
Abstract
CKD-related nutritional therapy (NT) is a crucial cornerstone of CKD patients’ treatment, but the role of NT has not been clearly investigated in autosomal dominant polycystic kidney disease (ADPKD). Several clinical studies have focused on new pharmacological approaches to delay cystic disease progression, but there are no data on dietary interventions in ADPKD patients. The aim of this paper is to analyze the evidence from the literature on the impact of five nutritional aspects (water, sodium, phosphorus, protein intake, and net acid load) in CKD-related ADPKD extrapolating—where information is unavailable—from what occurs in CKD non-ADPKD patients Sodium intake restriction could be useful in decreasing the growth rate of cysts. Although further evidence is needed, restriction of phosphorus and protein intake restriction represent cornerstones of the dietary support of renal non-ADPKD patients and common sense can guide their use. It could be also helpful to limit animal protein, increasing fruit and vegetables intake together with a full correction of metabolic acidosis. Finally, fluid intake may be recommended in the early stages of the disease, although it is not to be prescribed in the presence of moderate to severe reduction of renal function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited disorder caused by mutations of polycystin 1 (PKD1) and polycystin 2 (PKD2) genes. As a result, multiple cystic formations grow into kidney parenchyma causing compression and disruption of functional kidney tissue leading to chronic kidney disease (CKD) and ultimately to end-stage renal disease (ESRD) in almost 50% of patients older than 50 years old. Cysts and aneurysms can also develop in several tissues and organs such as the liver, pancreas and blood vessels (especially encephalic and aortic ones). Arterial hypertension and left ventricular hypertrophy are quite prevalent in ADPKD patients [1, 2]. Some clinical studies have focused on new pharmacological approaches to slow down ADPKD progression, but few of them have dealt with nutritional and dietary aspects in ADPKD patients. The aim of this paper is to analyze the evidence from the literature on the impact of five nutritional aspects (water, sodium, phosphorus, protein intake, and net acid load) in CKD-related ADPKD, extrapolating the information, where unavailable, from what occurs in CKD non-ADPKD patients.
Water intake
High fluid intake represents an appealing therapeutic strategy due to the pathophysiological background and to the lack of severe side effects; on the other side, this approach is not always accepted by patients. The recommended fluid intake in ADPKD patients is still an open debate within the scientific community. The rationale is linked to the attempt to suppress anti-diuretic hormone (ADH) secretion and consequently both adenylyl cyclase and cyclic adenosine monophosphate (cAMP) production through vasopressin V2 receptor activation, since adenylyl cyclase and cAMP secretion induces renal cysts growth [3, 4]. Several studies show that an increased fluid intake, with consequent ADH suppression, induces regression of cysts growth and also reduces kidney disease progression [5, 6].
Nagao et al. evaluated the consequences of increasing fluid intake in rats: by increasing fluid intake from 3.5 to 8.2-fold for 10 weeks, a 68% reduction in arginine vasopressin urinary excretion and a urine osmolality up to 290 mOsm/kg occurred. A significant reduction (29.8 and 27% in male and female mice, respectively) in kidney weight and serum urea (from 38 to 26 mg/dl) was found. Therefore, a 54 and 28% reduction in cystic area and kidney/body weight ratio was also observed [7].
Hopp et al. reported that over-hydration had protective effects in an ADPKD rat model [8]. Water intake reduced urinary vasopressin and cAMP levels together with delaying disease progression in PKD rats. Rats assigned to the experimental arm also presented higher phosphodiesterase activity, lower cAMP levels and they were less sensitive to cystogenic effects of arginine-vasopressin [8]. In another study in 13 ADPKD patients, water intake higher than 7 Ls/day for a week led to urine osmolality reduction but it did not affect urinary cAMP excretion [9]. In a non-randomized trial performed on 30 ADPKD patients (17 with high water intake and 13 with unforced water intake) no changes were observed at 1-year follow-up, despite higher diuresis rates. Osmolality reduction was observed only in the higher water intake ADPKD patients during the follow-up period, as well as a reduction in estimated glomerular filtration rate (eGFR) (p = 0.011) and an increase in kidney volume [10]. Two years later, Amro et al. demonstrated that a 12-month hypo-osmolality and high water intake diet led to a significant reduction in copeptin (an arginine-vasopressin surrogate biomarker) and urine osmolality [11].
