
Abstract
Objectives
The aim of the study was to investigate the clinicopathological characteristics and prognosis of proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) and determine the differences between PGNMID associated with extrarenal disease and without clear etiology as well as the differences between IgG1 and IgG3 subtypes.
Methods
Data from 46 patients with PGNMID observed from January 2014 to September 2021 in Peking University First Hospital were retrospectively analyzed, including 36 patients without clear etiology (Group A) and 10 patients with extrarenal disease (Group B).
Results
At presentation patients showed proteinuria (95.7%), hematuria (89.1%), renal insufficiency (73.9%), and hypocomplementemia of C3 or C4 (35.6%). Monoclonal immunoglobulin or cell clones were detected in 22.2% of patients (10/45). The monoclonal immunoglobulins deposited in kidney were IgG3 in 40 patients, IgG1 in 5, and IgM in one. Monoclonal IgG1 deposits were more common in Group B than in Group A (4/10 vs. 1/36, p = 0.006). The intensity of glomerular C3 deposition and the frequency of subendothelial deposits in IgG3 subtype were significantly higher than those in IgG1 subtype. During a median follow-up time of 12.2 (range 1–61) months, a higher level of serum creatinine at biopsy and a higher percentage of global glomerulosclerosis were independent predictors of end-stage kidney disease.
Conclusions
PGNMID associated with extrarenal disease was more likely to have monoclonal IgG1 deposits. PGNMID of IgG3 subtype differs from IgG1 subtype by higher intensity of glomerular C3 deposition and higher frequency of subendothelial deposits. Serum creatinine and global glomerulosclerosis were independent prognostic predictors of ESKD in PGNMID.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Monoclonal gammopathy of renal significance (MGRS) describes a group of hematologic disorders with monoclonal immunoglobulin-related renal injury [1]. The underlying plasma cell or B cell dyscrasia does not cause tumor complications or meet any current hematological criteria for immediate specific treatment of clonal diseases. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) is a glomerulus-limited injury and unique form of MGRS, first described in 2004 [2], with intact monoclonal immunoglobulin deposition. IgG3 is by far the most common subtype, constituting 60–68% of cases [3, 4]. IgG1 accounts for 24–29% of cases, more commonly in patients with a predominant membranous nephropathy pattern [5, 6] and in those with detectable monoclonal gammopathy [3, 4]. The most common pathologic pattern is membranoproliferative glomerulonephritis (MPGN) [3]. The immune nonorganized granular deposits observed by electron microscopy are similar to polyclonal immune complex-mediated glomerulonephritis. In contrast to patients with other types of MGRS, patients with PGNMID have a lower incidence of dysproteinemia and detectable underlying B- or plasma cell clones [3, 4, 7]. The etiology and pathogenesis of PGNMID remain uncertain. Several cases have reported that it is associated with other primary diseases, such as infection [8, 9], autoimmune diseases [10, 11], lung cancer [12] and pregnancy [13], apart from hematological malignancy [14,15,16]. Data on PGNMID in non-Caucasian populations are limited. In the current study, we retrospectively analyzed 46 PGNMID patients, including 10 patients with extrarenal disease, from a single institute in China to further understand the clinicopathologic features, prognosis and pathogenesis of PGNMID, and to determine the differences between PGNMID with extrarenal disease and without clear etiology as well as the differences between IgG1 and IgG3 subtype PGNMID.
Methods
Patients
Patients with renal biopsy-proven PGNMID from January 2014 to September 2021 at Peking University First Hospital were included. The diagnostic criteria of PGNMID were as follows: (1) presence of monoclonal immunoglobulin deposits on immunofluorescence (a single heavy chain and a single light chain); (2) proliferative or membranoproliferative glomerulonephritis on light microscopy; (3) immune complex-like granular electron dense deposits in glomeruli shown by electron microscopy; and (4) absence of clinical manifestation of cryoglobulinemia. The exclusion criterion was type I or II cryoglobulinemia in which the monoclonal component of the cryoprecipitate matched that of renal deposits. A total of 46 patients with PGNMID were enrolled in this study. According to the possible etiology, including infection, autoimmune diseases, solid tumors and hematological malignancy, patients with PGNMID were classified into Group A (etiology was unknown) and Group B (with associated underlying disease).
