic dermatitis – the fast and the furious?
As mentioned above, the T helper cell-dominated skin infiltrate is a hallmark of AD. Although CD8+ T cells have been de-scribed in dermis and epidermis of AD lesional skin as a source of IFN-γ and sometimes also IL-13 and IL-22 [54], the focus of research concentrated on the CD4+ cells, which are much more abundant in acute lesions. Recent studies investigating the development of skin lesions from the initiation to the flare-up describe a different picture: an early and transient presence of CD8+ T lymphocytes can be seen before clinical lesions occur [55]. Even more compelling, the absence of CD8+ cells suppressed the development of skin inflammation in a mouse model [56]. Therefore, the role of cytotoxic T cells may have been underestimated in the past.
Generating autoallergen-specific T cell clones, we observed in our experiments high frequencies of CD8+ T cells reactive to α-NAC. While from lesional skin only 10 % of T cell clones were CD8+ as expected, we surprisingly found 61 % clones generated from the circulation of sensitized donors to be cytotoxic T cells [57]. Investigating these circulating, autoreactive, cytotoxic T cells in more detail using MHC-tetramers, we could show that these cells are significantly more abundant in AD patients. Furthermore, these cells display more often an effector-memory or even a terminally differentiated effector-memory (“TEMRA”) phenotype, and do produce IFN-γ [Roesner et al., in revision]. So, α-NAC-specific cells may sustain the inflammation also in the absence of environmental allergens. Being present in the skin and with the ability to produce IFN-γ they may contribute to a weakened skin barrier and a shift in the inflammatory milieu towards the Th1 type, which is often observed in the chronic phase of AD. Adding to this, there is direct evidence for the influence of autoallergy on eczema severity from studies showing that skin application of the autoallergen induces eczema-like reactions in sensitized patients [4, 10, 58].
Furthermore, the link between autoallergy and the severity of AD underlines the importance of the phenomenon, which was found in two of 14 studies as a significant correlation and in further three studies as a tendency [2]. Although it cannot be stated by now whether autoallergy is a bystander effect of severe skin inflammation and cell death or whether autoimmune processes themselves contribute substantially to AD exacerbation, the preponderance of autoallergy in patients with a more severe and chronic disease [7] suggests a causal connection that deserves further investigation.
Abbreviations
- AD:
-
Atopic dermatitis
- cDNA:
-
Complementary deoxyribonucleic acid
- CIA:
-
Collegium Internationale Allergologicum
- DAMP:
-
Danger-associated molecular pattern
- Fc:
-
Fragment crystallizable
- hTrx:
-
Human thioredoxin
- IFN:
-
Interferon
- IgE:
-
Immunglobulin E
- IL:
-
Interleukin
- ILC:
-
Innate lymphoid cell
- IUIS:
-
International Union of Immunological Societies
- ML:
-
MD-2-related lipid recognition
- MnSOD:
-
Manganese superoxide dismutase
- NAC:
-
Nascent protein associated complex
- PAMP:
-
Pathogen-associated molecular patterns
- PAR:
-
Protease-activated receptors
- PBMC:
-
Peripheral blood mononuclear cells
- TEMRA:
-
CD45RA re-expressing effector/memory T cell
- Th:
-
T helper cell
- TLR:
-
Toll-like receptor
- Trx:
-
Thioredoxin
- TSLP:
-
Thymic stromal lymphopoietin
References
Matzinger P. The danger model: a renewed sense of self. Science 2002;296:301–5
Tang TS, Bieber T, Williams HC. Does „autoreactivity“ play a role in atopic dermatitis? J Allergy Clin Immunol 2012;129:1209–15.e2
Higashi N, Niimi Y, Aoki M, Kawana S. Clinical features of antinuclear antibody-positive patients with atopic dermatitis. J Nippon Med Sch 2009;76:300–7
Schmid-Grendelmeier P, Fluckiger S, Disch R, Trautmann A, Wuthrich B, Blaser K et al. IgE-mediated and T cell-mediated autoimmunity against manganese superoxide dismutase in atopic dermatitis. J Allergy Clin Immunol 2005;115:1068–75
Szakos E, Lakos G, Aleksza M, Gyimesi E, Pall G, Fodor B et al. Association between the occurrence of the anticardiolipin IgM and mite allergen-specific IgE antibodies in children with extrinsic type of atopic eczema/dermatitis syndrome. Allergy 2004;59:164–7
Seher T, Meyer JU, Traidl-Hoffmann C, Brockow K, Ollert M, Gutermuth J et al. A high-troughput screening assay for detection of auto-IgE (P009). Exp Dermatol 2012;21:e1–e58
