
Abstract
Purpose
The effect of growth hormone (GH) on adipose tissue and the role of adipokines in modulating metabolism are documented, but with discordant data. Our aim was to evaluate the impact of GH treatment on a series of selected adipokines known to have a metabolic role and poorly investigated in this setting.
Methods
This is a prospective study. Thirty-one prepubertal children (25 M, 6 F; aged 8.5 ± 1.6 years) with isolated GH deficiency treated with GH for at least 12 months and 30 matched controls were evaluated. Auxological and metabolic parameters, insulin sensitivity indexes, leptin, soluble leptin receptor, adiponectin, visfatin, resistin, omentin, adipocyte fatty acid-binding protein and retinol-binding protein-4 were evaluated before and after 12 months of treatment.
Results
At baseline, no significant difference in metabolic parameters was found between GHD children and controls, except for higher LDL cholesterol (p = 0.004) in the first group. At multivariate analysis, LDL cholesterol was independently associated with resistin (B 0.531; p = 0.002), while IGF-I was the only variable independently associated with visfatin (B 0.688; p < 0.001). After 12 months, a significant increase in fasting insulin (p = 0.008), Homa-IR (p = 0.007) and visfatin (p < 0.001) was found, with a concomitant decrease in LDL cholesterol (p = 0.015), QUICKI (p = 0.001), ISI Matsuda (p = 0.006), leptin (p = 0.015) and omentin (p = 0.003)]. At multivariate analysis, BMI was the only variable independently associated with leptin (B 0.485; p = 0.040).
Conclusions
GH treatment modifies adipokine secretion and the perturbation of some adipokine levels could contribute to the clinical and metabolic changes observed during the follow-up.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Growth hormone (GH) during childhood, in addition to promote linear growth, plays a key metabolic role and adipose tissue is known to be an important target for GH action [1]. Adipocytes secrete hormones known as adipokines, many of which regulate metabolism and influence insulin sensitivity and secretion [2]. Untreated GH deficiency (GHD) in children, as well as in adults, is associated with abnormalities in body composition, in addition to a cluster of cardiometabolic risk factors such as increased peripheral inflammatory markers and impairment in glucose and lipid metabolism, and GH treatment seems to exert beneficial effects on most of these alterations [3–5].
Given the known effect of GH on adipose tissue and the role of adipokines in modulating metabolism and insulin homeostasis, many studies have evaluated the effect of GHD and GH treatment on the circulating levels of the most common adipokines, such as leptin and adiponectin. A negative impact on these adipokine levels has been reported in untreated GHD children, with a partial improvement during GH treatment, but with very discordant data across the studies. GHD seems to be associated with elevated leptin levels which most likely reflect an increased fat mass in these patients [6] and a direct effect of GH on leptin production and metabolism is hypothesized [7] although this effect is not confirmed by all studies [8]. Conversely, adiponectin seems to be unaffected in untreated GHD, and GH treatment has been shown to have discordant effect on its levels [9, 10]. Similarly, the relationship between the modifications of adipokine levels and the metabolic changes that occur in GHD children has not been unequivocally demonstrated [11, 12]. Given the discrepancies of the existing data about the role of the most investigated adipokines, our aim was to analyze in children affected by GHD how GH replacement modifies a series of new selected adipokines, known to have a metabolic role and poorly investigated in this setting, and the relationship of these modifications with the metabolic changes.
Materials and methods
We prospectively studied 31 prepubertal children (25 M, 6 F; mean age 8.5 ± 1.6 years; range 5.3–10.3) with isolated GHD consecutively admitted to the Section of Endocrinology of the University of Palermo during the years 2013–2014, treated with GH for at least 12 months and never investigated before in other clinical studies. Thirty prepubertal healthy subjects, matched for sex (22 M, 8 F), age (mean age 8.9 ± 1.6 years; range 4.6–10.6) and BMI were recruited among children referred for assessment of short stature as a control group at baseline. All children, even the older ones, were in the first stage of sexual development to avoid any interference of the onset of puberty with insulin sensitivity and body composition and maintained the prepubertal hormonal status during the observation period. For the same reasons, we excluded children affected by multiple pituitary hormone deficiency or receiving other hormonal replacement treatment, to exclusively evaluate the effects of GH. The diagnosis of GHD was established by the clinical, auxological and biochemical criteria of the GH Research Society [13].