Informed consent was obtained from each patient. The research was in compliance with the Declaration of Helsinki and approved by the ethics committee of Peking University First Hospital.
Clinical and laboratory evaluation
Demographic, clinical, laboratory data, treatment and outcomes were obtained retrospectively from the electronic medical records. Patients were followed up in the outpatient clinic of our department. Clinical data and laboratory data were recorded at the time of kidney biopsy, after treatment, and at last follow-up. Acute kidney injury (AKI) was defined by an increase in serum creatinine (Scr) > 50% within 7 days, an increase in Scr ≥ 0.3 mg/dL (26.5 µmol/L) within 2 days, or oliguria (urine output < 0.5 ml/kg/h) > 6 h [17]. Nephrotic syndrome was defined as 24-h urinary protein ≥ 3.5 g/d and hypoalbuminemia (serum albumin < 30 g/L). Renal response was defined as described previously [3]. Complete remission (CR) was defined as remission of proteinuria to < 500 mg/d with normal renal function; partial remission (PR) was defined as reduction in proteinuria by at least 50% and to < 2 g/d with stable renal function (no more than a 20% increase of serum creatinine compared with the baseline); persistent renal dysfunction (PRD) was defined as failure to meet criteria for either CR or PR but not reaching end-stage kidney disease (ESKD), including patients with unremitting proteinuria or progressive chronic kidney disease; ESKD was defined as requiring renal replacement therapy or eGFR of < 15 mL/min/1.73 m2. Renal survival rates were calculated from the time of kidney biopsy to the last follow-up and ESKD.
Renal pathology
All renal biopsy samples were examined by routine light microscopy, immunofluorescence, and electron microscopy according to standard techniques. Paraffin tissue was stained with hematoxylin and eosin, periodic acid-Schiff, Masson trichrome, and Jones methenamine silver. Immunofluorescence was performed on 3-μm cryostat sections using a panel of FITC-conjugated rabbit anti-human antibodies to IgG, IgM, IgA, C3, C1q, fibrinogen, albumin, κ, λ, and IgG1-4 (Dako Corporation, Carpenteria, CA, USA). Immunofluorescence staining intensity was graded 0 to 4 + on a semiquantitative scale. Two renal pathologists made histopathologic diagnoses separately. Differences in diagnosis between them were resolved by rereviewing the biopsy slides to reach consensus.
Statistical analysis
Statistical analysis was performed using SPSS 21 for Windows (IBM Corp. in Armonk, NY). Data are expressed as the mean ± standard deviation or median with range for continuous variables and proportions for categorical variables. Analysis was performed using t tests, Mann–Whitney U tests, and χ2 tests, as appropriate for variable type. Correlation analysis was performed using Spearman's rank correlation coefficient. Survival analysis was performed by univariate survival analysis, multivariable Cox regression models, and Kaplan–Meier curves. Statistical significance was assumed at p < 0.05.
Results
Demographic and clinical data
Forty-six PGNMID patients were included, including 36 patients without clear etiology (Group A) and 10 patients with extrarenal disease (Group B): 2 cases associated with lymphoma, 3 with chronic hepatitis B, one with upper respiratory infection, 2 with systemic lupus erythematosus, one with Kimura disease, and one with pregnancy. Demographic and clinical characteristics are presented in Table 1, and no significant difference was observed between Group A and Group B. The average age at diagnosis was 51.8 years, and 54.3% (25/46) of patients were male. A total of 95.7% (44/46) had proteinuria (median proteinuria was 5.3 g), and 60.9% (28/46) had nephrotic syndrome. The median Scr at diagnosis was 152.3 µmol/L (range 47.2–788.0 µmol/L). Serum C3 or C4 decreased in 16 (35.6%) patients, and C1q decreased in 38.9% (7/18). Tests of serum cryoglobulin performed in 38 patients revealed that 52.6% (20/38) of patients were positive for serum cryoglobulin (including 15 type III cryoglobulinemia, 3 type II cryoglobulinemia and 2 unspecified). No significant difference was observed between patients with or without cryoglobulinemia in demographic and clinical features (Supplementary Table 1).
