
Abstract
Purpose of Review
Bone marrow adipose tissue (BMAT) is a distinct adipose tissue with diverse local and systemic effects, affecting both physiological processes and pathological conditions, including hematopoiesis, bone remodeling, osteoporosis, obesity, anorexia nervosa, diabetes, and cancer. BMAT increases with age and bone loss, while the significance of this phenomenon has been neglected until recently. Bone cells and BMAT are mutually connected in terms of bone remodeling and energy metabolism. It has been suggested that high BMAT is caused by a shift in bone marrow mesenchymal stromal cell (BMSC) differentiation in favor of adipogenesis, and BMAT promotes bone loss through direct or indirect interaction with bone cells. However, it remains unclear why osteoporosis accelerates BMAT accumulation and what is the role of BMAT in bone remodeling and particularly in bone loss. The purpose of this review is to present the latest published data on the role of BMAT in physiological bone processes and during osteoporosis progression.
Recent Findings
BMAT secretes numerous endocrine factors designated as adipokines as well as pro-inflammatory cytokines, which affect bone homeostasis through the regulation of osteoblast and osteoclast function. Most clinical data from osteoporotic patients demonstrate a negative relationship between BMAT and bone mass. Through technological advances in BMAT imaging, investigators are now able to quantify BMAT in humans and animal models. Pharmaceutical interventions targeting either bone loss or BMA expansion shed light in the understanding of the possible interactions between BMAT and bone cells.
Summary
A neglected feature of osteoporosis progression is BMAT development. BMAT appears as a “new tissue” with unique properties, which undoubtedly plays important physiological and pathological roles, but which remains insufficiently understood.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bone integrity is maintained through a dynamic process, known as bone remodeling, on trabecular and cortical bones, resulting from a continuous balanced interplay between bone resorption caused by osteoclasts and bone formation employed by osteoblasts. Osteoclasts initiate the remodeling cascade by removing bone matrix and subsequently osteoblasts refill these cavities with bone matrix [1]. Disorders of bone loss, such as osteoporosis, are associated with increased rates of bone remodeling resulting in bone loss due to an overwhelming osteoclastic activity. Receptor activator of NF-κB ligand (RANKL) is a central regulator of bone remodeling by mediating osteoclast-induced bone resorption, through binding to its transmembrane receptor RANK, while it is naturally inhibited by the soluble decoy receptor osteoprotegerin (OPG) [2]. Osteoblasts are derived from bone marrow mesenchymal stromal cells (BMSC), which are multipotent and can give rise to several other distinct cell types, including adipocytes. An imbalance in the regulation of osteoblast and adipocyte differentiation is commonly observed in osteoporosis, which is associated with increased rate of bone marrow adipose tissue (BMAT) formation.
BMAT, an adipose tissue that lies within bone marrow, has been surprisingly disregarded for many decades. In the past years, BMAT was considered as an inactive filler of the bone marrow (BM) cavity that substitutes hematopoietic cells in response to a decreasing demand for hematopoiesis. It is only in recent years that BMAT has become recognized as a specific and active fat depot, with recent advances suggesting that BMAT differs from other fat depots not only anatomically but also developmentally, functionally, and metabolically [3, 4]. Because of these unique characteristics, BMAT appears today as a “new tissue” with much potential for revealing new mechanisms on human health and disease. During the last two decades, several lines of evidence support the impact of BMAT expansion in bone, metabolic and nutritional diseases including osteoporosis, obesity, anorexia nervosa, diabetes and cancer, whereas the molecular mechanisms remain insufficiently understood [5].
BMAT Progression
In humans, BMAT is virtually absent at birth and bone cavities are mainly filled with red hematopoietic marrow, while BMAT is innately programmed to expand during life, leading to a conversion of the red marrow to fatty “yellow” marrow [6, 7]. Interestingly, a medium-aged, healthy lean adult is estimated to have BMAT that corresponds to approximately 10% of the total fat mass, and by the age of 25 BMAT occupies 50 to 70% of BM volume [8]. BMAT initially forms at terminal phalanges, and then expands towards other peripheral skeletal sites such as appendicular skeleton and eventually in the axial skeleton [9]. BMAT initiates at femur and tibiae bones at the age of 7 and continues until the age of 18. Thus, BMAT arises during normal human development, increases with age, and represents a major class of adipose tissue. In mice, BMAT begins at the distal tibia around day 7, and expands during body maturation and progression but with decreased rates compared to humans. Notably, 12-week-old C57BL/6J male mice have less than 1% BMAT volume in the tibia diaphysis, as shown by osmium tetroxide staining combined with micro-computed tomography (microCT) [10••].
Two distinct populations of marrow adipocytes have been identified, constitutive and regulated. Constitutive marrow adipocytes (cMAT) form a stable dense fat depot arising at early stages of life, occupy distal parts of the appendicular skeleton and are less responsive to stimuli [10••]. On the other hand, regulated marrow adipocytes (rMAT) are located scattered within the hematopoietic marrow of the axial skeleton and the proximal appendicular bones, and their formation and expansion occur at later phases of life in response to nutritional, hormonal, and temperature challenges [10••, 11].
