
Abstract
Introduction
Cushing’s disease (CD) results from uncontrolled hypercortisolism induced by ACTH-secreting corticotroph adenomas; accordingly, patients diagnosed with CD usually present several comorbidities and an increased risk of mortality. Hypothesis-driven screenings have led to identification of rare alterations in a low number of patients, although the genetic basis underlying CD has remained unclear until recently. Using whole-exome sequencing, recurrent mutations have been reported in the gene coding for the ubiquitin-specific protease 8 (USP8), a protein with deubiquitinase (DUB) activity that modulates the lysosomal turnover of the EGF receptor (EGFR) and other membrane proteins.
Methods
In this review, we summarize the recent genetic findings and discuss the clinical and pathological implications of USP8 deregulation in corticotroph adenomas.
Conclusions
Mutations in USP8 have been identified in 35–62 % of functional sporadic corticotroph adenomas causing Cushing’s disease, but not in any other type of pituitary tumor. These mutations are found mostly in adult female patients and lead to an aberrant DUB activation by impairing the regulation of USP8 by members of the 14-3-3 family of proteins. The consequence of this hyperactivation is a longer retention of EGFR at the plasma membrane which promotes an enhanced production of ACTH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Endogenous Cushing’s syndrome is the consequence of the chronic exposure to high levels of cortisol produced by the adrenal glands. Most cases of Cushing’s syndrome are caused by hypersecretion of adrenocorticotropic hormone (ACTH) from corticotroph adenomas of the anterior pituitary, known as Cushing’s disease (CD). Thus, the clinical manifestations of CD correspond to the typical signs and symptoms of hypercortisolism, such as accumulation of central fat, moon face, muscle weakness, skin lesions, bone fractures, impairment of immune function and the development of hyperlipidemia, hypertension and diabetes. When untreated, patients with Cushing’s syndrome have an increased risk of mortality, mainly due to their susceptibility to infections, cardiovascular failures and stroke [1, 2].
Pituitary tumors are relatively common lesions, as revealed by epidemiologic studies based on imaging techniques and autopsy examinations [3], despite most of them are silent and clinically irrelevant. Corticotroph adenomas account for ~10 % of them, although the prevalence might be higher due to the difficulties in the detection that frequently implies a delay in the diagnosis of CD. The incidence of Cushing’s disease varies with sex and age. Female patients are three to four times more likely to develop CD; however, the sex ratio is equal in children, with a male preponderance under the age of ten [4, 5] that turns into a female preponderance during the adolescence and becomes much more accentuated in the adulthood [6, 7].
Genetics of Cushing’s disease
Pituitary adenomas are considered benign lesions, most of them sporadic, that arise from the clonal expansion of a single cell containing one or few mutations that confer particular adaptive advantages [8–10]. Based on that premise, multiple efforts have been done to identify causative mutations in pituitary adenomas, including functional ACTH-producing corticotroph tumors.
Screening for known mutations associated with other endocrine pathologies and cancers has been useful to detect uncommon variants of CD but was unsuccessful for identifying recurrent mutations. Rarely, CD can appear as part of a familial disorder, such as the multiple endocrine neoplasia type 1, an autosomal dominant disease caused by germline inactivating mutations in the gene MEN1. Pituitary adenomas occur in ~40 % of cases and represent predominantly prolactinomas; corticotroph adenomas, conversely, are very infrequent in this context. In a cohort of 78 pediatric patients with CD, three cases with history of familial disease (two harboring MEN1 mutations and one with TSC2 mutations) were positive for germline mutations, but none of the sporadic cases [11]. Regarding the related syndrome of MEN4, a very rare condition caused by mutations in CDKN1B, so far only one patient with ACTH-producing corticotroph adenoma has been reported [12]. Another candidate gene has been the aryl hydrocarbon receptor interacting protein AIP, which has been found mutated in ~20 % of familial isolated pituitary adenomas predisposing to acromegaly and gigantism. In pituitary adenomas with sporadic origin, however, AIP mutations are rare and do not seem to be involved in CD [13]. In a large, prospective study including 443 patients with sporadic pituitary adenomas and without familial history, AIP germline mutations were found only in 3.6 % of all cases including three of 44 patients with CD, one pediatric male and two adult female patients [14]. Similarly, 3 % of mutations in AIP have been described in another study with 127 sporadic patients, but none of them had a corticotroph adenoma causing CD [15].