Hematologic evaluation
Monoclonal immunoglobulin was detected by serum or urine protein electrophoresis/immunofixation in 5 of 45 patients (11.1%), which matched glomerular deposited monoclonal proteins. Thirty-seven patients received a serum-free light chain assay, and no patient had an elevated serum-free light chain ratio with an extended renal range (0.3–3.1) [18]. Bone marrow aspirate and biopsy were performed in 36 patients and showed no obvious plasma cell infiltration. Flow cytometry of bone marrow revealed clonal plasma cells in seven patients with a median percentage of 0.10% (range: 0.03–0.60%) and 16.50% clonal B cells with λ restriction in one patient with lymphoma who had global deposition of IgM λ. No patient met the diagnostic criteria of multiple myeloma. Patients with hypocomplementemia of C4 were more likely to be identified as clones in serum, urine or bone marrow (4/7 vs. 6/37, p = 0.037).
Renal pathology
The pathological characteristics are detailed in Table 2. On immunofluorescence, the monoclonal immunoglobulins, present only in glomeruli, were IgG3 κ in 31 (67.4%) patients, IgG3 λ in 9 (19.6%), IgG1 κ in 5 (10.9%), and IgM λ in one (2.2%). All patients had glomerular C3 deposition, and 82.6% had C1q deposition. Monoclonal IgG1 deposits were more common in Group B (4/10 vs. 1/36, p = 0.006). The circulating levels of C3 and C1q were associated with the intensity of glomerular C3 and C1q deposition (r = −0.480, p = 0.001 and r = −0.611, p = 0.009, respectively). As shown in Table 3, the intensity of glomerular C3 deposition in IgG3 PGNMID was significantly higher than that in IgG1 PGNMID (median 3+, range: + to 3 + vs. median + − + + , range ± to 3+, p = 0.021).
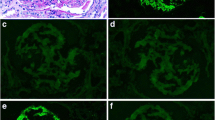
No significant difference in histological pattern was observed between Group A and Group B. Membranoproliferative glomerulonephritis (MPGN) was the most common histological pattern, observed in 36 (78.3%) patients. A total of 82.5% of patients (33/40) with IgG3 deposition had the MPGN pattern (Fig. 1). The proportion was higher than that of patients with IgG1 (40.0%, 2/5), although the difference was not significant (p = 0.065). Global glomerulosclerosis was observed in 82.4% of cases (affecting 2.6–38.3% of glomeruli). Focal cellular/fibrocellular crescents were present in 17 (38.6%) cases (affecting 3.6–32.3% of glomeruli), and in one case, crescents involved ≥ 50% of glomeruli.

Pathological findings. Immunofluorescence examination on frozen sections showed granular staining for IgG3 (a), κ (b), and C3 (c) with λ trace (d). Staining for IgG1, IgG2 and IgG4 was negative (not shown). e Light microscopy showed expansion of mesangial cell and matrix, and diffused endocapillary hypercellularity along with thickened glomerular basement membrane with double contours (periodic acid-silver methenamine, × 400). f Electron microscopy showed subendothelial and mesangial electron-dese deposits (× 8000)
On electron microscopy, glomeruli were available in 45 of 46 cases. Immune deposits were primarily identified in mesangial and subendothelial areas (seen in 93.3% and 84.4% of cases, respectively, Fig. 1). There were significantly more frequent subendothelial deposits in patients with monoclonal IgG3 than in those with IgG1 (89.7% vs. 40.0%, p = 0.023). The dense deposits showed no substructure, except two cases: fibrillar substructure was identified in subendothelial deposits in one case of MPGN pattern and in subepithelial deposits in one case with a pattern of atypical membranous nephropathy.
Treatment and prognosis
The median duration of follow-up in the 46 patients was 12.2 (range 1–61) months. Treatment and outcomes are presented in Fig. 2. In Group A, 18 patients who received prednisone and cyclophosphamide, two developed CR, one developed PR, four developed PRD, and 11 developed ESKD. Fourteen patients were treated with two to eight courses of bortezomib-based chemotherapy. Of these 14 patients, one developed CR, three developed PR, six developed PRD, and four developed ESKD. More patients who received prednisone and cyclophosphamide reached ESKD than patients who received bortezomib-based chemotherapy (61.1% vs. 28.6%, p = 0.087). One patient receiving rituximab developed PR. One patient with a high risk of infection received prednisone alone, progressing to ESKD. Two patients received renin angiotensin aldosterone system inhibitors alone. One of them developed CR, and the other patient progressed to ESKD. Two patients reached ESKD and died, between whom one patient died of intracerebral hemorrhage, and another died of undetermined cause.