Volumetric BMAT analysis is performed either with invasive or non-invasive imaging methods. Invasive methods include histological analysis of bone biopsies [12], while during the last decade, non-invasive imaging approaches were developed to visualize and quantify BMAT, including magnetic resonance imaging (MRI), which is the gold standard method to estimate BMAT [13, 14] and magnetic resonance spectroscopy (MRS), which determines the fat fraction and the fat composition distinguishing saturated from non-saturated lipids [15,16,17]. To further study the interactions between BMAT and bone remodeling, these methods are combined with measurements of bone mineral density (BMD) and structure with dual-energy x-ray absorptiometry (DEXA) and PET-microCT [18, 19]. Lately, the introduction of osmium tetroxide staining combined with micro-CT opens new horizons in the visualization and quantification of BMAT in rodents [20••]. Based on the abovementioned imaging methods, it has been recently shown that BMAT positively correlates with BMD in 4- to 10-year-old children [21], while a clear negative correlation between BMAT and bone mass was identified during aging and osteoporosis, suggesting an interplay between BMAT and bone remodeling.
BMAT in Osteoporosis
Osteoporosis is a multifactorial metabolic disease which is characterized by low bone density, reduced bone quality, and increased risk of fractures [22]. Osteoporosis is usually underdiagnosed and undertreated because of the lack of symptoms and it is often referred as the “silent epidemic” since one in three women and one in five men above the age of 50 will experience osteoporotic fractures. Apart from bone phenotype, osteoporotic patients also exhibit high BMAT. Various clinical studies have demonstrated that osteoporotic patients have 10% higher BMAT compared to age-matched healthy subjects [23,24,25,26]. All these clinical observations support a negative correlation between BMAT and bone mass. Numerous studies applying MRI methodology showed that healthy adults display an increase of BMAT in spinal vertebrae at a rate of 7% every 10 years and a comparison between sexes revealed that BMAT is 10% higher in men than in women the period before menopause, but the ratio reverses after menopause [27, 28].
Several clinical trials studying the effectiveness of anti-osteoporosis treatments have reported effects on BMAT. Various studies demonstrate that anti-resorptive drugs effectively reduce BMAT expansion in osteoporotic women. For example, postmenopausal osteoporotic women treated with estrogen either at a long or at a short term showed a decrease in BMAT as assessed by bone biopsies or MRI [29,30,31]. Similar results were shown upon treatment with the parathyroid hormone teriparatide [32••]. In addition, osteoporotic women treated with the bisphosphonate risedronate showed a reduction in BMAT and improved BMD compared to the control group [33,34,35,36, 37•, 38•]. The new anti-osteoporotic drug romosozumab, an antibody that targets the anti-anabolic protein sclerostin, efficiently improves bone mass and ameliorates BMAT [39]. Even though most intervention studies targeting improvement in bone quality manage to ameliorate BMAT, there are limited data regarding the opposite, i.e., the effect of BMAT treatment in bone mass.
The spatiotemporal pattern of BMAT formation in rodents is considered to be quite similar to humans. Through histological analysis and osmium tetroxide staining, it has been demonstrated that the BMAT accumulation in long bones substantially increases with aging. However, the percentage of BMAT in rodents is lower compared to humans and varies among different mouse strains [10••]. The negative association between high BMAT and low BMD in osteoporotic patients [40] was reproduced in ovariectomized mice [41, 42•] and other genetic osteoporotic mouse models [43••]. The use of animal models of osteoporosis can substantially improve our understanding on the pathophysiological mechanisms involved in bone resorption and BMAT expansion. Our group has recently established two distinct genetic mouse models of osteoporosis through the expression of human RANKL (huRANKL) in transgenic mice (TgRANKL). The mild osteoporosis Tg5516 model expressing huRANKL at low levels develops trabecular bone loss and adjacent increase in BMAT, while the severe osteoporosis model Tg5519 overexpressing huRANKL displays trabecular bone loss, cortical porosity, and extended BMAT throughout the BM cavity [43••]. Based on these observations, it seems that BMAT recruitment is linked with sites of active bone resorption. However, it is still unclear how BMAT and bone loss are linked during osteoporosis and which process precedes the other.
The impact of bone resorption on BMAT formation has been confirmed in various clinical and animal studies through pharmaceutical inhibition of bone resorption [29, 30, 33, 37•, 44]. However, it remains unclear whether BMAT affects bone resorption in vivo since the current data are limited [41]. Paradoxically, pharmaceutical inhibition of BMAT with a PPARγ2 antagonist bisphenol-A-diglycidyl-ether (BADGE) in normal and diabetic male mice as well as genetic studies in PPARγ knockout mice demonstrated that the loss of BMAT induced osteogenesis due to increased osteoblast activity without affecting osteoclasts and bone loss [45, 46]. Therefore, more efforts must be made in the understanding of BMAT effect on bone resorption and in osteoporosis.
Molecular Basis of BMAT Interaction with Bone Cells
First of all, BMAT could contribute as an energy supply, through lipid release, to neighboring cells like osteoblasts and hematopoietic stem cells (HSCs). The increased adipocyte numbers observed during aging and osteoporosis, may contribute to the energy maintenance of bone cells and HSCs. This hypothesis is mainly supported by in vitro experiments, where it is demonstrated that adipocytes can transfer free fatty lipids (FFAs) to hematopoietic cells through a controlled process termed lipolysis to support proliferation and survival [47, 48].