Somatic mosaicism for gain-of-function of the Gα subunit GNAS is associated with McCune–Albright syndrome. Several recurrent mutations in GNAS1 have been found in ~40 % of sporadic somatotroph adenomas and occasionally other phenotypes, but so far only two patients from a series of 32 corticotroph adenomas had a somatic mutation in the GNAS1 gene [16]. In addition, other rare germline mutations found in CD include succinate dehydrogenase subunit [17], DAX-1 mutations in the context of X-linked congenital adrenal hypoplasia [18] and DICER-1 mutations in children presenting with pituitary blastoma [19]. Rare somatic events have been found in one patient with Nelson’s syndrome and TP53 mutations [20] and glucocorticoid receptor [21]. PRKAR1A, PDE11A, PDE8B or the recently described PRKACA have been screened in corticotroph adenomas but no mutation has been found.
In summary, the identification of recurrent causative mutations has been unsuccessful for the vast majority of corticotroph tumors with sporadic origin and the genetic basis underlying CD has remained elusive for a long time.
USP8 is recurrently mutated in Cushing’s disease
Next-generation sequencing technologies are very useful tools to decipher the genetic events driving multiple diseases; for example, we have taken advantage of whole-exome sequencing to detect causative genetic alterations in different endocrine diseases, such as primary aldosteronism [22] or Cushing’s syndrome produced by sporadic adrenal adenomas [23]. Using this approach, we have search for somatic mutations in corticotroph adenomas from patients with CD and we have identified recurrent somatic mutations in the gene encoding the ubiquitin-specific protease 8 (USP8) in four in an initial set of ten tumors. These mutations were validated in a small group of seven patients, with a final prevalence of 35 % (6/17 tumors) [24]. Of note, all of them were located in exon 14, defining a hotspot region that overlaps with the sequence that codes for the 14-3-3 binding motif, highly conserved between different species [24].
Subsequently, two different retrospective studies have analyzed the prevalence of USP8 mutations in corticotroph adenomas in two large cohorts of patients with CD [25, 26]. We have designed a multicentric study to analyze the rate of USP8 mutations including 134 patients with CD from seven different participating centers in Europe, Brazil and the United States. Similar to our first report, we have found a prevalence of 36 % (48/134) [25]. Ma and coworkers have reported a higher frequency of mutations using exome sequencing (8/12 corticotroph adenomas, 67 %) and Sanger sequencing in 108 additional adenomas from Chinese patients with CD, with a total prevalence of 62 % [26]. In addition to possible effects of ethnic diversity in the genetic background, more restrictive diagnostic and inclusion criteria could be an explanation for this difference.
Together with the high prevalence of USP8 alterations, we should consider that (a) the average number of non-synonymous mutations per case was low in both exome-sequencing studies (median 7, range 3–23, reported in Reincke et al.; and median 5, range 1–9, reported in Ma et al.), (b) mutations in genes previously associated with adrenal Cushing’s syndrome were not identified, (c) recurrent mutations were detected only in USP8, (d) excluding synonymous changes, USP8 was the only mutated gene in some adenomas [26], and (e) changes in USP8 were absent in 11 silent (non-secreting) corticotroph adenomas [25] as well as 36 [24] and 150 [26] samples of other types of pituitary adenomas. Altogether, these observations strongly support the evidence that USP8 plays a starring role in the development of the CD.