An updated survey has highlighted that, although 61% of patients had water intake higher than 2 Ls/day, only 4% had a fluid intake higher than 4 Ls/day [12]. To date, it is still unclear what patients mean by “high fluid intake” and no evidence about the efficacy has been published: randomized clinical trials are probably needed [13]. Data on over-hydration as a method to delay and reduce the cystogenic pathway are still ineffective at present. However, over-hydration is often recommended in ADPKD patients, especially in the early phases of the disease. But a common complication in ADPKD is renal calculus, and in these cases hydroponic therapy is useful. Therefore, the use of such therapy, as there are no papers showing a deterioration of residual renal function after hydroponic therapy, is not to be excluded a priori, but rather should be carefully evaluated case by case [14, 15].
The use of vasopressin V2-receptor antagonists (Tolvaptan) to slow the increase in total kidney volume has recently been demonstrated by a major phase 3 multicenter double-blind placebo-controlled study [16]. In fact, the TEMPO 3:4 trial demonstrated, in 1445 patients aged 18–50 years, that the use of Tolvaptan led to a reduced increase in kidney volume, of 2.8% compared to 5.5% per year (p < 0.001) in controls [16]. Tolvaptan antagonizes the V2 receptors in the collecting duct of the kidney, causing an elimination of electrolyte-free water (aquaresis) and a consequent increase in plasma sodium. The action of Tolvaptan regarding the 24-h urinary volume increase is dose-dependent and appears to affect the same mechanisms as high water intake action in antagonizing the V2 receptors in the collecting duct and suppressing ADH secretion and consequently both adenylyl cyclase and cAMP production [17].
Sodium intake
HALT PKD is a perspective randomized double-blind study designed to verify whether intensive blood pressure control, through a combination of angiotensin converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs), can delay kidney disease progression compared to monotherapy with ACEi only, in ADPKD patients [18]. A post-hoc analysis of HALT PKD was performed to assess if lowering urine osmolality by a reduction of sodium intake (< 2.4 g/day) could lead to better results than over-hydration [19]. Torres et al. analyzed data from 558 ADPKD patients with normal renal function (study-A with eGFR 91 ml/min on average) and 486 stage 3 CKD patients (study-B with eGFR 48 ml/min on average) both on low sodium diet. Patients showed a mild urinary sodium reduction from 178 to 166 mmol/die (study A) and from 177 to 152 mmol/die (study B). In the study A cohort, changes in urinary sodium excretion were significantly associated with kidney volume growth (0.43 and 0.09%/year respectively, for each 18 mEq of urinary sodium excretion). At the same time, changes in urinary sodium excretion were not associated with a delay of renal function loss (− 0.07 ml/min/1.73 m2/year for each 18 mEq of urinary sodium excretion). In the study B cohort, urinary sodium excretion was related to composite end-points (hazard ratio 1.08 for each 18 mEq urinary sodium excretion) and to progression of kidney disease (–0.09 ml/min/1.73 m2/year for each 18 mEq of urinary sodium excretion). Torres et al.’s findings suggest that dietary sodium restriction may play a beneficial role in ADPKD patients [19].
Amro and coll. [11], as already mentioned, have demonstrated that a reduced dietary intake of sodium can produce a significant reduction in vasopressin secretion in patients with ADPKD, in the same way as an increased intake of water. This finding has also been confirmed by Taylor et al. [20]. Based on these reported findings, dietary sodium intake restriction may be helpful in the clinical management of ADPKD patients, as well as in essential hypertensive patients and/or non-ADPKD CKD patients [21, 22].
Phosphorus intake and role of phosphorus level in ADPKD patients
A cross-link between phosphorus levels and cardiovascular mortality in a non-CKD population has been reported [23,24,25], as well as in CKD and ESRD patients on renal replacement therapy [26, 27]. However, there is no prospective randomized or retrospective study investigating the effect of phosphorus intake and cardiovascular and renal outcomes in patients with ADPKD. On the other hand, there are no data actually reported in the literature that link the dietary phosphorus intake and ADPKD progression.
A very recent paper [28] has highlighted, in a pediatric population with ADPKD and preserved renal function, the presence of hypophosphatemia and suppressed bone formation. The study showed that although there was no difference with respect to controls in the high fibroblast growth factor 23 (FGF23) levels the level should nevertheless be considered inappropriate given the concomitant hypophosphatemia [28]. The pathophysiology and potential clinical consequences of aspects such as FGF23 and fractional phosphorus excretion need to be further investigated in patients with ADPKD.