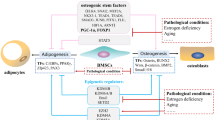
As both BM adipocytes and osteoblasts are derived from BMSCs, also known as skeletal stem cells (SSCs), it is reasonable to assume that increased BMAT formation is associated with a reciprocal suppression of osteogenesis [49,50,51]. Distinct sets of transcription factors are activated during the commitment of precursor cells into osteoblasts or adipocytes. Runt-related transcription factor 2 (RUNX2) and transcription factor Sp7 (Osterix) regulate osteoblast differentiation [52, 53], while CCAAT/enhancer binding proteins (C/EBP) and peroxisome proliferative activated receptors gamma (PPARγ) promote adipogenesis [54,55,56,57]. In animal model studies, it has been demonstrated that upregulation of PPARγ results in high BMAT and low bone mass, while downregulation of PPARγ leads to a low BMAT and high bone mass phenotype [45, 58,59,60,61].
The differentiation of BMSCs to either adipocytes or osteoblasts is a two-step process, including lineage commitment and maturation. The lineage commitment of BMSCs is fine-tuned by the action of various extracellular matrix components, growth factors, cytokines, and chemokines, which in turn activate a cascade of signaling events regulating the key transcription factors such as PPARγ and C/EBPs or Runx2 and Osterix for adipogenesis or osteogenesis, respectively. The lineage commitment of BMSCs towards adipocytes or osteoblasts is regulated by a complex network of signaling pathways including transforming growth factor-beta (TGFβ)/bone morphogenic protein (BMP) signaling, wingless-type MMTV integration site family (Wnt) signaling, Hedgehogs (Hh), Notch, and fibroblast growth factors (FGFs) [62,63,64,65,66,67,68,69,70]. In general, the activation of Wnt signaling and Hedgehogs induces osteogenic differentiation, while activation of TGFβ/BMP, Notch, and FGFs signaling may exert a dual effect either favoring osteogenesis or adipogenesis depending on the ligand. These observations clearly show that all signaling pathways do not take place individually but rather act synergistically to promote BMSC’s shift depending on the stimuli. Estrogen deficiency, increased glucocorticoid levels, oxidative stress, and immobilization favor adipogenesis over osteogenesis. Thus, treatment with estrogen, or intermittent PTH results in increased bone mass and reduced BMAT [29, 32••, 40]. Similarly, sclerostin, an inhibitor of bone formation expressed by osteocytes, stimulates adipogenesis [39], while OPG, an inhibitor of RANKL and bone resorption, inhibits adipocyte differentiation in vitro [71]. Considering that OPG functions as a blockage of RANKL activity, it is possible that RANKL regulates BMAT formation either directly or indirectly.
BMAT can interact with its microenvironment through the secretion of numerous factors, while the secretion profile of BMAT and its functional endocrine and paracrine implications remain largely unexplored. So far, it has been shown that BMAT secretes endocrine factors designated as adipokines such as adiponectin, as well as other pro-inflammatory molecules, such as tumor necrosis factor (TNF) and interleukin-6, which affect bone homeostasis through the regulation of osteoblast and osteoclast functions. In vitro studies report that human adipocytes derived from BMSCs secrete cytokines, including macrophage inflammatory protein (MIP-1), granulocyte colony-stimulating factor (G-CSF), and granulocyte macrophage colony-stimulating factor (GM-CSF) [72], whereas adipocytes from mouse BMSCs may also secrete chemokines such as chemokine (C-X-C motif) ligand 1 (CXCL1) and chemokine (C-X-C motif) ligand 2 (CXCL2) [73]. Adipose tissue secretes also a series of cytokines, which are termed adipokines including leptin, adiponectin, chemerin, omentin, and resistin. These have profound effects on surrounding and/or remote cell types [72, 74,75,76,77,78,79]. In the BM of osteoporotic postmenopausal women, the levels of leptin and adiponectin were significantly lower, whereas the effects of leptin on bone are not conclusive [74, 80,81,82,83].
The coexistence of increased BMAT and bone destruction with aging and osteoporosis, also suggests a mechanistic link between adipogenesis and osteoclastogenesis. Indeed, BM adipocytes produce RANKL, and thus can promote osteoclastogenesis [84,85,86]. A subpopulation of Pref-1+ pre-adipocytes was recently identified in BM that notably expresses RANKL and increased numbers of these cells coincide with aging [87]. A working hypothesis could be that RANKL+/Pref-1+ pre-adipocytes may contribute to bone loss through stimulation of osteoclastogenesis. In a recent study, mice lacking PTH receptors in mesenchymal stem cells develop high BMAT and reduced bone mass. In this model, BMAT was shown to produce high levels of RANKL, and there was also abundance of the RANKL+/Pref-1+ pre-adipocytes, suggesting that pre-adipocytes may contribute to bone loss [32••]. Therefore, BMAT could regulate osteoclast formation either directly through RANKL production or indirectly through adiponectin secretion, which stimulates osteoblasts to produce RANKL [77, 88,89,90]. Paradoxically, the impact of BMAT on bone resorption in vivo remains unclear and further studies are needed to take this further. In addition, the positive effect of numerous anti-resorptive therapies in BMAT attenuation suggests that osteoclasts are associated with BMAT formation. However, the underlying molecular mechanisms that connect osteoclasts with BMAT progression remain unknown.