As auxological data, we considered height and growth velocity 1 year before the diagnosis. Clinical and auxological criteria included height more than 2 standard deviations (SDS) below the mean and a growth velocity over 1 year more than 1 SDS below the mean for age, or a decrease in height SDS of more than 0.5 over 1 year or, without severe short stature, a growth velocity more than 2 SDS below the mean over 1 year or, finally, height more than 1.5 SDS below the midparental height.
As a radiological criterion, we considered a bone age delay, estimated from an X-ray of the left wrist and hand and evaluated according to the methods of Greulich and Pyle, of at least 1 year with respect to the chronological age [14]. Biochemically, GHD was demonstrated by failure of GH to respond to an arginine and glucagon stimulation test, performed on two different days, with GH peaks below 7 µg/l. Neuroimaging, with magnetic resonance imaging of the hypothalamic-pituitary region, in line with our protocol, was performed in children with signs pointing to multiple pituitary hormone deficiency or indicative of an intracranial lesion and in children with more severe GHD, like those with height more than 3 SDS below the mean, GH peak ≤ 3 μg/l or IGF-I levels below 2 SDS (No. 18 children). Among them, three patients showed a pituitary hypoplasia and 2 a partial empty sella. The patients received GH once daily at bedtime with a pen injection system. During the entire follow-up, insulin-like growth factor (IGF)-I levels allowed us to determine the GH dose. Specifically, the target was IGF-I levels between 0.5 and 1.5 SDS. The initial daily dose of GH was 0.025 mg/Kg, increased to 0.028 mg/Kg from month 6 to 12.
Study protocol
In all patients, after the diagnosis of GHD was made, the auxological and metabolic evaluation was performed at baseline and after 12 months of GH treatment, while in the control subjects these evaluations were performed only at baseline.
In addition to the measurement of body height, body mass index (BMI) and waist circumference (WC), in all children a blood sample was drawn after an overnight fast for the measurement of IGF-I, fasting glucose, fasting insulin, Hemoglobin A1c (HbA1c) and lipid profile [total, high-density lipoprotein (HDL) and low-density lipoprotein (LDL) cholesterol]. This sample also served as the baseline sample for an oral glucose tolerance test (OGTT). Blood samples were collected every 30 min for 2 h for glucose and insulin measurements.
Height, growth velocity, BMI and IGF-I were expressed as SDS due to the wide age range of patients.
As surrogate estimates of insulin sensitivity, we used the homeostasis model assessment estimate of insulin resistance (Homa-IR) [15], the quantitative insulin sensitivity check index (QUICKI) [16] and the insulin sensitivity index (ISI), a composite index derived from the OGTT and validated by Matsuda and DeFronzo [17].
To evaluate the adipose function, in all children we measured the serum levels of leptin, soluble leptin receptor (sOB-R), adiponectin, visfatin, resistin, omentin, adipocyte fatty acid-binding protein (AFABP) and retinol-binding protein-4 (RBP4) after an overnight fast.
The institutional Ethics Committee of the University of Palermo approved this study. At the time of hospitalization, an informed consent for the scientific use of the data was obtained from both the participants and their parents.