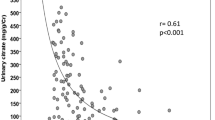
We must therefore use experimental data drawn from randomized prospective studies designed for other purposes. We used the Independent study [29, 30] to investigate a general population with CKD to search for data to evaluate the phosphorus level in ADPKD patients and mortality. Obviously, such analysis allows only to generate hypotheses, which would then need to be confirmed by prospective randomized prospective studies. In the Independent–HD study, 33 ADPKD patients are enrolled [29]. Table 1 shows baseline data based on phosphorus serum levels according to the following tertiles: the 1st tertile was 3–4.6 mg/dl (parathormone [PTH] 145 ± 95 pg/ml and creatinine 9 ± 2 mg/dl); the 2nd tertile was 4.6–5.5 mg/dl (PTH 261 ± 98 pg/ml and creatinine 10.3 ± 3 mg/dl) and the 3rd tertile was 5.5–6.8 mg/dl (PTH 284 ± 110 pg/ml and creatinine 12.3 mg/dl). Kt/V did not differ among the three groups. In the follow-up period, 12 patients died due to cardiovascular disease, as shown in Fig. 1—the mortality rate according to serum phosphorus levels was 8% in the lowest tertile, 34% in the medium tertile, and 58% in the highest tertile (p < 0.001).

Deaths due to cardiovascular causes by tertile of phosphorus in APKD patients who participated in the Independent-HD study (data from [23])
We also evaluated data of the Independent-CKD study [30]: 15 ADPKD patients were found (7 patients with P < 4.5 mg/dl and 8 with P > 4.6 mg/dl) (Table 2) with medium eGFR 34 ± 6 ml/min. Of these 15 patients, 9 died from cardiovascular disease and/or started renal replacement therapy: 2 (28%) had P < 4.5 mg/dl and 7 (87%) had P > 4.6 mg/dl.
Post-hoc analyses involving 33 and 15 patients cannot provide scientific proof; however, the results mentioned above suggest that hyperphosphatemia could be accountable for a higher mortality risk also in ADPKD-ESRD patients. To date, it still remains to be demonstrated that a reduction of phosphorus serum levels can impact on cardiovascular mortality in ADPKD patients: this could represent a new challenge to be faced in the coming future.
Protein intake
Experimental data have highlighted that an early restriction of protein intake can delay CKD progression in non-ADPKD patients, and it has been repeatedly shown that the reduction of protein intake must be accompanied by an energy intake of 30–35 kcal/kg/BW/day to avoid dangerous episodes of caloric-protein malnutrition [31, 32]. Back in 1992, Aukema et al. showed a lower increase of cystic volume, namely a 46% reduction in cyst area in mice on low protein intake (LP) compared to mice with normal protein intake, or casein and sunflower seed oil (SO) and fish oil (FO) [33]. The authors also demonstrated that a vegetable protein-enriched diet led to a 28% reduction in kidney weight (p = 0.0037), a 37% reduction (p = 0.0089) in cyst score (cystic area % x relative body weight) and a 25% reduction in kidney water content (p = 0.0144) compared to an animal protein-enriched diet [34].
Lower protein intake (6 vs. 17.4 g %) was associated with a 30% reduction of kidney weight (p = 0.001), a 25% reduction in cyst score (p = 0.0327) and a 35% reduction in kidney water content [34]. Similar results were reported by Tomobe et al. in a 105 day follow-up period with vegetable protein intake in mice [35]. In a small sample study on genetic polycystic rats, a reduction in the protein intake caused a reduction of kidney and glomerular volume, but it did not affect size and/or growth of the kidney cysts [36].
Finally, Ogborn et al. evaluated two dietetic schedules (with the same protein intake) administered to mice during a period of 6 weeks comparing casein vs. soil seeds. The authors found a cystic volume reduction (from 7 to 4.3 ml/kg) associated to vegetable protein intake (p < 0.001), together with a reduction of epithelial cells (from 21 to 15.7 cells/mm, p < 0.001), of macrophage infiltration (from 43.5 to 25.3, p < 0.001) and of fibrosis (from 1.06 to 0.6 ml/kg, p < 0.001) [37].
More recently, Yamaguchi et al. did not find any clinical benefit in Pkd2WS25 mice (ADPKD mice) after 13 weeks of soil protein nutrition compared to casein intake in the control population [38]. In ARPKD (autosomic recessive polycystic kidney disease) mice, an animal protein-based diet led to an increase of kidney and liver weight together with an increase of kidney water content and cystic area [38]. Kidney and liver histology did not improve but lower proteinuria and higher urine pH suggested that a soil seeds diet could be favorable in the long-term period, independently of its effects on cystogenesis [38].