Conclusion
A “neglected” feature of osteoporosis progression is BMAT development. BMAT is a “new tissue” with unique properties, which remains insufficiently understood. Animal and clinical studies have revealed that BMAT increases during aging and is further enhanced in osteoporosis, emphasizing its potential impact in bone remodeling. The detrimental role of BMAT has been highlighted through the identification of secreted endocrine and/or paracrine factors (RANKL, pro-inflammatory cytokines and adipokines) that regulate bone metabolism. However, interventions targeting BMAT are limited and as a result the impact of BMAT on bone remodeling is far from conclusive. On the other hand, a positive effect of anti-resorptive therapies on BMAT progression has been established, while the underlying mechanisms have not been defined yet. Therefore, further studies are needed in order to shed light on the mechanistic basis of BMAT formation during osteoporosis and its pathophysiological role in bone remodeling.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2(4):389–406. https://doi.org/10.1016/S1534-5807(02)00157-0.
Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142(12):5050–5. https://doi.org/10.1210/endo.142.12.8536.
de Paula FJA, Rosen CJ. Structure and function of bone marrow adipocytes. Compr Physiol. 2017;8(1):315–49. https://doi.org/10.1002/cphy.c170010.
Hardouin P, Rharass T, Lucas S. Bone marrow adipose tissue: to be or not to be a typical adipose tissue? Front Endocrinol. 2016;7:1–11. https://doi.org/10.3389/fendo.2016.00085.
Veldhuis-Vlug AG, Rosen CJ. Clinical implications of bone marrow adiposity. ARPN J Eng Appl Sci. 2017;12(10):3218–21. https://doi.org/10.1111/joim.12718.
Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology. 2001;2(3):165–71. https://doi.org/10.1023/A:1011513223894.
Ecklund K, Vajapeyam S, Feldman HA, Buzney CD, Mulkern RV, Kleinman PK, et al. Bone marrow changes in adolescent girls with anorexia nervosa. J Bone Miner Res. 2010;25(2):298–304. https://doi.org/10.1359/jbmr.090805.
Blebea JS, Houseni M, Torigian DA, Fan C, Mavi A, Zhuge Y, et al. Structural and functional imaging of normal bone marrow and evaluation of its age-related changes. Semin Nucl Med. 2007;37(3):185–94. https://doi.org/10.1053/j.semnuclmed.2007.01.002.
Scheller EL, Rosen CJ. What’s the matter with MAT? Marrow adipose tissue, metabolism, and skeletal health. Ann N Y Acad Sci. 2014;1311(1):14–30. https://doi.org/10.1111/nyas.12327.
•• Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B, et al. Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat. Commun. 2015;6:1–13. https://doi.org/10.1038/ncomms8808. A study that provides a thorough characterization of regulated and constitutive marrow adipocyte tissues.
Scheller EL, Cawthorn WP, Burr AA, Horowitz MC, MacDougald OA. Marrow adipose tissue: trimming the fat. Trends Endocrinol Metab. 2016;27(6):392–403. https://doi.org/10.1016/j.tem.2016.03.016.
Meunier P, Aaron J, Edouard C, Vignon G. Osteoporosis and the replacement of cell populations of the marrow by adipose tissue. A quantitative study of 84 iliac bone biopsies. Clin Orthop Relat Res. 1971;80:147–54.
Li G, Xu Z, Fan J, Yuan W, Zhang L, Hou L, et al. To assess differential features of marrow adiposity between postmenopausal women with osteoarthritis and osteoporosis using water/fat MRI. Menopause. 2017;24(1):105–11. https://doi.org/10.1097/GME.0000000000000732.
Baum T, Yap SP, Dieckmeyer M, Ruschke S, Eggers H, Kooijman H, et al. Assessment of whole spine vertebral bone marrow fat using chemical shift-encoding based water-fat MRI. J Magn Reson Imaging. 2015;42(4):1018–23. https://doi.org/10.1002/jmri.24854.
Pansini V, Monnet A, Salleron J, Hardouin P, Cortet B, Cotten A. 3 Tesla 1H MR spectroscopy of hip bone marrow in a healthy population, assessment of normal fat content values and influence of age and sex. J Magn Reson Imaging. 2014;39(2):369–76. https://doi.org/10.1002/jmri.24176.
Pansini VM, Monnet A, Salleron J, Penel G, Migaud H, Cotten A. Reproducibility of 1 H MR spectroscopy of hip bone marrow at 3 tesla. J Magn Reson Imaging. 2012;36(6):1445–9. https://doi.org/10.1002/jmri.23783.
Li X, Shet K, Xu K, Rodríguez JP, Pino AM, Kurhanewicz J, et al. Unsaturation level decreased in bone marrow fat of postmenopausal women with low bone density using high resolution magic angle spinning (HRMAS)1H NMR spectroscopy. Bone. 2017;105:87–92. https://doi.org/10.1016/j.bone.2017.08.014.