Hormone and biochemical assays
All biochemical data were collected after overnight fasting. Glycaemia and lipids were measured by standard methods (Modular P800, Roche, Milan). HbA1c levels were determined by HPLC (Bio-Rad Laboratories, Milan, Italy). Serum insulin was measured by ELISA (DRG Instruments GmbH, Germany). The sensitivity of the method was 1 IU/ml. The normal insulin range (IU/ml) was 5–19. GH levels were measured by ELISA assay using commercially available kits (hGH SENSITIVE ELISA Mediagnost E022, Germany). The sensitivity yields 0.0115 μg/l, with intra- and inter-assay coefficients of variation (CV) 3.7–7.9 and 3.1–5.9 %, respectively. The 2nd International Standard NIBSC Code 98/574 was used as standard material. Serum IGF-I was assayed in the same laboratory with ELISA assay (OCTEIA IGF-I kit, IDS Inc., Fountain Hills, AZ, USA). The sensitivity of the method was 1.9 µg/l. The inter- and intra-assay CV values were 7–7.1 and 2.3–3.5 %, respectively, at IGF-I levels of 90.7–186 and 66.7–120.9 µg/l, respectively. The normal ranges (males and females combined) of total IGF-I levels (µg/l) were the following: 12–108 (0–1 years); 13–100 (1–3 years); 26–280 (3–6 years); 85–230 (6–9 years); 98–404 (9–12 years); 142–525 (12–15 years); 146–415 (15–20 years). Values were expressed as SDS according to the normative data provided by the manufacturer. Human leptin (ng/ml), sOB-R (ng/ml), adiponectin (µg/ml), resistin (ng/ml), visfatin (ng/ml), RBP4 (µg/ml), AFABP (ng/ml) and omentin-1 (ng/ml) were assayed using an ELISA sandwich enzyme immuno-assay (BioVendor, Heidelberg, Germany).
Statistical analysis
The Statistical Packages for Social Sciences SPSS version 17 was used for data analysis. Baseline characteristics were presented as mean ± SDS or as median values ± interquartile range (IR) for continuous variables, when appropriate (i.e. for the variables without normal distribution). Normality of distribution for quantitative variables was assessed with the Kolmogorov–Smirnov test. The differences between groups were evaluated with the t test when with normal distribution or with the Mann–Whitney test (nonparametric test) when without normal distribution. Pearson’s correlation was performed among continuous variables with normal distribution; correlations among continuous variables without normal distribution were determined using the Spearman’s test (nonparametric equivalent for Pearson test). To identify the independent variables which influence the adipokine levels, a linear regression model was performed. A p value <0.05 was considered statistically significant.
Results
Clinical and hormonal parameters
The clinical and hormonal features of control subjects and of GHD children at diagnosis and after 12 months of GH treatment are shown in Table 1.
No significant difference in height, BMI SDS and WC SDS between GHD children at baseline and control subjects was found. As expected, GHD children at baseline showed significantly lower growth velocity (−2.7 ± 0.3 vs. −1.5 ± 0.2 SDS; p < 0.001), IGF-I [−2.31 (−3.67 to −0.95) vs. 1.47 (−1.19–2.11) SDS; p < 0.001)] and GH peak after both arginine (4.3 ± 1.1 vs. 10.9 ± 2.6 µg/l; p < 0.004) and glucagon test (5.5 ± 1.1 vs. 16.2 ± 3.7 µg/l; p < 0.001) than controls.
In GHD children after 12 months, we observed a significant increase in growth (height: −1.6 ± 0.6 vs. −2 ± 0.7 SDS; p < 0.001; growth velocity: 1.3 ± 0.4 vs. −2.7 ± 0.3 SDS; p < 0.001), with a concomitant significant increase in IGF-I [0.54 (0.07–1.15) vs. −2.31 (−3.67 to −0.95) SDS; p < 0.001)] and a decrease in BMI [− 0.80 (−2.64–1.25) vs. −0.39 (−2.26–1.81) SDS; p = 0.047)], without significant change in WC SDS (Table 1).
Metabolic parameters
No significant difference was found in fasting glucose (4.6 ± 0.6 vs. 4.4 ± 0.2 mmol/l; p = 0.206), fasting insulin [(2.4 (0.5–15.1) vs. 2.4 (0.8–6.6) IU/ml; p = 0.735)], HbA1c (5.2 ± 0.3 vs. 5.2 ± 0.3 %; p = 0.961) and insulin sensitivity indexes between GHD children at baseline and control subjects. GHD children showed higher LDL cholesterol (2.4 ± 0.6 vs. 1.9 ± 0.1 mmol/l; p = 0.004) than controls, without any difference in triglycerides, total and HDL cholesterol (Table 1).