Mothes N, Niggemann B, 5Jenneck C, Hagemann T, Weidinger S, Bieber T et al. The cradle of IgE autoreactivity in atopic eczema lies in early infancy. J Allergy Clin Immunol 2005;116:706–9
Storm van Leuwen W, Bien Z, Varekamp H. Über die Hautreaktion mit Extrakten menschlicher Hautschuppen bei allergischen Krankheiten. Klin Wochenschr 1926;5:1023–5
Spitzauer S, Schweiger C, Sperr WR, Pandjaitan B, Valent P, Muhl S et al. Molecular characterization of dog albumin as a cross-reactive allergen. J Allergy Clin Immunol 1994;93:614–27
Natter S, Seiberler S, Hufnagl P, Binder BR, Hirschl AM, Ring J et al. Isolation of cDNA clones coding for IgE autoantigens with serum IgE from atopic dermatitis patients. Faseb J 1998;12:1559–69
Valenta R, Mittermann I, Werfel T, Garn H, Renz H. Linking allergy to autoimmune disease. Trends Immunol 2009;30:109–16
Valenta R, Duchene M, Pettenburger K, Sillaber C, Valent P, Bettelheim P et al. Identification of profilin as a novel pollen allergen; IgE autoreactivity in sensitized individuals. Science 1991;253:557–60
Werfel T. The role of leukocytes, keratinocytes, and allergen- specific IgE in the development of atopic dermatitis. J Invest Dermatol 2009;129:1878–91
Nugent CT, Morgan DJ, Biggs JA, Ko A, Pilip IM, Pamer EG et al. Characterization of CD8+ T lymphocytes that persist after peripheral tolerance to a self antigen expressed in the pancreas. J Immunol 2000;164:191–200
Herrath MG von, Dockter J, Nerenberg M, Gairin JE, Oldstone MB. Thymic selection and adaptability of cytotoxic T lymphocyte responses in transgenic mice expressing a viral protein in the thymus. J Exp Med 1994;180:1901–10
Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity 2006;25:261–70
Enouz S, Carrie L, Merkler D, Bevan MJ, Zehn D. Autoreactive T cells bypass negative selection and respond to selfantigen stimulation during infection. J Exp Med 2012;209:1769–79
Wambre E, DeLong JH, James EA, LaFond RE, Robinson D, Kwok WW. Differentiation stage determines pathologic and protective allergen-specific CD4+ T-cell outcomes during specific immunotherapy. J Allergy Clin Immunol 2012;129:544–51,51.e1-7
Aichberger KJ, Mittermann I, Reininger R, Seiberler S, Swoboda I, Spitzauer S et al. Hom s 4, an IgE-reactive autoantigen belonging to a new subfamily of calcium-binding proteins, can induce Th cell type 1-mediated autoreactivity. J Immunol 2005;175:1286–94
Mittermann I, Reininger R, Zimmermann M, Gangl K, Reisinger J, Aichberger KJ et al. The IgE-reactive autoantigen Hom s 2 induces damage of respiratory epithelial cells and keratinocytes via induction of IFN-gamma. J Invest Dermatol 2008;128:1451–9
Sokol CL, Barton GM, Farr AG, Medzhitov R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol 2008;9:310–8
McKerrow JH, Caffrey C, Kelly B, Loke P, Sajid M. Proteases in parasitic diseases. Annu Rev Pathol 2006;1:497–536
Wills-Karp M, Nathan A, Page K, Karp CL. New insights into innate immune mechanisms underlying allergenicity. Mucosal Immunol 2010;3:104–10
Bessot JC, Pauli G. Mite allergens: an overview. Eur Ann Allergy Clin Immunol 2011;43:141–56
Brandt EB, Sivaprasad U. Th2 Cytokines and Atopic Dermatitis. J Clin Cell Immunol 2011;2:110
Lund S, Walford HH, Doherty TA. Type 2 innate lymphoid cells in allergic disease. Curr Immunol Rev 2013;9:214–21
Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature 2009;457:585–8
Raghavan B, Martin SF, Esser PR, Goebeler M, Schmidt M. Metal allergens nickel and cobalt facilitate TLR4 homodimerization independently of MD2. EMBO Rep 2012;13:1109–15
Inohara N, Nunez G. ML - a conserved domain involved in innate immunity and lipid metabolism. Trends Biochem Sci 2002;27:219–21
Gilles S, Behrendt H, Ring J, Traidl-Hoffmann C. The pollen enigma: modulation of the allergic immune response by non-allergenic, pollen-derived compounds. Curr Pharm Des 2012;18:2314–9
Traidl-Hoffmann C, Jakob T, Behrendt H. Determinants of allergenicity. J Allergy Clin Immunol 2009;123:558–66
Kucuksezer UC, Palomares O, Ruckert B, Jartti T, Puhakka T, Nandy A et al. Triggering of specific Toll-like receptors and proinflammatory cytokines breaks allergen-specific T-cell tolerance in human tonsils and peripheral blood. J Allergy Clin Immunol 2013;131: 875–85
Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol 2010;87:989–99