Arentsen L, Hansen KE, Yagi M, Takahashi Y, Shanley R, McArthur A, et al. Use of dual-energy computed tomography to measure skeletal-wide marrow composition and cancellous bone mineral density. J Bone Miner Metab. 2017;35(4):428–36. https://doi.org/10.1007/s00774-016-0796-1.
Patsch JM, Li X, Baum T, Yap SP, Karampinos DC, Schwartz AV, et al. Bone marrow fat composition as a novel imaging biomarker in postmenopausal women with prevalent fragility fractures. J Bone Miner Res. 2013;28(8):1721–8. https://doi.org/10.1002/jbmr.1950.
•• Scheller EL, Troiano N, Vanhoutan JN, Bouxsein MA, Fretz JA, Xi Y, et al. Use of osmium tetroxide staining with microcomputerized tomography to visualize and quantify bone marrow adipose tissue in vivo. Methods Enzymol. 2014;537:123–39. https://doi.org/10.1016/B978-0-12-411619-1.00007-0. This paper for the first time provides a new technical approach for the quantification of BMAT in rodents with osmium tetroxide and micro-CT.
Gao Y, Zong K, Gao Z, Rubin MR, Chen J, Heymsfield SB, et al. Magnetic resonance imaging–measured bone marrow adipose tissue area is inversely related to cortical bone area in children and adolescents aged 5–18 years. JClin Densitom. 2015;18(2):203–8. https://doi.org/10.1016/j.jocd.2015.03.002.
Gullberg B, Johnell O, Kanis JA. International original article world-wide projections for hip fracture. Osteoporos Int. 1997;7(5):407–13. https://doi.org/10.1007/PL00004148.
Yeung DKW, Griffith JF, Antonio GE, Lee FKH, Woo J, Leung PC. Osteoporosis is associated with increased marrow fat content and decreased marrow fat unsaturation: a proton MR spectroscopy study. J Magn Reson Imaging. 2005;22(2):279–85. https://doi.org/10.1002/jmri.20367.
Tang GY, Lv ZW, Tang RB, Liu Y, Peng YF, Li W, et al. Evaluation of MR spectroscopy and diffusion-weighted MRI in detecting bone marrow changes in postmenopausal women with osteoporosis. Clin Radiol. 2010;65(5):377–81. https://doi.org/10.1016/j.crad.2009.12.011.
Li GW, Xu Z, Chen QW, Tian YN, Wang XY, Zhou L, et al. Quantitative evaluation of vertebral marrow adipose tissue in postmenopausal female using MRI chemical shift-based water-fat separation. Clin Radiol. 2014;69(3):254–62. https://doi.org/10.1016/j.crad.2013.10.005.
Cordes C, Baum T, Dieckmeyer M, Ruschke S, Diefenbach MN, Hauner H, et al. MR-based assessment of bone marrow fat in osteoporosis, diabetes, and obesity. Front Endocrinol (Lausanne). 2016;7:1–7. https://doi.org/10.3389/fendo.2016.00074.
Kugel H, Jung C, Schulte O, Heindel W. Age- and sex-specific differences in the 1H-spectrum of vertebral bone marrow. J Magn Reson Imaging. 2001;13:263–8. https://doi.org/10.1002/1522-2586(200102)13:2<263::AID-JMRI1038>3.0.CO;2-M.
Griffith JF, Yeung DKW, Ma HT, Leung JCS, Kwok TCY, Leung PC. Bone marrow fat content in the elderly: a reversal of sex difference seen in younger subjects. J Magn Reson Imaging. 2012;36(1):225–30. https://doi.org/10.1002/jmri.23619.
Syed FA, Oursler MJ, Hefferanm TE, Peterson JM, Riggs BL, Khosla S. Effects of estrogen therapy on bone marrow adipocytes in postmenopausal osteoporotic women. Osteoporos Int. 2008;19(9):1323–30. https://doi.org/10.1007/s00198-008-0574-6.
Limonard EJ, Veldhuis-Vlug AG, Van Dussen L, Runge JH, Tanck MW, Endert E, et al. Short-term effect of estrogen on human bone marrow fat. J Bone Miner Res. 2015;30(11):2058–66. https://doi.org/10.1002/jbmr.2557.
Bredella MA, Gerweck AV, Barber LA, Breggia A, Rosen CJ, Torriani M, et al. Effects of growth hormone administration for 6 months on bone turnover and bone marrow fat in obese premenopausal women. Bone. 2014;62:29–35. https://doi.org/10.1016/j.bone.2014.01.022.
•• Fan Y, Hanai J, Le PT BR, Maridas D, De Mambro V, et al. Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab. 2017;25(3):661–72. https://doi.org/10.1016/j.cmet.2017.01.001. This paper shows that PTH regulates bone marrow mesenchymal stem cell fate between bone and adipocytes.
Jin J, Wang L, Wang XK, Lai PL, Huang MJ, J D Di, et al. Risedronate inhibits bone marrow mesenchymal stem cell adipogenesis and switches RANKL/OPG ratio to impair osteoclast differentiation 2013;180(1):21–29. doi:https://doi.org/10.1016/j.jss.2012.03.018.
Watts NB, Harris ST, Genant HK, Wasnich RD, Miller PD, Jackson RD, et al. Intermittent cyclical etidronate treatment of postmenopausal osteoporosis. N Engl J Med. 1990;323(2):73–9. https://doi.org/10.1056/NEJM199007123230201.