After 12 months of GH treatment, a significant increase in fasting insulin [6 (1.5–17.4) vs. 2.4 (0.5–15.1) IU/ml; p = 0.008)], and Homa-IR [1.3 (0.2–5.4) vs. 0.5 (0.1–4.2); p = 0.007)] was documented, with a concomitant decrease in LDL cholesterol (2.1 ± 0.6 vs. 2.4 ± 0.6 mmol/l; p = 0.015), QUICKI (0.36 ± 0.05 vs. 0.44 ± 0.08; p = 0.001) and ISI Matsuda [7.1 (2.6–22) vs. 17.2 (6.2–37.7); p = 0.006)]. No significant change was observed in fasting glucose, HbA1c, triglycerides, total and HDL cholesterol (Table 1).
Adipokine levels
Serum adipokine levels of control subjects, GHD children at baseline and after 12 months of GH treatment are shown in Table 2.
At baseline GHD children showed significantly higher resistin [3.7 (1.4–7.8) vs. 3 (1–4.3) ng/ml; p = 0.007)] and lower visfatin [0.4 (0–10.1) vs. 2.9 (0.1–15.1) ng/ml; p = 0.011)] than controls, without significant difference in other adipokine levels.
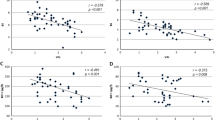
At univariate analysis, resistin was found to be directly correlated with BMI (rho 0.318; p = 0.018) and LDL cholesterol (rho 0.535; p < 0.001) and multivariate analysis confirmed the independent correlation between resistin and LDL cholesterol (B 0.531; p = 0.002) (Fig. 1).

Independent variables influencing resistin, visfatin and leptin levels at multivariate analysis
Conversely, visfatin was found to have a significant inverse correlation with fasting glucose (rho −0.334; p = 0.019) and a direct correlation with IGF-I (rho 0.394; p = 0.021) and total cholesterol (rho 0.325; p = 0.027). IGF-I (B 0.688; p < 0.001) was the only variable independently associated with visfatin at multivariate analysis (Fig. 1).
Adiponectin was directly correlated with ISI Matsuda (rho 0.341; p = 0.045) and QUICKI (rho 0.443; p = 0.005) and negatively with fasting insulin (rho −0.419; p = 0.008) and Homa-IR (rho −0.443; p = 0.005), but none of these variables was independently correlated with adiponectin at multivariate analysis. No significant correlation with metabolic parameters was found for the other adipokines evaluated (data not shown).
After 12 months of treatment, we found a significant decrease in leptin [1 (0.01–7.4) vs. 1.5 (0.1–11.2) ng/ml; p = 0.015)] and omentin levels [283 (168–395) vs. 304 (214–427) ng/ml; p = 0.003)], with a concomitant increase in visfatin (3.4 (0–14.6) vs. 0.4 (0–10.1) ng/ml; p < 0.001). A trend in increase in resistin, although not statistically significant, was found [4.5 (2.4–20.1) vs. 3.7 (1.4–7.8) ng/ml; p = 0.075)], while no significant change was found in sOB-R, adiponectin, resistin, AFABP and RBP4 (Table 2).
At univariate analysis, leptin levels at 12 months were found to be directly correlated with BMI (rho 0.683; p < 0.001), WC (rho 0.670; p < 0.001), fasting insulin (rho 0.449; p = 0.013), Homa-IR (rho 0.591; p = 0.001) and inversely correlated with QUICKI (rho −0.432; p = 0.017). At multivariate analysis, BMI (B 0.485; p = 0.040) was the only variable independently associated with leptin (Fig. 1).
Visfatin at 12 months was found to be correlated with IGF-I (rho 0.572; p = 0.002), while omentin was negatively correlated with fasting insulin (rho −0.484; p = 0.007) and Homa-IR (rho −0.582 p = 0.001) and positively with ISI Matsuda (rho 0.648; p = 0.017). At multivariate analysis, none of the variables was independently associated with omentin. No significant correlation with metabolic parameters was found for the other adipokines evaluated (data not shown).
Discussion
Overall, our results suggest that GH treatment exerts its metabolic effects in different ways. If on the one hand GH favorably affects adipose metabolism, as demonstrated by the improvement in lipid profile, reduction in leptin and increase in visfatin levels, on the other it acts by negatively altering the insulin sensitivity.