a Treatment and outcomes of Group A. b Outcomes of Group A and Group B. CR: Remission of proteinuria to < 500 mg/d with normal renal function; PR: Reduction in proteinuria by at least 50% and to < 2 g/d with stable renal function (no more than a 20% increase in serum creatinine); PRD: Failure to meet criteria for either CR or PR but not reaching ESKD, including patients with unremitting proteinuria, or progressive chronic kidney disease; ESKD: End-stage kidney disease. Chemo: Chemotherapy; Pred: Prednisone; CTX: cyclophosphamide; RTX: Rituximab; RAAS-I: Renin angiotensin aldosterone system inhibitors
In Group B, four patients developed CR, including one patient with lymphoma treated with chemotherapy for lymphoma, one patient with upper respiratory infection receiving prednisone and cyclophosphamide after infection, one patient with Kimura disease treated with lymphadenectomy and one pregnant patient who had been reported [13]. The pregnant patient was treated with prednisone and achieved CR after delivery. Another patient with lymphoma received renin angiotensin aldosterone system inhibitors alone due to the poor condition progressing to ESKD. Three patients with chronic hepatitis B received antiviral therapy with or without prednisone, but replication of the virus continued. Of these three patients, two developed PRD, and one progressed to ESKD. Two patients with SLE treated with prednisone combined with immunosuppressive therapy developed PRD and ESKD.
Prognostic factors associated with renal survival are presented in Table 4. The independent predictors of progression to ESKD on multivariate analysis were a higher level of Scr at biopsy and a higher percentage of global glomerulosclerosis. In an Receiver Operation Characteristic Curve analysis based on progression to ESKD, the best cutoffs were 120.6 µmol/L for Scr (sensitivity 95.0%; specificity 50.0%) and 6.2% for the percentage of global glomerulosclerosis (sensitivity 75.0%; specificity 76.9%). For convenience, the cutoffs were designed as 120 µmol/L for Scr and 6% for the percentage of global glomerulosclerosis, with no significant decline in sensitivity and specificity. According to the Kaplan–Meier survival analysis, renal survival was remarkably poorer in the subgroup with Scr ≥ 120 µmol/L (p = 0.004) (Fig. 3a) and global glomerulosclerosis ≥ 6% (p = 0.001) (Fig. 3b).

Kaplan–Meier curves demonstrating differences in renal survival. a Renal survival according to serum creatinine (p = 0.004). b Renal survival according to the percent of global glomerulosclerosis (p = 0.001)
Discussion
In the current study, we retrospectively analyzed the clinicopathological features and outcomes of 46 patients with PGNMID. To the best of our knowledge, this is the largest case series of PGNMID in the Chinese population.
In our study, the clinical manifestations were similar to those in prior studies except for the higher frequency of cryoglobulinemia (53%) [3, 4, 7, 19]. These patients with cryoglobulinemia were lack of clinical manifestation of cryoglobulinemic vasculitis such as purpura, fatigue, arthralgia or peripheral neuropathy [20]. Histologic findings specific for cryoglobulinemic glomerulonephritis such as intraluminal thrombi or fibrillary or microtubular deposits on electron microscopy [21] were also absent in renal biopsy. Besides, the monoclonal component of serum cryoprecipitate did not match that of renal deposits. Few cases of cryoglobulinemic glomerulonephritis that renal immunoglobulin deposits did not correspond to the monoclonal serum cryoglobulin were reported up to now [22]. Thus, diagnosis of cryoglobulinemic glomerulonephritis was excluded.