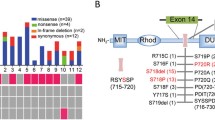
In total, 22 different USP8 mutations have been reported in 129 functional corticotroph adenomas of 271 patients in these three studies (summarized in Fig. 1), comprising eight single missense, eight deletions and six different double mutations. All of them were in frame mutations found in heterozygosis and located in the exon 14, either in the hotspot previously described or very close to it. The analysis of available paired blood samples revealed that mutations were somatic. Strikingly, 94.5 % (122/129) of all the events targeted only two residues recurrently, Ser718 and Pro720, with two substitutions (p.Pro720Arg and p.Ser718Pro) and one deletion (p.Ser718del) accounting for 85.2 % of them (104/122).

Mutations in USP8 identified in corticotroph adenomas. Diagram showing the hotspot region including the changes of the different mutations so far identified in the amino acid sequence (white letters over red background) in the context of the complete protein. On the right, the overall mutation frequency of the three series is shown. Yellow box amino acid sequence of the 14-3-3 binding motif. MIT microtubule-interacting and trafficking domain, RHOD rhodanese-like domain, SBM SH3-binding motif, DUB deubiquitinase catalytic domain
The molecular effects as well as the clinical and pathologic implications of these mutations will be discussed in the next sections.
How does USP8 promote Cushing’s disease?
USP8 regulates the recycling of EGFR
The gene USP8 codes for an enzyme of ~130 kDa with deubiquitinase (DUB) activity, i.e., cleaves ubiquitin peptides from target proteins. This activity is tightly modulated by members of the 14-3-3 family of proteins [27], highly conserved proteins involved in many intracellular processes. The 14-3-3 binding motif RSYpSSP present in USP8—where pS is a phosphorylated serine corresponding to Ser718 in humans and Ser680 in mice—is critical for the recognition and interaction between both proteins. Phosphorylation of this residue enables binding and retains USP8 inactive in the cytosol. On the contrary, its dephosphorylation leads to the release of 14-3-3 and the consequent activation of USP8 [27]. Similarly, mutation of the 14-3-3 binding motif reduces or even abolishes the interaction between USP8 and 14-3-3.
USP8 was originally postulated as a regulator of cell growth because its expression increases upon serum stimulation and the depletion of USP8 activity results in embryonic lethality in knock-out mice [28, 29]. Indeed, the pSer680 remains phosphorylated during interphase in mice cells and undergoes dephosphorylation in the M phase [27]. But the best known function of USP8 is the regulation of endosome sorting of different membrane proteins, mainly tyrosine-kinase receptors (RTKs). Several substrates of USP8 have been identified, including Nrdp1, c-Met, HER2 and the epidermal growth factor receptor (EGFR) [29–32]. EGFR and other RTKs are subject to fine negative regulatory mechanisms to prevent sustained activation that could lead to potentially damaging consequences [33]. Thus, in addition to trigger receptor activation, ligand binding induces EGFR internalization and polyubiquitination, a post-translational modification that targets the receptor to be degraded in the lysosome [34]. Conversely, USP8-mediated deubiquitination abrogates this process and directs EGFR back to the plasma membrane, where it can further contribute to signal transduction [35]. When hyperactivation of USP8 occurs by overexpression or mutation, ligand-induced degradation of EGFR is delayed and residence of potentially active receptor in the plasma membrane is prolonged [35].
EGFR promotes ACTH synthesis in corticotroph cells
Because EGFR is involved in cell growth and proliferation, and because uncontrolled EGFR activation is associated with different malignancies, several authors have investigated the presence of EGFR in normal pituitary gland, adenomas and pituitary carcinomas with the aim of finding a causative link between EGFR expression and tumorigenesis in the pituitary. EGFR is expressed in the normal pituitary, although at low levels [36–38]. Likewise, EGFR has been detected in ~60 % of adenomas at either the mRNA or the protein level. Its expression is not restricted to any specific tumor type and a variable immunoreactivity has been observed within each type [38] with the highest expression in corticotroph adenomas [37].