In our study, GHD children at diagnosis showed a worse metabolic panel, characterized by an unfavorable lipid profile and higher resistin levels. Several studies report an improvement in lipid profile after GH treatment [4], and our study also confirms this. Moreover, in our study, LDL cholesterol independently correlates with resistin levels, which are higher in GHD children than controls. Indeed, a possible mediator of the GH-modulated insulin sensitivity may be resistin, which has been shown to be linked to obesity and insulin resistance [18]. Higher resistin levels in untreated GHD children than controls have already been documented [19], while the effect of GH treatment is controversial. Our data are in agreement with some studies that have documented that GH treatment does not seem able to strongly modify resistin levels in GHD adults [10, 20], although we found a trend to an increase in resistin levels after 12 months, though not statistically significant. Partially in line with our results, Nozue et al. [21] demonstrated a rise in resistin after short-term GH therapy in GHD children, while López-Siguero et al. [12] showed a decline in resistin at 6 months of GH treatment and these data are in line with those of Meazza et al. [19] who showed a decrease in resistin levels. However, despite the lack of a statistically significant change, the trend to an increase in resistin in our patients during GH treatment is concomitant with a significant reduction in insulin sensitivity, as demonstrated by the decline in QUICKI and ISI Matsuda, in line with previous studies. We already demonstrated through euglycemic hyperinsulinemic clamp a decrease in insulin sensitivity in GHD children after GH treatment, even without evident changes in glucose tolerance [22] as well as a trend toward reduced insulin sensitivity with a compensatory hyperinsulinemic response or increased insulin levels, but with normal glucose levels, after GH treatment have been demonstrated [23]. In our opinion, the lack of changes in resistin levels, despite the increase in insulin resistance indexes, can be explained by the other significant changes that occur during GH treatment and that may impact the metabolic balance, such as the beneficial effects of GH on body composition and leptin levels. In our study, despite the lack of difference in leptin, sOB-R, BMI and WC between GHD children at baseline and controls, we found a significant decrease in leptin after GH treatment with an independent correlation with BMI, as demonstrated by other studies [24]. Notably, in this study, visfatin has proved to be the only adipokine to directly correlate with IGF-I levels. Visfatin is thought to have insulin-mimetic effects in various tissues [25] and it seems to be affected by weight loss, as demonstrated by Petelin et al. [26] in overweight subjects. Li et al. [27] showed higher visfatin levels in adults with GHD than controls, but to date data on visfatin during GH treatment are not available. We found lower visfatin in GHD children than controls at baseline and a rise in its levels after GH treatment, concomitantly with the increase in IGF-I. These data are partially in agreement with our previous data. Indeed, in acromegalic patients, we recently showed a correlation between visfatin, insulin sensitivity and IGF-1 levels, concluding that visfatin in acromegaly could be considered a useful index of disease activity [28]. Therefore, the increase in visfatin in GHD children could represent a favorable metabolic effect of GH treatment, correlated with the IGF-I increase and independent of the deterioration in insulin sensitivity. Conversely, we found no difference in AFABP and RBP4 levels, parameters known to be related with metabolic syndrome, cardiovascular risk factors and insulin resistance [29, 30], between GHD children and controls, and no significant change during GH treatment. These findings could be in line with some clinical studies which failed to show a relationship with metabolic parameters [31].
As regards adiponectin levels in GHD patients, the existing data are quite controversial. Adiponectin seems to be unaffected in untreated GHD [20], and GH treatment seems to have discordant effects on adiponectin, increasing it [12] or leaving it unchanged or slightly modifying it [10, 11]. We found a significant correlation between adiponectin and insulin sensitivity indexes at baseline, in line with other studies [32]. However, our data also confirm the unchanged adiponectin levels after GH treatment.
Finally, although no significant difference was found at baseline between GHD and controls, we surprisingly found a significant reduction in omentin levels after 12 months of treatment. It has been determined that omentin enhances insulin-stimulated glucose uptake in adipose tissue and many studies have shown that omentin is negatively correlated with BMI and fat mass, insulin resistance and metabolic syndrome [33, 34]. Our data are partially in agreement with these findings. Indeed, a negative correlation between omentin and insulin resistance indexes was found after GH treatment, although it was not confirmed at multivariate analysis.