We found some differences in clinicopathological features between IgG1 and IgG3 subtypes of PGNMID. First, patients with extrarenal disease were more likely to have monoclonal IgG1 deposition in glomeruli. Second, IgG3 has the greatest complement-fixing capacity and a high ability to activate the classic complement pathway, followed by IgG1 [23]. Glomerular monoclonal IgG3 was identified in 87% of our patients and was accompanied by co-deposition of C3 in all patients and C1q in 85% of patients. The intensity of glomerular C3 deposition and the frequency of hypocomplementemia of C3 in IgG3 subtype PGNMID were higher than those in IgG1 subtype PGNMID. These findings suggest that these heavy chains are sufficient to act on the classical pathway locally and systemically, which in turn activates downstream inflammatory mediators that promote glomerular leukocyte infiltration and proliferation, leading to glomerulonephritis. For patients with IgG1 subtype PGNMID, 60% of patients had C1q deposition apart from C3 deposition. No patient had hypocomplementemia of C3, and only one patient had a low level of serum C4, suggesting that only the local complement classical pathway was active. The weak capacity to fix C1q of IgG2 and inability to bind complement of IgG4 could explain the low incidence of IgG2 subtype PGNMID (no patients with IgG2 in our cohort and 3–16% in prior studies [3, 4]) and IgG4 subtype [5]. Third, the MPGN pattern was more common in patients with monoclonal IgG3, although the difference did not reach statistical significance. Last, there were significantly more frequent subendothelial deposits in patients with monoclonal IgG3 than in those with IgG1.
It was reported that IgG1 accounts for 24–29% of cases, more commonly in patients with a predominant membranous nephropathy pattern [5, 6] and in those with detectable monoclonal gammopathy [3, 4]. In our series, patients with the IgG1 subtype were also more likely to have monoclonal immunoglobulin (25%) than those with the IgG3 subtype (8%), but this difference did not reach statistical significance. In particular, patients with hypocomplementemia of C3 or C4 in our study were more likely to have detectable monoclonal immunoglobulin and clones, highlighting the relevance of B- or plasma cell clones and monoclonal immunoglobulin to the complement system.
Histories of underlying extrarenal disease, such as carcinoma, infection, autoimmune disease and hematologic diseases, in patients with PGNMID are not rare [3, 4, 24]. It seems that PGNMID is a special pathological feature and actually encompasses several entities with distinct origins. In our study, no significant difference was observed in the demographic and clinical characteristics between cases with or without extrarenal disease. However, interestingly, patients with extrarenal disease were more likely to have monoclonal IgG1 deposition in glomeruli.
In the study of Gumber et al. [7], with treatment directing at the underlying clone, 88% of patients (14/16) had a renal response (CR or PR), and no one reached ESKD. In our study, for patients with clone detection in Group A, two of three (67%) reached ESKD in the no-chemotherapy group, while only one of five patients (20%) reached ESKD in the chemotherapy group. This may suggest that for patients with PGNMID who cannot identify underlying etiology, plasma cell clone-directed therapy for detectable plasma cell clones of monoclonal IgG could improve the renal outcome. Recently, a clinical trial evaluated the efficacy of daratumumab in patients with PGNMID and showed that daratumumab was effective in treating patients with PGNMID [25]. However, we had no patients treated with daratumumab. For patients with extrarenal disease, patients were more likely to achieve CR than patients without clear etiology, although the difference did not reach statistical significance (p = 0.055). Thus, thorough investigation to determine the underlying cause is crucial, particularly in patients with monoclonal IgG1 deposits, and anti-etiology therapy may improve the renal outcome of patients with PGNMID associated with extrarenal disease.
There are three limitations of our study. First, serum-free light chain assay or bone marrow examination was to some extent lacking, declined by some patients. Second, the small sample size for IgG1 subtype PGNMID may limit the potential of finding differences between IgG3 and IgG1 subtype PGNMID and introduce random statistical effects. Thus, our findings on statistical analysis should be interpreted with caution, and further studies with larger sample sizes are needed to confirm these findings. Lastly, as a retrospective design, the small overall number of cases, the lack of standardized treatment, and the relatively short follow-up time may have led to the lack of statistical benefit of therapy in PGNMID in our study. Prospective, multicenter and controlled studies are required to define the optimal therapy for PGNMID.
In summary, some patients with PGNMID were associated with extrarenal disease and were more likely to have monoclonal IgG1 deposits. PGNMID of IgG3 subtype differs from IgG1 subtype by higher intensity of glomerular C3 deposition and higher frequency of subendothelial deposits. Serum creatinine and global glomerulosclerosis were independent prognostic predictors of ESKD in PGNMID.