Liberman UA, Weiss SR, Bröll J, Minne HW, Quan H, Bell NH, et al. Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis. N Engl J Med. 1995;333(22):1437–43. https://doi.org/10.1056/NEJM199511303332201.
Black DM, Thompson DE, Bauer DC, Ensrud K, Musliner T, Hochberg MC, et al. Fracture risk reduction with alendronate in women with osteoporosis: the Fracture Intervention Trial. FIT Research Group. J Clin Endocrinol Metab. 2000;85(11):4118–24. https://doi.org/10.1210/jcem.85.11.6953.
• Yang Y, Luo X, Yan F, Jiang Z, Li Y, Fang C, et al. Effect of zoledronic acid on vertebral marrow adiposity in postmenopausal osteoporosis assessed by MR spectroscopy. Skeletal Radiol. 2015;44(10):1499–505. https://doi.org/10.1007/s00256-015-2200-y. This study describes the beneficial anti-resorptive effects of zoledronic acid on vertebral marrow adiposity in postmenopausal osteoporosis.
Duque G, Li W, Adams M, Xu S, Phipps R. Effects of risedronate on bone marrow adipocytes in postmenopausal women. Osteoporos Int. 2011;22(5):1547–53. https://doi.org/10.1007/s00198-010-1353-8.
Chandra A, Lin T, Young T, Tong W, Ma X, Tseng WJ, et al. Suppression of sclerostin alleviates radiation-induced bone loss by protecting bone-forming cells and their progenitors through distinct mechanisms. J Bone Miner Res. 2017;32(2):360–72. https://doi.org/10.1002/jbmr.2996.
Gambacciani M, Ciaponi M, Cappagli B, Piaggesi L, De Simone L, Orlandi R, et al. Body weight, body fat distribution, and hormonal replacement therapy in early postmenopausal women. J Clin Endocrinol Metab. 1997;82(2):414–7. https://doi.org/10.1210/jcem.82.2.3735.
Iwaniec UT, Turner RT. Failure to generate bone marrow adipocytes does not protect mice from ovariectomy-induced osteopenia. Bone. 2013;53(1):145–53. https://doi.org/10.1016/j.bone.2012.11.034.
• Sui B, Hu C, Liao L, Chen Y, Zhang X, Fu X, et al. Mesenchymal progenitors in osteopenias of diverse pathologies: differential characteristics in the common shift from osteoblastogenesis to adipogenesis. Sci Rep. 2016;6(1):30186. https://doi.org/10.1038/srep30186. This study gives a comparative analysis of the adipogenic and osteogenic potential of mesenchymal progenitors from various models of osteopenia.
•• Rinotas V, Niti A, Dacquin R, Bonnet N, Stolina M, Han C-Y, et al. Novel genetic models of osteoporosis by overexpression of human RANKL in transgenic mice. J Bone Miner Res. 2014;29(5):1158–69. https://doi.org/10.1002/jbmr.2112. This paper describes two genetic mouse models of osteoporosis through the expression of human RANKL in transgenic mice, which develop an outstanding progressive BMAT phenotype.
Li G-W, Chang S-X, Fan J-Z, Tian Y-N, Xu Z, He Y-M. Marrow adiposity recovery after early zoledronic acid treatment of glucocorticoid-induced bone loss in rabbits assessed by magnetic resonance spectroscopy. Bone. 2013;52(2):668–75. https://doi.org/10.1016/j.bone.2012.11.002.
Cao J, Ou G, Yang N, Ding K, Kream BE, Hamrick MW, et al. Impact of targeted PPARγ disruption on bone remodeling. Mol Cell Endocrinol. 2015;410:27–34. https://doi.org/10.1016/j.mce.2015.01.045.
Sun H, Kim JK, Mortensen R, Mutyaba LP, Hankenson KD, Krebsbach PH. Osteoblast-targeted suppression of PPARc increases osteogenesis through activation of mTOR signaling. Stem Cells. 2013;31:2183–92. https://doi.org/10.1002/stem.1455.
Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, et al. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. 2017;77(6):1453–64. https://doi.org/10.1158/0008-5472.CAN-16-1645.
Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood. 2017;129(10):1320–32. https://doi.org/10.1182/blood-2016-08-734798.
Sivasubramaniyan K, Lehnen D, Ghazanfari R, Sobiesiak M, Harichandan A, Mortha E, et al. Phenotypic and functional heterogeneity of human bone marrow- and amnion-derived MSC subsets. Ann N Y Acad Sci. 2012;1266(1):94–106. https://doi.org/10.1111/j.1749-6632.2012.06551.x.
Abdallah BM, Kassem M. New factors controlling the balance between osteoblastogenesis and adipogenesis. Bone. 2012;50(2):540–5. https://doi.org/10.1016/j.bone.2011.06.030.
Rosen CJ, Bouxsein ML. Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol. 2006;2(1):35–43. https://doi.org/10.1038/ncprheum0070.
Komori T. Regulation of osteoblast differentiation by transcription factors. J Cell Biochem. 2006;99(5):1233–9. https://doi.org/10.1002/jcb.20958.
Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger-containing transcription factor Osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. https://doi.org/10.1016/S0092-8674(01)00622-5.
Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, et al. PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008;22(21):2941–52. https://doi.org/10.1101/gad.1709008.
Cao Z, Umek RM, McKnight SL. Regulated expression of three C / EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5(9):1538–52. https://doi.org/10.1101/gad.5.9.1538.
Kushwaha P, Khedgikar V, Gautam J, Dixit P, Chillara R, Verma A, et al. A novel therapeutic approach with Caviunin-based isoflavonoid that en routes bone marrow cells to bone formation via BMP2/Wnt-β-catenin signaling. Cell Death Dis. 2014;5(9):e1422. https://doi.org/10.1038/cddis.2014.350.
Kim J, Ko J. A novel PPARγ 2 modulator sLZIP controls the balance between adipogenesis and osteogenesis during mesenchymal stem cell differentiation. Cell Death Differ. 2014;21(10):1642–55. https://doi.org/10.1038/cdd.2014.80.
Cho SW, Yang JY, Her SJ, Choi HJ, Jung JY, Sun HJ, et al. Osteoblast-targeted overexpression of PPARγ inhibited bone mass gain in male mice and accelerated ovariectomy-induced bone loss in female mice. J Bone Miner Res. 2011;26(8):1939–52. https://doi.org/10.1002/jbmr.366.
Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, et al. PPARγ insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113(6):846–55. https://doi.org/10.1172/JCI200419900.
Li M, Pan LC, Simmons HA, Li Y, Healy DR, Robinson BS, et al. Surface-specific effects of a PPARγ agonist, darglitazone, on bone in mice. Bone. 2006;39(4):796–806. https://doi.org/10.1016/j.bone.2006.04.008.
Wan Y, Chong LW, Evans RM. PPAR-γ regulates osteoclastogenesis in mice. Nat Med. 2007;13(12):1496–503. https://doi.org/10.1038/nm1672.
Kang Q, Song W-X, Luo Q, Tang N, Luo J, Luo X, et al. A comprehensive analysis of the dual roles of BMPs in regulating adipogenic and osteogenic differentiation of mesenchymal progenitor cells. Stem Cells Dev. 2009;18(4):545–58. https://doi.org/10.1089/scd.2008.0130.
Tang Q-Q, Otto TC, Lane MD. Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci U S A. 2004;101(26):9607–11. https://doi.org/10.1073/pnas.0403100101.
Bennett CN, Ouyang H, Ma YL, Zeng Q, Gerin I, Sousa KM, et al. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J Bone Miner Res. 2007;22(12):1924–32. https://doi.org/10.1359/jbmr.070810.
Stevens JR, Miranda-Carboni GA, Singer MA, Brugger SM, Lyons KM, Lane TF. Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J Bone Miner Res. 2010;25(10):2138–47. https://doi.org/10.1002/jbmr.118.
Song BQ, Chi Y, Li X, Du WJ, Han ZB, Tian JJ, et al. Inhibition of notch signaling promotes the adipogenic differentiation of mesenchymal stem cells through autophagy activation and PTEN-PI3K/AKT/mTOR pathway. Cell Physiol Biochem. 2015;36(5):1991–2002. https://doi.org/10.1159/000430167.
Shimizu T, Tanaka T, Iso T, Matsui H, Ooyama Y, Kawai-Kowase K, et al. Notch signaling pathway enhances bone morphogenetic protein 2 (BMP2) responsiveness of Msx2 gene to induce osteogenic differentiation and mineralization of vascular smooth muscle cells. J Biol Chem. 2011;286(21):19138–48. https://doi.org/10.1074/jbc.M110.175786.
Kim W-K, Meliton V, Bourquard N, Hahn TJ, Parhami F. Hedgehog signaling and osteogenic differentiation in multipotent bone marrow stromal cells are inhibited by oxidative stress. J Cell Biochem [Internet]. 2010;111(5):1199–209. https://doi.org/10.1002/jcb.22846.
James AW, Pang S, Askarinam A, Corselli M, Zara JN, Goyal R, et al. Additive effects of sonic hedgehog and Nell-1 signaling in osteogenic versus adipogenic differentiation of human adipose-derived stromal cells. Stem Cells Dev. 2012;21(12):2170–8. https://doi.org/10.1089/scd.2011.0461.
Li L, Dong Q, Wang Y, Feng Q, Zhou P, Ou X, et al. Hedgehog signaling is involved in the BMP9-induced osteogenic differentiation of mesenchymal stem cells. Int J Mol Med. 2015;35(6):1641–50. https://doi.org/10.3892/ijmm.2015.2172.
Zhang L, Liu M, Zhou X, Liu Y, Jing B, Wang X, et al. Role of osteoprotegerin (OPG) in bone marrow adipogenesis. Cell Physiol Biochem. 2016;40:681–91. https://doi.org/10.1159/000452580.
Muruganandan S, Roman AA, Sinal CJ. Role of chemerin/CMKLR1 signaling in adipogenesis and osteoblastogenesis of bone marrow stem cells. J Bone Min Res. 2010;25(2):222–34. https://doi.org/10.1359/jbmr.091106.
Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin Exp Metastasis. 2015;32(4):353–68. https://doi.org/10.1007/s10585-015-9714-5.