Another paper reported that food restriction may delay ADPKD progression in experimental models [39]. Cyst volume was reduced by moderate food restriction (10–40%), while renal fibrosis and inflammation were not affected [39]. Molecular and biochemical investigations showed that food restriction improves ADPKD progression by suppression of the rapamycin pathway and liver B1/AMP kinase activation [39].
Kipp and coll reported that a 23% food restriction is able to impact on polycystic disease progression [40]; without affecting physiological growth or causing malnutrition or other side effects, it can postpone the disease’s progression [40]. In fact, the relative increase in kidney weight was 41 vs. 151% and cystic proliferation was 7.7 vs. 15.9% compared to controls [40].
Food restriction was able to preserve renal function in experimental PKD models suggesting that polycystic kidneys are susceptible to food and calorie restrictions [40, 41].
The above-mentioned experimental data were not confirmed in a human population [42, 43]. Klahr et al. evaluated 200 ADPKD patients from the MDRD study: CKD progression was faster in ADPKD patients than other patients; disease progression was found to be related to the disease per se, to higher serum creatinine levels (independently of GFR), to higher proteinuria and to higher mean blood pressure [42]. Therefore, in ADPKD patients with GFR between 25 and 55 ml/min/1.73 m2, neither lower protein intake, nor lower blood pressure levels significantly delayed renal disease progression [42]. On the other hand, in stage 4–5 CKD patients (GFR between 13 and 24 ml/min/1.73 m2), patients with a lower mean arterial pressure showed the fastest decline in GFR values. However, faster GFR decline did not appear to be related to the anti-hypertensive treatment. Reduced protein intake, but not keto-analogues prescription was associated to the slowest CKD progression [42].
In a retrospective study enrolling 109 ADPKD patients, Choukroun et al. demonstrated a weak positive relationship between eGFR changes and mean arterial blood pressure; no relation with dietary protein intake (r = 0.10; p = 0.33) or proteinuria was found (r = 0.10; p = 0.28) [43]. The authors’ conclusion was that lower CKD progression rate in ADPKD patients is mainly linked to genetic predisposition, female sex and aging, while blood pressure control and protein intake have no effect on CKD progression rate in ADPKD patients with advanced renal disease [43].
More recently, the role has been investigated of branched chain amino acids (BCAA: 0.2 g isoleucine, 0.4 g leucine, and 0.2 g valine) on cyst growth in an experimental model. The results showed that BCAA stimulated the cyst growth by cellular proliferation and mTOR/cAMP pathway activation [39]; however, it is important to underline that the BCAA group had a 10% higher protein intake with respect to controls (977 vs. 885 mg/day) [44].
In summary, no clear evidence concerning the role of a low-protein diet in delaying ADPKD progression is available at present. At the same time, nephrologists can only provide some recommendations on low-protein intake based on what occurs in non–ADPKD CKD patients.
A correct nutritional therapy has to take into consideration a proper intake of sodium, phosphate, protein (quantity and quality), amino acid supplements, and fluid intake [39] to ensure a better control of CKD signs and symptoms. This nutritional support has been described in a feasibility study by Taylor and coll [20]. including 12 ADPKD patients. Patients consumed their standard diet for a week and then shifted to a low sodium and protein diet together with higher fruit, vegetables and water intake for 4 weeks [20]. As a consequence, the patients enrolled consumed 36% sodium, 28% proteins and 99% amino acid precursors and increased by 42% fruit and vegetables intake. Urinary sodium, urea, net acid excretion and osmolality decreased by 20, 28, 20 and 15%, respectively; urinary volume increased by 35% [44]. Based on these preliminary findings, an interventional study protocol needs to be designed. Obviously, also in ADPKD the protein restriction has to be prescribed whilst at the same time maintaining an adequate energy intake, although experimental data do not exist in the literature.
Net acid load
In recent years, metabolic acidosis complications have been extensively studied in CKD patients. Consequently, correction of acid–base imbalance with sodium bicarbonate administration and/or vegetables and fruit intake is a mainstay of CKD treatment [13, 44,45,46,47,48].
Although this topic is crucial for CKD patients, no randomized controlled trial actually exists for ADPKD [44].