References
Leung N, Bridoux F, Batuman V, Chaidos A, Cockwell P, D’Agati VD, Dispenzieri A, Fervenza FC, Fermand JP, Gibbs S, Gillmore JD, Herrera GA, Jaccard A, Jevremovic D, Kastritis E, Kukreti V, Kyle RA, Lachmann HJ, Larsen CP, Ludwig H, Markowitz GS, Merlini G, Mollee P, Picken MM, Rajkumar VS, Royal V, Sanders PW, Sethi S, Venner CP, Voorhees PM, Wechalekar AD, Weiss BM, Nasr SH (2019) The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol 1:45–59. https://doi.org/10.1038/s41581-018-0077-4
Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, Appel GB, Aucouturier P, D’Agati VD (2004) Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int 1:85–96. https://doi.org/10.1111/j.1523-1755.2004.00365.x
Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, Stokes MB, Nadasdy T, D’Agati VD (2009) Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol 9:2055–2064. https://doi.org/10.1681/asn.2009010110
Bhutani G, Nasr SH, Said SM, Sethi S, Fervenza FC, Morice WG, Kurtin PJ, Buadi FK, Dingli D, Dispenzieri A, Gertz MA, Lacy MQ, Kapoor P, Kumar S, Kyle RA, Rajkumar SV, Leung N (2015) Hematologic characteristics of proliferative glomerulonephritides with nonorganized monoclonal immunoglobulin deposits. Mayo Clin Proc 5:587–596. https://doi.org/10.1016/j.mayocp.2015.01.024
Best Rocha A, Larsen CP (2017) Membranous glomerulopathy with light chain-restricted deposits: a clinicopathological analysis of 28 cases. Kidney Int Rep 6:1141–1148. https://doi.org/10.1016/j.ekir.2017.07.008
Guiard E, Karras A, Plaisier E, Duong Van Huyen JP, Fakhouri F, Rougier JP, Noel LH, Callard P, Delahousse M, Ronco P (2011) Patterns of noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol 7:1609–1616. https://doi.org/10.2215/cjn.10611110
Gumber R, Cohen JB, Palmer MB, Kobrin SM, Vogl DT, Wasserstein AG, Nasta SD, Bleicher MB, Bloom RD, Dember L, Cohen A, Weiss BM, Hogan JJ (2018) A clone-directed approach may improve diagnosis and treatment of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Kidney Int 1:199–205. https://doi.org/10.1016/j.kint.2018.02.020
Fujita E, Shimizu A, Kaneko T, Masuda Y, Ishihara C, Mii A, Higo S, Kajimoto Y, Kanzaki G, Nagasaka S, Iino Y, Katayama Y, Fukuda Y (2012) Proliferative glomerulonephritis with monoclonal immunoglobulin G3κ deposits in association with parvovirus B19 infection. Hum Pathol 12:2326–2333. https://doi.org/10.1016/j.humpath.2012.04.004
Santana de Roberts R, Batal I, Aljareh A, Jim B (2021) Proliferative glomerulonephritis with monoclonal immunoglobulin deposits associated with parvovirus B19. BMJ Case Rep. https://doi.org/10.1136/bcr-2021-243061
Dahan K, Albert C, Arlet JB, Callard P, Ronco P (2010) Non-Randall proliferative glomerulonephritis with humps and monotypic IgG deposits in primary Sjögren’s syndrome: a first case report. NDT Plus 6:558–563. https://doi.org/10.1093/ndtplus/sfq147
Fujiwara T, Komatsuda A, Ohtani H, Togashi M, Sawada K, Wakui H (2013) Proliferative glomerulonephritis with monoclonal IgG deposits in a patient with autoimmune hemolytic anemia. Clin Nephrol 6:494–498. https://doi.org/10.5414/cn107267
Higashihara T, Okada A, Nakamura Y, Saigusa H, Homma S, Matsumura M, Kusano T, Shimizu A, Takano H (2020) Proliferative glomerulonephritis with monoclonal immunoglobulin deposits without conspicuous mesangial proliferation, complicated with squamous cell lung carcinoma. Intern Med 4:557–562. https://doi.org/10.2169/internalmedicine.2993-19
Liu M-Y, Wang S-X, Dong Y, Zhou F-D, Zhao M-H (2021) Pregnancy-associated proliferative glomerulonephritis with monoclonal immunoglobulin deposits. J Nephrol 5:1669–1672. https://doi.org/10.1007/s40620-020-00894-y