In conclusion, if this study is quite confirmatory for many of the parameters investigated, such as insulin sensitivity and leptin levels, we showed for the first time the behavior of new selected adipokines, known to have a metabolic role and never investigated before in this setting, and their correlation with some metabolic and hormonal parameters. The action of GH could modify adipokine secretion, and the perturbation of some adipokine levels correlates with metabolic impairment. If resistin and omentin resulted differently correlated with some metabolic parameters, respectively, at baseline and after GH treatment, conversely visfatin proved to be strongly correlated with the hormonal target in GHD subjects (as IGF-I), and leptin with body mass. In addition, during GH treatment, the favorable metabolic effects seem to be represented by a decrease in leptin and an increase in visfatin, while the negative effects seem to be represented by a decrease in omentin.
The main limitation of this study may be related to the small size of the population studied and to the short-term follow-up. In our opinion, these factors could be responsible for the lack of statistical significance in the evaluation of some adipokines. To better understand whether all the above-mentioned adipokines may represent a metabolic biomarker useful to identify the progression toward metabolic abnormalities associated with GH treatment in GHD children, we believe that these data must be validated in additional larger prospective studies with longer follow-up, where patients are randomized to different GH doses.
References
Vijayakumar A, Novosyadlyy R, Wu Y, Yakar S et al (2010) Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm IGF Res 20(1):1–7
Cantley J (2014) The control of insulin secretion by adipokines: current evidence for adipocyte-beta cell endocrine signalling in metabolic homeostasis. Mamm Genome 2:442–454
Capalbo D, Esposito A, Di Mase R et al (2012) Update on early cardiovascular and metabolic risk factors in children and adolescents affected with growth hormone deficiency. Minerva Endocrinol 37:379–389
Salerno M, Esposito V, Farina V et al (2006) Improvement of cardiac performance and cardiovascular risk factors in children with GH deficiency after 2 years of GH replacement therapy: an observational, open, prospective, case-control study. J Clin Endocrinol Metab 91:1288–1295
van der Sluis IM, Boot AM, Hop WC et al (2002) Long-term effects of growth hormone therapy on bone mineral density, body composition, and serum lipid levels in growth hormone deficient children: a 6-year follow-up study. Horm Res 58:207–214
Nørrelund H, Gravholt CH, Englaro P et al (1998) Increased levels but preserved diurnal variation of serum leptin in GH-deficient patients: lack of impact of different modes of GH administration. Eur J Endocrinol 138:644–652
Elimam A, Lindgren AC, Norgren S et al (1999) Growth hormone treatment downregulates serum leptin levels in children independent of changes in body mass index. Horm Res 52:66–72
Gill MS, Toogood AA, Jones J et al (1999) Serum leptin response to the acute and chronic administration of growth hormone (GH) to elderly subjects with GH deficiency. J Clin Endocrinol Metab 84:1288–1295
Edén Engström B, Burman P, Holdstock C et al (2003) Effects of growth hormone (GH) on ghrelin, leptin, and adiponectin in GH-deficient patients. J Clin Endocrinol Metab 88:5193–5198
Hana V, Silha JV, Justova V et al (2004) The effects of GH replacement in adult GH-deficient patients: changes in body composition without concomitant changes in the adipokines and insulin resistance. Clin Endocrinol (Oxf) 60:442–450
Ciresi A, Amato MC, Criscimanna A et al (2007) Metabolic parameters and adipokine profile during GH replacement therapy in children with GH deficiency. Eur J Endocrinol 156:353–360
López-Siguero JP, López-Canti LF, Espino R et al (2011) Effect of recombinant growth hormone on leptin, adiponectin, resistin, interleukin-6, tumor necrosis factor-α and ghrelin levels in growth hormone-deficient children. J Endocrinol Invest 34:300–306
Growth Hormone Research Society (2000) Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin Endocrinol Metab 85:3990–3993