Our group has designed a prospective multicenter randomized controlled study to evaluate the effects of metabolic acidosis correction on CKD progression and renal death (UBI study, ClinicalTrials.gov Identifier: NCT01640119); definitive data will soon be available [49]. The UBI study included 84 ADPKD patients, 38 in the experimental group and 46 in control group. Eleven patients dropped out or were lost to follow-up. Table 3 shows the baseline data of the 73 active patients. Baseline bicarbonate levels did not differ between the experimental (22.0 ± 1.9 mmol/l) and control groups (22.3 ± 2.6 mmol/l). During the 3-year follow-up, bicarbonate levels increased in the experimental group: from 25.4 ± 2.6 at 1 year, to 26.4 ± 2.6 at 2 years, and 26.3 ± 1.8 mmol/l at 3 years, whereas no changes were observed in the control group. Twenty-four hour urinary urea, potassium and phosphate excretion were used to calculate the potential renal acid load (PRAL) and net endogenous acid production (NEAP). NEAP represents the non-volatile acid load derived from diet, estimated by the production of non-volatile acids and bases produced during digestion, calculated by the known nutritional content. PRAL and NEAP were calculated as previously described [45] by the Remer and Manz formula and the Frassetto formula, respectively [50, 51]. Figures 2 and 3 show changes in NEAP and PRAL in the ADPKD patients taking sodium bicarbonate orally vs. the control group [Fig. 4 shows changes in eGFR for both groups: eGFR declined during 36 months of follow-up by 1.5 ml/min/year in the experimental ADPKD group vs. 3.3 ml/min/year in the ADPKD control group (p < 0.001)]. In the control group, 19% of patients doubled their serum creatinine in the follow-up period vs. 9.6% of patients in the experimental group. Renal replacement therapy had to be started in 24% of control patients vs. 10% in the experimental group, while 2 patients (one for each group) died.

Variation of net endogenous acid production (NEAP) in ADPKD subjects who participated in the UBI study and underwent oral administration of sodium bicarbonate compared to the control group. The plot box represents the mean ± SD, the dotted lines the maximum value and the minimum value. *p < 0.001 vs. NO-bicarbonate use

Variation of potential renal acid load (PRAL) in ADPKD subjects who participated in the UBI study and underwent oral administration of sodium bicarbonate, compared to the control group. The plot box represents the mean ± SD, the dotted lines the maximum value and the minimum value. *p < 0.001 vs. NO-bicarbonate use

GFR loss during the 36-month follow-up of patients with ADPKD who participated in the UBI study. The plot box represents the mean ± SD, the dotted lines the maximum value and the minimum value. #p < 0.01 vs. NO-bicarbonate use. *p < 0.001 vs. NO-bicarbonate use
Sodium bicarbonate administration represents one of the treatments of metabolic acidosis in CKD patients; our group [45] has already reported that a vegetarian low-protein diet makes possible a 50% decrease of administered sodium bicarbonate, in line with Goraya et al.’s data [46]. In 1998, Tanner demonstrated, in experimental models, that potassium citrate administration improved eGFR levels in PKD mice and he concluded that further data are needed to better understand the role of nutritional interventions in ADPKD models and patients [52]. Four years before, Torres had demonstrated a renal acidification defect following ammonium chloride load in ADPKD patients without renal insufficiency in comparison to healthy subjects [53].
Conclusions
Evidence exists that dietetic interventions cannot modify the ADPKD course with the exception of sodium intake reduction. At the same time, we cannot renounce the provision of nutritional support to ADPKD patients [54]. Dietary support makes it possible to prevent and treat several signs, symptoms and complications closely linked to renal insufficiency progression which are addictive effects of conventional pharmacological treatment. A dietary support could improve metabolic, nutritional and clinical conditions of CKD and ADPKD patients together with their quality of life, postponing the need for renal replacement treatment.
In conclusion, even though data on nutritional therapy in ADPKD are scant, common sense can be a guide in suggesting that sodium intake restriction may be useful to decrease cyst growth rate [17]; although further evidence is needed, a restriction of phosphorus and protein intake restriction represent cornerstones of the dietary support in renal patients. It could be also helpful to limit animal protein, increasing the intake of fruit and vegetables together with a full correction of metabolic acidosis. Finally, fluid intake may be recommended in the early stages of the disease, but it is not to be prescribed in the presence of a moderate to severe reduction of renal function. In conclusion, there is currently little evidence testifying the importance of nutritional treatment as a therapy to slow the growth of the volume of the kidneys and the progression of CKD in ADPKD subjects. Therefore, studies focused on this issue need to be performed in coming years [55, 56].