Evidence confirms the presence and functionality of EGFR in corticotroph cells. For example, sustained stimulation by EGF—that can be also be detected in ACTH-producing adenomas—induces the replication of corticotroph and mammotroph cells in primary cultures of mouse pituitary [39]. IHC staining using two antibodies against intracellular and extracellular epitopes of EGFR revealed that this receptor presents a moderate-to-high expression in corticotroph adenomas that also co-localizes with ACTH [37]. Moreover, Fukouka et al. demonstrated that EGFR can enhance POMC expression (the precursor of ACTH) and ACTH secretion in corticotroph tumors using canine and murine models. Likewise, treatment with gefitinib abolished EGFR signaling and blocked POMC expression in corticotroph cells, reduced corticotroph tumor mass and reversed the signs of hypercortisolism in mice [40].
Completing the puzzle: USP8 increases ACTH synthesis in an EGFR-depending fashion
Functional assays have provided solid evidences on how mutated USP8 can be involved in the development of CD [24–26]. As mentioned before, all the mutations detected so far in the sequence of USP8 by our group and by Ma and coworkers clustered in a hotspot region containing the highly conserved 14-3-3 binding motif (Fig. 2). Consequently, these mutants exhibit a reduced interaction with 14-3-3 proteins and higher DUB activity than the wild-type form, although at different degrees [24–26]. Likewise, EGFR deubiquitination is increased in cells expressing USP8 mutants, resulting in recycling of activated EGFR from early endosomes and the accumulation of the receptor in the plasma membrane upon stimulation by EGF [24–26].

USP8 modulates EGFR signaling and ACTH production. a In normal corticotroph cells, the stimulation of EGFR signaling (1) by EGFR ligands (such as EGF) promotes Erk1/2-mediated activation of transcription factors and the subsequent transcription of POMC (2), the precursor of ACTH. After activation, EGFR is internalized in endosome vesicles and covalently linked to ubiquitin chains (3); this ubiquitination targets EGFR to lysosomal degradation (4). USP8 regulates EGFR turnover by removing the chains of ubiquitin from the receptor; then, untargeted EGFR turns back to the plasma membrane via recycling endosomes (5). USP8 activity is controlled by the interaction with 14-3-3 proteins, which retain USP8 in an inactive state that is reversible (6). b Mutations found in corticotroph adenomas affect the binding of USP8 to 14-3-3. Thus, mutant USP8 proteins are constitutively active and reduce the amount of ubiquitin-ligated EGFR in the endosomes (7). Consequently, EGFR degradation is impaired (8) and the receptor is accumulated in the plasma membrane (9), increasing signal transduction and POMC transcription (10)
The mechanism underlying EGFR-dependent POMC transcription has been suggested by Fukuoka et al. and further supported by Ma et al. and our data. The stabilization of the receptor in the plasma membrane enhances the EGFR-dependent signal transduction in the presence of EGF, leading to the activation of downstream kinases, such as Erk1/2 and Akt. Experiments with kinase inhibitors have shown that POMC transcription is triggered by Erk1/2-mediated activation of transcription factors that bind to the AP-1 response element on the POMC promoter [24, 40]. The final evidence linking ACTH secretion to USP8 activity has been provided by experiments in primary cultures of human corticotroph cells. Silencing USP8 expression using shRNA significantly reduced the amount of secreted ACTH in cells originated from adenomas with mutated USP8 but not in those from a wild-type adenoma [26]. In the same line, ACTH secretion is reduced after treatment with gefitinb only in corticotroph cells from mutated human adenomas and not from those with wild-type USP8 [26]. These experiments demonstrate the dependence of corticotroph adenomas cells on USP8 hyperactivation via EGFR signaling and might provide a rationale for personalized treatment strategies for these patients in the future.
From genotype to phenotype: clinical and pathologic implications
The development of CD is the logical consequence of sustained EGFR activation and ACTH production in the context of activating USP8 mutations in corticotroph cells. To better understand the pathophysiological effects of these mutations, we will summarize the most relevant associations reported in the three series of patients published so far. Nevertheless, one should be aware that the retrospective nature of the studies, the lack of complete registries of pathologic and hormonal data, the differences in the diagnostic protocols and postoperative management, and intrinsic features of the study cohorts constitute important limitations.