Greulich W, Pyle S (1959) Radiographic atlas of skeletal development of the hand and wrist, 2nd edn. Stanford University Press, Stanford
Matthews DR, Hosker JP, Rudenski AS et al (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Katz A, Nambi SS, Mather K et al (2000) Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab 85:2402–2410
Matsuda M, DeFronzo RA (1999) Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22:1462–1470
Shuldiner AR, Yang R, Gong DW (2001) Resistin, obesity and insulin resistance–the emerging role of the adipocyte as an endocrine organ. N Engl J Med 345:1345–1346
Meazza C, Elsedfy HH, Pagani S et al (2014) Metabolic parameters and adipokine profile in growth hormone deficient (GHD) children before and after 12-month GH treatment. Horm Metab Res 46:219–223
Svensson J, Herlitz H, Lundberg PA et al (2005) Adiponectin, leptin, and erythrocyte sodium/lithium countertransport activity, but not resistin, are related to glucose metabolism in growth hormone-deficient adults. J Clin Endocrinol Metab 90:2290–2296
Nozue H, Kamoda T, Matsui A (2007) Serum resistin concentrations in growth hormone-deficient children during growth hormone replacement therapy. Metabolism 56:1514–1517
Ciresi A, Amato MC, Giordano C (2015) Reduction in insulin sensitivity and inadequate β-cell capacity to counteract the increase in insulin resistance in children with idiopathic growth hormone deficiency during 12 months of growth hormone treatment. J Endocrinol Invest 38:351–359
Heptulla RA, Boulware SD, Caprio S et al (1997) Decreased insulin sensitivity and compensatory hyperinsulinemia after hormone treatment in children with short stature. J Clin Endocrinol Metab 82:3234–3238
Rauch F, Westermann F, Englaro P et al (1998) Serum leptin is suppressed by growth hormone therapy in growth hormone-deficient children. Horm Res 50:18–21
Fukuhara A, Matsuda M, Nishizawa M et al (2005) Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 307:426–430
Petelin A, Bizjak M, Černelič-Bizjak M et al (2014) Low-grade inflammation in overweight and obese adults is affected by weight loss program. J Endocrinol Invest 37(8):745–755
Li ZP, Zhang M, Gao J et al (2014) Study of the correlation between growth hormone deficiency and serum leptin, adiponectin, and visfatin levels in adults. Genet Mol Res 13:4050–4056
Ciresi A, Amato MC, Pizzolanti G et al (2015) Serum visfatin levels in acromegaly: correlation with disease activity and metabolic alterations. Growth Horm IGF Res 25:240–246
Kralisch S, Fasshauer M (2013) Adipocyte fatty acid binding protein: a novel adipokine involved in the pathogenesis of metabolic and vascular disease? Diabetologia 56(1):10–21
Graham TE, Yang Q, Blüher M et al (2006) Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med 354:2552–2563
von Eynatten M, Humpert PM (2008) Retinol-binding protein-4 in experimental and clinical metabolic disease. Expert Rev Mol Diagn 8:289–299
Lanes R, Soros A, Gunczler P et al (2006) Growth hormone deficiency, low levels of adiponectin, and unfavorable plasma lipid and lipoproteins. J Pediatr 149:324–329
Jaikanth C, Gurumurthy P, Cherian KM et al (2013) Emergence of omentin as a pleiotropic adipocytokine. Exp Clin Endocrinol Diabetes 121:377–383
Li XP, Zeng S, Wang M et al (2014) Relationships between serum omentin-1, body fat mass and bone mineral density in healthy Chinese male adults in Changsha area. J Endocrinol Invest 37(10):991–1000
Acknowledgments
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Ethical approval
All procedures performed were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent for the scientific use of the data was obtained from both the participants included in the study and their parents.
Rights and permissions
About this article

Cite this article
Ciresi, A., Pizzolanti, G., Leotta, M. et al. Resistin, visfatin, leptin and omentin are differently related to hormonal and metabolic parameters in growth hormone-deficient children. J Endocrinol Invest 39, 1023–1030 (2016). https://doi.org/10.1007/s40618-016-0475-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-016-0475-z