References
Liut F, Izzi C, Dallera N, Scolari F (2017) ADPKD e cuore. G Ital Nefrol 34(Suppl 69):119–130
Churchill DN, Bear JC, Morgan J, Payne RH, McManamon PJ, Gault MH (1984) Prognosis of adult onset polycystic kidney disease re-evaluated. Kidney Int 26:190–193
Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ (2003) Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant poly- cystic kidneys. Kidney Int 63:1983–1994
Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP (2004) Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 279:40419–40430
Gattone VH 2nd, Wang X, Harris PC, Torres VE (2003) Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9:1323–1326
Yamaguchi T, Nagao S, Kasahara M, Takahashi H, Grantham JJ (1997) Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am J Kidney Dis 30:703–709
Nagao S (2006) Increased Water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 17:2220–2227
Hopp K, Wang X, Ye H, Irazabal MV, Harris PC, Torres VE (2015) Effects of hydration in rats and mice with polycystic kidney disease. Am J Physiol Renal Physiol 308(3):F261–F266
Barash I, Ponda MP, Goldfarb DS, Skolnik EY (2010) A pilot clinical study to evaluate changes in urine osmolality and urine camp in response to acute and chronic water loading in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 5:693–697
Higashihara E, Kikuo Nutahara K, Tanbo M, Hara H, Miyazaki I, Kobayashi H, Nitatori T (2014) Does increased water intake prevent disease progression in autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 29:1710–1719
Amro OW, Paulus JK, Noubary F, Perrone RD (2016) Low-osmolar diet and adjusted water intake for vasopressin reduction in autosomal dominant polycystic kidney disease: a pilot randomized controlled trial. AM J Kidney Dis 68:882–891
El-Damanawi R, Harris T, Sandford RN, Karet Frankl FE, Hiemstra TF (2017) Patient survey of current water Intake practices in autosomal dominant Polycystic kidney disease: the SIPs survey. Clin Kidney J 10:305–309
Taylor JM, Hamilton-Reeves JM, Sullivan DK, Gibson CA, Creed C, Carlson SE, Wesson DE, Grantham JJ (2017) Diet and polycystic kidney disease: a pilot intervention study. Clin Nutr 36(2):458–466
Wang CJ, Grantham JJ, Wetmore JB (2013) The medicinal use of water in renal disease. Kidney Int 84:45–53
Clark WF, Sontrop JM, Huang SH, Moist L, Bouby N, Bankir L (2016) Hydration and chronic kidney disease progression: a critical review of the evidence. Am J Nephrol 43:281–292
Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS; TEMPO 3:4 Trial Investigators (2012) Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367(25):2407–2418
Gansevoort RT, Arici M, Benzing T, Birn H, Capasso G, Covic A, Devuyst O, Drechsler C, Eckardt KU, Emma F, Knebelmann B, Le Meur Y, Massy ZA, Ong AC, Ortiz A, Schaefer F, Torra R, Vanholder R, Więcek A, Zoccali C, Van Biesen W (2016). Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol Dial Transplant 31(3):337–348
Chapman AB, Torres VE, Perrone RD, Steinman TI, Bae KT, Miller JP, Miskulin DC, Rahbari Oskoui F, Masoumi A, Hogan MC, Winklhofer FT, Braun W, Thompson PA, Meyers CM, Kelleher C, Schrier RW (2010) HALT T polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol 5(1):102–109
Torres VE, Abebe KZ, Schrier RW, Perrone RD, Chapman AB, Yu AS, Braun WE, Steinman TI, Brosnahan G, Hogan MC, Rahbari FF, Grantham JJ, Bae KT, Moore CG, Flessner MF (2017) Dietary salt restriction is beneficial to the management of autosomal dominant polycystic kidney disease. Kidney Int 91(2):493–500
Taylor JM, Ptomey L, Hamilton-Reeves JM, Sullivan DK, Creed C, Carlson SE, Wesson DE, Grantham JJ, Gibson CA (2016) Experiences and perspectives of polycystic kidney disease patients following a diet of reduced osmoles, protein, and acid precursors supplemented with water: a qualitative study. PLoS One 11(8):e0161043