Barbour SJ, Beaulieu MC, Zalunardo NY, Magil AB (2011) Proliferative glomerulonephritis with monoclonal IgG deposits secondary to chronic lymphocytic leukemia. Report of two cases. Nephrol Dial Transplant 8:2712–2714. https://doi.org/10.1093/ndt/gfr251
Bhat P, Weiss S, Appel GB, Radhakrishnan J (2007) Rituximab treatment of dysproteinemias affecting the kidney: a review of three cases. Am J Kidney Dis 4:641–644. https://doi.org/10.1053/j.ajkd.2007.05.027
Noto R, Kamiura N, Ono Y, Tabata S, Hara S, Yokoi H, Yoshimoto A, Yanagita M (2017) Successful treatment with bortezomib and dexamethasone for proliferative glomerulonephritis with monoclonal IgG deposits in multiple myeloma: a case report. BMC Nephrol 1:127. https://doi.org/10.1186/s12882-017-0524-7
Barry R, James MT (2015) Guidelines for classification of acute kidney diseases and disorders. Nephron 4:221–226. https://doi.org/10.1159/000441425
Hutchison CA, Harding S, Hewins P, Mead GP, Townsend J, Bradwell AR, Cockwell P (2008) Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol 6:1684–1690. https://doi.org/10.2215/cjn.02290508
Bridoux F, Javaugue V, Nasr SH, Leung N (2021) Proliferative glomerulonephritis with monoclonal immunoglobulin deposits: a nephrologist perspective. Nephrol Dialysis Transplant 2:208–215. https://doi.org/10.1093/ndt/gfz176
Desbois AC, Cacoub P, Saadoun D (2019) Cryoglobulinemia: an update in 2019. Joint Bone Spine 6:707–713. https://doi.org/10.1016/j.jbspin.2019.01.016
Fogo AB, Lusco MA, Najafian B, Alpers CE (2016) AJKD atlas of renal pathology: cryoglobulinemic glomerulonephritis. Am J Kidney Dis 2:e5–e7. https://doi.org/10.1053/j.ajkd.2015.12.007
Karras A, Noël L-H, Droz D, Delansorne D, Saint-André J-P, Aucouturier P, Alyanakian M-A, Grünfeld J-P, Lesavre P (2002) Renal involvement in monoclonal (type I) cryoglobulinemia: two cases associated with IgG3 kappa cryoglobulin. Am J Kidney Dis 5:1091–1096
Vidarsson G, Dekkers G, Rispens T (2014) IgG subclasses and allotypes: from structure to effector functions. Front Immunol. https://doi.org/10.3389/fimmu.2014.00520
Nasr SH, Sethi S, Cornell LD, Fidler ME, Boelkins M, Fervenza FC, Cosio FG, D’Agati VD (2011) Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol 1:122–132. https://doi.org/10.2215/cjn.05750710
Zand L, Rajkumar SV, Leung N, Sethi S, El Ters M, Fervenza FC (2021) Safety and efficacy of daratumumab in patients with proliferative gn with monoclonal immunoglobulin deposits. J Am Soc Nephrol 5:1163–1173. https://doi.org/10.1681/ASN.2020101541
Acknowledgements
We would like to thank the patients for their participation in this study.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 82070747).
Author information
Authors and Affiliations
Contributions
ML contributed to the study design, data collecting and analysis, and draft of the manuscript; XY reviewed the renal pathologies and interpreted the pathological data; SW reviewed the renal pathologies. AQ collected the data; FZ designed, supervised the study, reviewed and edit the manuscript; MZ reviewed and edit the manuscript. All authors approved of submitting the manuscript for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
This study was performed in accordance with the Helsinki Declaration, and was approved by the ethics committee of the Peking University first hospital.
Informed consent
Written informed consent were obtained from the patient.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, M., Yu, X., Wang, S. et al. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits: an entity associated with distinct diseases and comparison between IgG1 and IgG3 subtypes. J Nephrol 35, 2363–2372 (2022). https://doi.org/10.1007/s40620-022-01317-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-022-01317-w