Some observations are strongly consistent among the three publications. For example, the frequency of mutations in corticotroph adenomas is surprisingly high in all series. Moreover, these mutations have only been detected in functional ACTH-producing corticotroph adenomas, but not in any other pituitary tumor (in total, n = 197), including silent corticotroph adenomas [24–26]. These facts clearly demonstrate the relevance of USP8 mutations, which constitute a specific trait of CD.
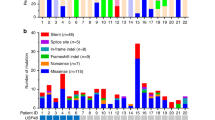
Patients who develop tumors with USP8 mutations are mainly female. The analysis of the data from the three series shows that the risk of having an USP8-mutant adenoma is two times higher in females: 55.3 % (115/208) in female vs. 22.2 % (14/63) in male patients. In addition, most mutations in females were found at the ages of 20–40 years old, while tumors in children and adults diagnosed >50 years were mostly wild type (Fig. 3). It is tempting to speculate that tumors harboring USP8 mutations could be more sensitive to the effect of sex hormones, but this aspect requires further research.

Sex and age prevalence of USP8 mutations in patients diagnosed with CD. Distribution of cases according to sex (females, top graph; males, bottom graph) and age at diagnosis. Data are represented as % of cases diagnosed in each category in 5-year intervals and have been obtained from Perez-Rivas et al. [25] and the supplementary tables of Reincke et al. [24] and Ma et al. [26]
Average tumor size in each series was different and it is not clear whether adenomas with mutations in USP8 are smaller or larger than those with wild-type sequence. In our series, tumors in females with USP8 mutations were larger that their wild-type counterpart (8 vs. 10 mm, respectively) [25], while Ma and coworkers showed a predominance of mutations in smaller adenomas (<5 mm) [26]. However, large tumors (>20 mm) in both series were found mostly wild type, suggesting that USP8 hyperactivation influences hormonal production rather than tumor growth. In line with this hypothesis, we have observed that the proliferative effects of USP8 in AtT-20 mouse cells coexpressing EGFR are not further enhanced by the mutant proteins [24]. Even though the involvement of USP8 in this process is apparent—cell growth is increased after USP8 transfection in AtT-20 expressing EGFR, and the lack of USP8 activity leads to cell cycle arrest and a strong reduction of EGFR levels [24, 28, 29]—the indirect upregulation of EGFR signaling by altering a modulator protein might result in less mitogenic activity than direct alterations of the receptor by either overexpression or aggressive EGFR-activating mutations commonly found in other types of tumors. This hypothesis could feasibly explain the low mitotic index of most corticotroph adenomas despite exhibiting aberrant EGFR signaling, and the lack of oncogenic mutations in these tumors, including those affecting genes downstream to EGFR, usually related to a malignant phenotype.
Regarding hormonal features, few associations have been reported and none of them has been corroborated by the other studies. Basal plasma ACTH and morning serum cortisol do not seem to correlate with USP8 mutations, even though we found lower levels of ACTH in our discovery study [24]. Ma and coworkers have shown that USP8 tumors have a higher ratio of ACHT/tumor size, suggesting that these tumors are hormonally more active [26]. In our last study, we have reported that postoperative 24 h urinary-free cortisol levels are higher in patients that had adenomas with mutations in USP8 and are less likely to develop adrenal insufficiency than those with wild-type adenomas. Because postoperative hypocortisolemia is considered a predictive marker of long-term remission, we hypothesize that the former patients could be on a higher risk of recurrence [25]. However, all these observations still need to be validated by further prospective, controlled studies.
Conclusions
In summary, recurrent somatic hotspot mutations in the gene of the ubiquitin-specific protease 8 (USP8) are frequently observed in corticotroph adenomas causing CD, but not in any other pituitary tumor entity. Mutations usually appear in young adult females and only occasionally in men, but further studies are needed to clarify their clinical implication. All the mutations described so far target the 14-3-3 binding domain of USP8 and generate proteins with constitutive DUB activity leading to EGFR stabilization in the plasma membrane. In consequence, mutant USP8 enhances EGFR signaling, which promotes ACTH synthesis in corticotroph cells. Over time, this mechanism could lead to the development of hormonal active adenomas causing CD.