Rebholz CM, Crews DC, Grams ME, Steffen LM, Levey AS, Miller ER 3rd, Appel LJ, Coresh J (2016) DASH (dietary approaches to stop hypertension) diet and risk of subsequent kidney disease. Am J Kidney Dis 68(6):853–861
Nomura K, Asayama K, Jacobs L, Thijs L, Staessen JA (2017) Renal function in relation to sodium intake: a quantitative review of the literature. Kidney Int 92(1):67–78
Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G; Cholesterol And Recurrent Events Trial Investigators (2005) Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 112(17):2627–2633
Chang AR, Lazo M, Appel LJ, Gutiérrez OM, Grams ME (2014) High dietary phosphorus intake is associated with all-cause mortality: results from NHANES III. Am J Clin Nutr 99(2):320–327
Gutiérrez OM (2013) The connection between dietary phosphorus, cardiovascular disease, and mortality: where we stand and what we need to know. Adv Nutr. 4(6):723–729
Tonelli M (2013) Serum phosphorus in people with chronic kidney disease: you are what you eat. Kidney Int 84(5):871–873
O’Seaghdha CM, Hwang SJ, Muntner P, Melamed ML, Fox CS (2011) Serum phosphorus predicts incident chronic kidney disease and end-stage renal disease. Nephrol Dial Transplant 26(9):2885–2890
De Rechter S, Bacchetta J, Godefroid N, Dubourg L, Cochat P, Maquet J, Raes A, De Schepper J, Vermeersch P, Van Dyck M, Levtchenko E, D’Haese P, Evenepoel P, Mekahli D (2017) Evidence for bone and mineral metabolism alterations in children with autosomal dominant polycystic kidney disease. J Clin Endocrinol Metab 102(11):4210–4217
Di Iorio B, Molony D, Bell C, Cucciniello E, Bellizzi V, Russo D, Bellasi A; INDEPENDENT Study Investigators (2013) Sevelamer versus calcium carbonate in incident hemodialysis patients: results of an open-label 24-month randomized clinical trial. Am J Kidney Dis 62(4):771–778
Di Iorio B, Bellasi A, Russo D; INDEPENDENT Study Investigators (2012) Mortality in kidney disease patients treated with phosphate binders: a randomized study. Clin J Am Soc Nephrol. 7(3):487–493
Pérez-Torres A, González Garcia ME, San José-Valiente B, Bajo Rubio MA, Celadilla Diez O, López-Sobaler AM, Selgas R (2017) Protein-energy wasting syndrome in advanced chronic kidney disease: prevalence and specific clinical characteristics. Nefrologia Jul 26. pii:S0211–6995(17)30141-8. https://doi.org/10.1016/j.nefro.2017.06.004 [Epub ahead of print]
Cupisti A, D’Alessandro C, Finato V, Del Corso C, Catania B, Caselli GM, Egidi MF (2017) Assessment of physical activity, capacity and nutritional status in elderly peritoneal dialysis patients. BMC Nephrol 18(1):180
Aukema HM, Ogborn MR, Tomobe K, Takahashi H, Hibino T, Holub BJ. (1992) Effects of dietary protein restriction and oil type on the early progression of murine polycystic kidney disease. Kidney Int. 42(4):837–842
Aukema HM, Housini I, Rawling JM (1999) Dietary soy protein effects on inherited polycystic kidney disease are influenced by gender and protein level. J Am Soc Nephrol 10(2):300–308
Tomobe K, Philbrick D, Aukema HM, Clark WF, Ogborn MR, Parbtani A, Takahashi H, Holub BJ (1994) Early dietary protein restriction slows disease progression and lengthens survival in mice with polycystic kidney disease. J Am Soc Nephrol 5(6):1355–1360
Barsotti G, Cupisti A, Moriconi L, Cozza V, Falbo E, Gattai V, Pozzolini L, Meola M (1995) Effects of reduced protein intake in rats with congenital polycystic kidney without renal failure. Contrib Nephrol 115:137–140
Ogborn MR, Nitschmann E, Weiler HA, Bankovic-Calic N (2000) Modification of polycystic kidney disease and fatty acid status by soy protein diet. Kidney Int 57(1):159–166
Yamaguchi T, Devassy JG, Monirujjaman M, Gabbs M, Aukema HM (2016) Lack of Benefit of early intervention with dietary flax and fish oil and soy protein in orthologous rodent models of human hereditary polycystic kidney disease. PLoS One 11(5):e0155790