Hradetzky S, Balaji H, Roesner LM, Heratizadeh A, Mittermann I, Valenta R et al. The human skin-associated autoantigen alpha-NAC activates monocytes and dendritic cells via TLR-2 and primes an IL-12-dependent Th1 response. J Invest Dermatol 2013;133:2289–92
Miller LS, Modlin RL. Toll-like receptors in the skin. Semin Immunopathol 2007;29:15–26
Mills KH. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol 2011;11:807–22
Kaufmann SH. Heat shock proteins and the immune response. Immunol Today 1990;11:129–36
Lamb JR, Bal V, Mendez-Samperio P, Mehlert A, So A, Rothbard J et al. Stress proteins may provide a link between the immune response to infection and autoimmunity. Int Immunol 1989;1:191–6
Pockley AG. Heat shock proteins as regulators of the immune response. Lancet 2003;362:469–76
Young RA. Stress proteins and immunology. Annu Rev Immunol 1990;8:401–20
Aalberse RC. Structural biology of allergens. J Allergy Clin Immunol 2000;106:228–38
Hauser M, Roulias A, Ferreira F, Egger M. Panallergens and their impact on the allergic patient. Allergy Asthma Clin Immunol 2010;6:1
Valenta R, Duchene M, Ebner C, Valent P, Sillaber C, Deviller P et al. Profilins constitute a novel family of functional plant pan-allergens. J Exp Med 1992;175:377–85
Crameri R, Faith A, Hemmann S, Jaussi R, Ismail C, Menz G et al. Humoral and cell-mediated autoimmunity in allergy to Aspergillus fumigatus. J Exp Med 1996;184:265–70
Casagrande BF, Fluckiger S, Linder MT, Johansson C, Scheynius A, Crameri R et al. Sensitization to the yeast Malassezia sympodialis is specific for extrinsic and intrinsic atopic eczema. J Invest Dermatol 2006;126: 2414–21
Gaitanis G, Velegraki A, Mayser P, Bassukas ID. Skin diseases associated with Malassezia yeasts: facts and controversies. Clin Dermatol 2013;31:455–63
Limacher A, Glaser AG, Meier C, Schmid-Grendelmeier P, Zeller S, Scapozza L et al. Cross-reactivity and 1.4-A crystal structure of Malassezia sympodialis thioredoxin (Mala s 13), a member of a new pan-allergen family. J Immunol 2007;178:389–96
Balaji H, Heratizadeh A, Wichmann K, Niebuhr M, Crameri R, Scheynius A et al. Malassezia sympodialis thioredoxinspecific T cells are highly cross-reactive to human thioredoxin in atopic dermatitis. J Allergy Clin Immunol 2011;128:92–9.e4
Scheynius A, Johansson C, Buentke E, Zargari A, Linder MT. Atopic eczema/dermatitis syndrome and Malassezia. Int Arch Allergy Immunol 2002;127:161–9
Selander C, Zargari A, Mollby R, Rasool O, Scheynius A. Higher pH level, corresponding to that on the skin of patients with atopic eczema, stimulates the release of Malassezia sympodialis allergens. Allergy 2006;61:1002–8
Kumar V, Sercarz E. Induction or protection from experimental autoimmune encephalomyelitis depends on the cytokine secretion profile of TCR peptide-specific regulatory CD4 T cells. J Immunol 1998;161:6585–91
Ong PY, Leung DY. Immune dysregulation in atopic dermatitis. Curr Allergy Asthma Rep 2006;6:384–9
Hradetzky S, Roesner LM, Balaji H, Heratizadeh A, Mittermann I, Valenta R et al. Cytokine effects induced by the human autoallergen alpha-NAC. J Invest Dermatol 2014; 134:1570–8
Hijnen D, Knol EF, Gent YY, Giovannone B, Beijn SJ, Kupper TS et al. CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-gamma, IL-13, IL-17, and IL-22. J Invest Dermatol 2013;133:973–9
Hennino A, Jean-Decoster C, Giordano-Labadie F, Debeer S, Vanbervliet B, Rozieres A et al. CD8+ T cells are recruited early to allergen exposure sites in atopy patch test reactions in human atopic dermatitis. J Allergy Clin Immunol 2011;127:1064–7
Hennino A, Vocanson M, Toussaint Y, Rodet K, Benetiere J, Schmitt AM et al. Skin-infiltrating CD8+ T cells initiate atopic dermatitis lesions. J Immunol 2007;178:5571–7
Heratizadeh A, Mittermann I, Balaji H, Wichmann K, Niebuhr M, Valenta R et al. The role of T-cell reactivity towards the autoantigen alpha-NAC in atopic dermatitis. Br J Dermatol 2011;164:316–24
Mayer C, Appenzeller U, Seelbach H, Achatz G, Oberkofler H, Breitenbach M et al. Humoral and cell-mediated autoimmune reactions to human acidic ribosomal P2 protein in individuals sensitized to Aspergillus fumigatus P2 protein. J Exp Med 1999;189:1507–12
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
The authors state that there are no conflict of interest.
Rights and permissions
About this article

Cite this article
Hradetzky, S., Werfel, T. & Rösner, L.M. Autoallergy in atopic dermatitis. Allergo J Int 24, 16–22 (2015). https://doi.org/10.1007/s40629-015-0037-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40629-015-0037-5