Abbreviations
- CD:
-
Cushing’s disease
- ACTH:
-
Adrenocorticotropic hormone
- POMC:
-
Pro-opiomelacortin
- USP8:
-
Ubiquitin-specific protease 8
- DUB:
-
Deubiquitinase
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
References
Lacroix A, Feelders RA, Stratakis CA, Nieman LK (2015) Cushing’s syndrome. Lancet 6736:1–15
Sharma ST, Nieman LK, Feelders RA (2015) Comorbidities in Cushing’s disease. Pituitary 18:188–194
Ezzat S, Asa SL, Couldwell WT et al (2004) The prevalence of pituitary adenomas: a systematic review. Cancer 101:613–619
Libuit LG, Karageorgiadis AS, Sinaii N et al (2015) A gender-dependent analysis of Cushing’s disease in childhood: pre-and postoperative follow-up. Clin Endocrinol 83:72–77
Storr HL, Isidori AM, Monson JP et al (2004) Prepubertal Cushing’s disease is more common in males, but there is no increase in severity at diagnosis. J Clin Endocrinol Metab 89:3818–3820
Storr HL, Alexandraki KI, Martin L et al (2011) Comparisons in the epidemiology, diagnostic features and cure rate by transsphenoidal surgery between paediatric and adult-onset Cushing’s disease. Eur J Endocrinol 164:667–674
Lonser RR, Wind JJ, Nieman LK et al (2013) Outcome of surgical treatment of 200 children with Cushing’s disease. J Clin Endocrinol Metab 98:892–901
Gicquel C, Le Bouc Y, Luton JP et al (1992) Monoclonality of corticotroph macroadenomas in Cushing’s disease. J Clin Endocrinol Metab 75:472–475
Biller BM, Alexander JM, Zervas NT et al (1992) Clonal origins of adrenocorticotropin-secreting pituitary tissue in Cushing’s disease. J Clin Endocrinol Metab 75:1303–1309
Schulte HM, Oldfield EH, Allolio B et al (1991) Clonal composition of pituitary adenomas in patients with Cushing’s disease: determination by X-chromosome inactivation analysis. J Clin Endocrinol Metab 73:1302–1308
Stratakis CA, Tichomirowa MA, Boikos S et al (2010) The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin Genet 78:457–463
Georgitsi M, Raitila A, Karhu A et al (2007) Brief report: germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab 92:3321–3325
Korbonits M, Storr HL, Kumar AV (2012) Familial pituitary adenomas—who should be tested for AIP mutations? Clin Endocrinol (Oxf) 77:351–356
Cazabat L, Bouligand J, Salenave S et al (2012) Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab 97:663–670
Preda V, Korbonits M, Cudlip S et al (2014) Low rate of germline AIP mutations in patients with apparently sporadic pituitary adenomas before the age of 40: a single-centre adult cohort. Eur J Endocrinol 171:659–666
Williamson EA, Ince PG, Harrison D et al (1995) G-protein mutations in human pituitary adrenocorticotrophic hormone-secreting adenomas. Eur J Clin Invest 25:128–131
Xekouki P, Stratakis CA (2012) Succinate dehydrogenase (SDHx) mutations in pituitary tumors: could this be a new role for mitochondrial complex II and/or Krebs cycle defects? Endocr Relat Cancer 19:33–40
De Menis E, Roncaroli F, Calvari V et al (2005) Corticotroph adenoma of the pituitary in a patient with X-linked adrenal hypoplasia congenita due to a novel mutation of the DAX-1 gene. Eur J Endocrinol 153:211–215
De Kock L, Sabbaghian N, Plourde F et al (2014) Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol 128:111–122
Pinto EM, Siqueira SA, Cukier P et al (2011) Possible role of a radiation-induced p53 mutation in a Nelson’s syndrome patient with a fatal outcome. Pituitary 14:400–404
Karl M, Lamberts SW, Koper JW et al (1996) Cushing’s disease preceded by generalized glucocorticoid resistance: clinical consequences of a novel, dominant-negative glucocorticoid receptor mutation. Proc Assoc Am Physicians 108:296–307