Warner G, Hein KZ, Nin V, Edwards M, Chini CC, Hopp K, Harris PC, Torres VE, Chini EN (2016) Food restriction ameliorates the development of polycystic kidney disease. J Am Soc Nephrol 27(5):1437–1447
Kipp KR, Rezaei M, Lin L, Dewey EC, Weimbs T (2016) A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am J Physiol Renal Physiol 310(8):F726-F731
Peng CY, Sankaran D, Ogborn MR, Aukema HM (2009) Dietary soy protein selectively reduces renal prostanoids and cyclooxygenases in polycystic kidney disease. Exp Biol Med (Maywood) 234(7):737–743
Klahr S, Breyer JA, Beck GJ, Dennis VW, Hartman JA, Roth D, Steinman TI, Wang SR, Yamamoto ME (1995) Dietary protein restriction, blood pressure control, and the progression of polycystic kidney disease. Modification of Diet in Renal Disease Study Group. J Am Soc Nephrol 5(12)2037–2034
Choukroun G, Itakura Y, Albouze G, Christophe JL, Man NK, Grünfeld JP, Jungers P (1995) Factors influencing progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 6(6):1634–1642
Yamamoto J, Nishio S, Hattanda F, Nakazawa D, Kimura T, Sata M, Makita M, Ishikawa Y, Atsumi T (2017) Branched-chain amino acids enhance cyst development in autosomal dominant polycystic kidney disease. Kidney Int 92(2):377–387
Di Iorio BR, Di Micco L, Marzocco S, De Simone E, De Blasio A, Sirico ML, Nardone L, On Behalf Of Ubi Study Group (2017) Very low-protein diet (VLPD) reduces metabolic acidosis in subjects with chronic kidney disease: the “Nutritional Light Signal” of the Renal Acid Load. Nutrients 9(1)
Goraya N, Simoni J, Jo CH, Wesson DE. (2013) A comparison of treating metabolic acidosis in CKD stage 4 hypertensive kidney disease with fruits and vegetables or sodium bicarbonate. Clin J Am Soc Nephrol. 8(3):371–381
Goraya N, Wesson DE (2013) Does correction of metabolic acidosis slow chronic kidney disease progression? Curr Opin Nephrol Hypertens 22(2):193–197
Bissler JJ. (2015) Therapies for polycystic kidney disease. Curr Opin Pediatr. 27(2):227–232
Di Iorio B, Aucella F, Conte G, Cupisti A, Santoro D (2012) A prospective, multicenter, randomized, controlled study: the correction of metabolic acidosis with use of bicarbonate in Chronic Renal Insufficiency (UBI) Study. J Nephrol. 25(3):437–440
Remer T, Manz F (1994) Estimation of the renal net acid excretion by adults consuming diets containing variable amounts of protein. Am J Clin Nutr 59:1356–1361
Frassetto LA, Todd KM, Morris RJC, Sebastian A (1998) Estimation of net endogenous noncarbonic acid production in humans from diet potassium and protein contents. Am J Clin Nutr 68:576–583
Tanner GA (1998) Potassium citrate/citric acid intake improves renal function in rats with polycystic kidney disease. J Am Soc Nephrol 9(7):1242–1248
Torres VE, Keith DS, Offord KP, Kon SP, Wilson DM (1994) Renal ammonia in autosomal dominant polycystic kidney disease. Kidney Int 45(6):1745–1753
D’Alessandro C, Piccoli GB, Calella P, Brunori G, Pasticci F, Egidi MF, Capizzi I, Bellizzi V, Cupisti A (2016) “Dietaly”: practical issues for the nutritional management of CKD patients in Italy. BMC Nephrol 17(1):102
Magistroni R, Boletta A (2017) Defective glycolysis and the use of 2-deoxy-D-glucose in polycystic kidney disease: from animal models to humans. J Nephrol 30(4):511–519
Girardat-Rotar L, Puhan MA, Braun J, Serra AL. (2017).Long-term effect of coffee consumption on autosomal dominant polycystic kidneys disease progression: results from the Suisse ADPKD, a Prospective Longitudinal Cohort Study. J Nephrol. https://doi.org/10.1007/s40620-017-0396-8
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical approval
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Rights and permissions
About this article

Cite this article
Di Iorio, B.R., Cupisti, A., D’Alessandro, C. et al. Nutritional therapy in autosomal dominant polycystic kidney disease. J Nephrol 31, 635–643 (2018). https://doi.org/10.1007/s40620-018-0470-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-018-0470-x