Holloway WR, Collier FM, Aitken CJ, Myers DE, Hodge JM, Malakellis M, et al. Leptin inhibits osteoclast generation. J Bone Miner Res. 2002;17(2):200–9. https://doi.org/10.1359/jbmr.2002.17.2.200.
Thommesen L, Stunes AK, Monjo M, Grøsvik K, Tamburstuen MV, Kjøbli E, et al. Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem. 2006;99(3):824–34. https://doi.org/10.1002/jcb.20915.
Xie H, Tang SY, Luo XH, Huang J, Cui RR, Yuan LQ, et al. Insulin-like effects of visfatin on human osteoblasts. Calcif Tissue Int. 2007;80(3):201–10. https://doi.org/10.1007/s00223-006-0155-7.
Tu Q, Zhang J, Dong LQ, Saunders E, Luo E, Tang J, et al. Adiponectin inhibits osteoclastogenesis and bone resorption via APPL1-mediated suppression of Akt1. J Biol Chem. 2011;286(14):12542–53. https://doi.org/10.1074/jbc.M110.152405.
Muruganandan S, Dranse HJ, Rourke JL, Mcmullen NM, Sinal CJ. Chemerin neutralization blocks hematopoietic stem cell osteoclastogenesis. Stem Cells. 2013;31(10):2172–82. https://doi.org/10.1002/stem.1450.
Liu LF, Shen WJ, Ueno M, Patel S, Kraemer FB. Characterization of age-related gene expression profiling in bone marrow and epididymal adipocytes. BMC Genomics. 2011;12:212. https://doi.org/10.1186/1471-2164-12-212.
Idelevich A, Sato K, Baron R. What are the effects of leptin on bone and where are they exerted? J Bone Miner Res. 2013;28(1):18–21. https://doi.org/10.1002/jbmr.1812.
Lindenmaier LB, Philbrick KA, Branscum AJ, Kalra SP, Turner RT, Iwaniec UT. Hypothalamic leptin gene therapy reduces bone marrow adiposity in ob/ob mice fed regular and high-fat diets. Front Endocrinol (Lausanne). 2016;7:1–9. https://doi.org/10.3389/fendo.2016.00110.
Hamrick MW, Della-Fera MA, Choi YH, Pennington C, Hartzell D, Baile CA. Leptin treatment induces loss of bone marrow adipocytes and increases bone formation in leptin-deficient ob/ob mice. J Bone Miner Res. 2005;20(6):994–1001. https://doi.org/10.1359/JBMR.050103.
Turner RT, Kalra SP, Wong CP, Philbrick KA, Lindenmaier LB, Boghossian S, et al. Peripheral leptin regulates bone formation. J Bone Miner Res. 2013;28(1):22–34. https://doi.org/10.1002/jbmr.1734.
Gasparrini M, Rivas D, Elbaz A, Duque G. Differential expression of cytokines in subcutaneous and marrow fat of aging C57BL/6J mice. Exp Gerontol. 2009;44(9):613–8. https://doi.org/10.1016/j.exger.2009.05.009.
Goto H, Osaki M, Fukushima T, Sakamoto K, Hozumi A, Baba H, et al. Human bone marrow adipocytes support dexamethasone-induced osteoclast differentiation and function through RANKL expression. Biomed Res. 2011;32(1):37–44.
Goto H, Hozumi A, Osaki M, Fukushima T, Sakamoto K, Yonekura A, et al. Primary human bone marrow adipocytes support TNF-α-induced osteoclast differentiation and function through RANKL expression. Cytokine. 2011;56(3):662–8. https://doi.org/10.1016/j.cyto.2011.09.005.
• Takeshita S, Fumoto T, Naoe Y, Ikeda K. Age-related marrow adipogenesis is linked to increased expression of RANKL. J Biol Chem. 2014;289(4):16699–710. https://doi.org/10.1074/jbc.M114.547919. This is the first study which describes the identification of a new subpopulation of pre-adipocytes that express RANKL and Pref1 which may have a role in bone loss during aging.
Wang Q-P, Li X-P, Wang M, Zhao L-L, Li H, Xie H, et al. Adiponectin exerts its negative effect on bone metabolism via OPG/RANKL pathway: an in vivo study. Endocrine. 2014;47(3):845–53. https://doi.org/10.1007/s12020-014-0216-z.
Ealey KN, Kaludjerovic J, Archer MC, Ward WE. Adiponectin is a negative regulator of bone mineral and bone strength in growing mice. Exp Biol Med. 2008;233(12):1546–53. https://doi.org/10.3181/0806-RM-192.
Luo X-H, Guo L-J, Xie H, Yuan L-Q, Wu X-P, Zhou H-D, et al. Adiponectin stimulates RANKL and inhibits OPG expression in human osteoblasts through the MAPK signaling pathway. J Bone Miner Res. 2006;21(10):1648–56. https://doi.org/10.1359/jbmr.060707.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Vagelis Rinotas and Eleni Douni declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Molecular Biology of Bone Marrow Fat Adiposity
Rights and permissions
About this article

Cite this article
Rinotas, V., Douni, E. Molecular Interaction of BMAT with Bone. Curr Mol Bio Rep 4, 34–40 (2018). https://doi.org/10.1007/s40610-018-0093-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-018-0093-y