Beuschlein F, Boulkroun S, Osswald A et al (2013) Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 45:440–444
Beuschlein F, Fassnacht M, Assié G et al (2014) Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med 370:1019–1028
Reincke M, Sbiera S, Hayakawa A et al (2014) Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet 47:31–38
Pérez-Rivas LG, Theodoropoulou M, Ferraù F et al (2015) The gene of the ubiquitin-specific protease 8 is frequently mutated in adenomas causing Cushing’s disease. J Clin Endocrinol Metab 100:E997–E1004
Ma ZY, Song ZJ, Chen JH et al (2015) Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res 25:306–317
Mizuno E, Kitamura N, Komada M (2007) 14-3-3-dependent inhibition of the deubiquitinating activity of UBPY and its cancellation in the M phase. Exp Cell Res 313:3624–3634
Naviglio S (1998) UBPY: a growth-regulated human ubiquitin isopeptidase. EMBO J 17:3241–3250
Niendorf S, Oksche A, Kisser A et al (2007) Essential role of ubiquitin-specific protease 8 for receptor tyrosine kinase stability and endocytic trafficking in vivo. Mol Cell Biol 27:5029–5039
Cao Z, Wu X, Yen L et al (2007) Neuregulin-induced ErbB3 downregulation is mediated by a protein stability cascade involving the E3 ubiquitin ligase Nrdp1. Mol Cell Biol 27:2180–2188
Byun S, Lee SYY, Lee J et al (2013) USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin Cancer Res 19:3894–3904
Meijer IMJ, van Leeuwen JEM (2011) ERBB2 is a target for USP8-mediated deubiquitination. Cell Signal 23:458–467
Goh LK, Sorkin A (2013) Endocytosis of receptor tyrosine kinases. Cold Spring Harb Perspect Biol 5:a017459
Avraham R, Yarden Y (2011) Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol 12:104–117
Mizuno E, Iura T, Mukai A et al (2005) Regulation of epidermal growth factor receptor down-regulation by UBPY-mediated deubiquitination at endosomes. Mol Biol Cell 16:5163–5174
Kontogeorgos G, Stefaneanu L, Kovacs K, Cheng Z (1996) Localization of epidermal growth factor (EGF) and epidermal growth factor receptor (EGFr) in human pituitary adenomas and nontumorous pituitaries: an immunocytochemical study. Endocr Pathol 7:63–70
Theodoropoulou M, Arzberger T, Gruebler Y et al (2004) Expression of epidermal growth factor receptor in neoplastic pituitary cells: evidence for a role in corticotropinoma cells. J Endocrinol 183:385–394
Onguru O, Scheithauer BW, Kovacs K et al (2004) Analysis of epidermal growth factor receptor and activated epidermal growth factor receptor expression in pituitary adenomas and carcinomas. Mod Pathol 17:772–780
Honda J, Oomizu S, Kiuchi Y et al (2000) Identification of epidermal growth factor mRNA-expressing cells in the mouse anterior pituitary. Neuroendocrinology 71:155–162
Fukuoka H, Cooper O, Ben-Shlomo A et al (2011) EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J Clin Invest 121:4712–4721
Acknowledgments
M.R. is supported by the Else Kröner-Fresenius-Stiftung (Grant # 2012_A103) and the German Research Foundation (DFG, Grant # RE 752/20-1). L.G.P.R. is supported by funds from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007–2013) under REA Grant Agreement No. 608765.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have nothing to disclose.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Informed consent
For this type of study formal consent is not required.
Rights and permissions
About this article

Cite this article
Perez-Rivas, L.G., Reincke, M. Genetics of Cushing’s disease: an update. J Endocrinol Invest 39, 29–35 (2016). https://doi.org/10.1007/s40618-015-0353-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-015-0353-0