
Abstract
Purpose of Review
This article provides a brief overview of mechanisms of inflammatory liver injury and how this applies to drug hepatotoxicity with a particular emphasis on the role of inflammation in acetaminophen-induced liver injury.
Recent Findings
Significant progress has been made in the last decade in our understanding of the initiation of sterile inflammation after necrotic cell death by the release of damage-associated molecular patterns and their recognition by toll-like receptors and others on macrophages. These events trigger the formation of cytokines and chemokines directly or with assistance of inflammasome activation thereby activating and recruiting leukocytes including neutrophils and monocyte-derived macrophages into the necrotic areas. Although this sterile inflammatory response is mainly geared towards the removal of necrotic cell debris and preparation of regeneration, there are conditions where these innate immune cells can aggravate the initial injury. The mechanisms and controversial findings of the innate immunity are being discussed in detail. In contrast, drug metabolism and formation of a reactive metabolite that binds to proteins in the absence of extensive cell death can induce an adaptive immune response, which eventually also results in severe liver injury. However, the initiating event appears to be the formation of protein adducts, which act as haptens to activate an adaptive immune response. Overall, these mechanisms are less well understood.
Summary
The past decade has revolutionized our understanding of the mechanisms that control the interplay between cell death and innate or adaptive immune responses. This report provides an update on these mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The liver is a versatile organ with a variety of functions. Because of its large endogenous macrophage population, i.e., Kupffer cells, and the easy recruitment of circulating leukocytes, the liver is one of the primary organs responsible for innate immunity [1]. Increased intestinal permeability during various disease states can result in exposure of the liver to gut-derived viruses, bacteria, and other pathological material through the portal vein blood. As the liver is the first organ with a substantial innate immune component to be exposed to these materials, Kupffer cells are programmed to be highly responsive to these agents, and they are very effective at their clearance and removal [1, 2]. Moreover, Kupffer cells can recruit other immune populations, many of which have been proposed to provoke further hepatic damage due to their ability to secrete toxic compounds, such as proteases and reactive oxygen species (ROS). Because of its diverse array of metabolizing genes, the liver is also the primary organ responsible for drug and xenobiotic metabolism. This includes phase I oxidation and phase II conjugation reactions, both of which are responsible for increasing the hydrophilicity of drugs and enhancing their excretion and clearance. While this function is important for the elimination of xenobiotics, occasionally these reactions result in the formation of a reactive metabolite, which can cause liver damage through a variety of mechanisms [3, 4••]. Drug-induced liver injury (DILI), which remains a major cause of drug development failure due to preclinical toxicity or being pulled from the market due to toxicity in the clinic, is generally divided into 2 principal categories: (1) Direct, dose-dependent hepatotoxins, which induce intracellular signaling pathways leading to cell death (for example, acetaminophen, APAP) [5]. This direct liver injury can be further aggravated by an innate immune response. (2) Idiosyncratic DILI, which generally is not dose-dependent, delayed in onset, and affects only very few individuals exposed to therapeutic doses. Although the innate immune system can be involved in the initiation of the response, it is generally established that the injury is caused by the adaptive immune system [6, 7].
The combination of the presence of a potent innate immune population along with the propensity for hepatocytes to be routinely exposed to toxic compounds can result in substantial liver inflammation after exposure to hepatotoxic compounds. This chapter will focus on established mechanisms that mediate this interaction. We will provide an initial overview of mechanisms of inflammation in the liver and then focus on relevant laboratory models where the primary mechanisms were initially established.
Mechanisms of Liver Inflammation
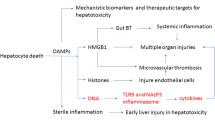
The inflammatory response in the absence of overt infection (sterile inflammation) has been a topic of considerable interest in hepatology in recent years. The initiating signal for sterile inflammation is likely the result of release of intracellular content during necrosis [8, 9]. When cells undergo necrosis, they lose membrane integrity and intracellular molecules spill out into the surrounding area. Surprisingly, many proteins, nucleic acids, and other cellular components act as pro-inflammatory signals when outside of their normal environment [8, 9]. A number of different signals have been established, that when released from dying or damaged hepatocytes, activate local immune cells and initiate the sterile inflammatory response. This includes a diverse and growing array of molecules, such as high mobility group box 1 (HMGB1) protein, ATP, mitochondrial DNA (mtDNA), nuclear DNA fragments, RNA, purines, uric acid, heat-shock proteins, and bile acids [10]. These molecules are collectively referred to as damage-associated molecular patterns (DAMPs), and their activity is mediated by various classes of pattern recognition receptors present on immune cell populations, including toll-like receptors (TLRs), purinergic receptors, and the receptor for advanced glycation end-products (RAGE) (reviewed in [11]) (Fig. 1a). Ligation of these receptors by DAMPs results in fundamental changes in immune cells that shifts them towards a pro-inflammatory phenotype and results in initiation of the inflammatory signal through release of cytokines, such as interleukins and chemokines [12]. These chemokines and cytokines then further amplify the signaling to sustain an inflammatory response. Importantly, this inflammatory response can provoke further injury if not controlled appropriately but is typically required for both normal regeneration of the liver and prevention of acute infection, which can be deadly during acute liver failure [1]. As such, the immune system plays an imperative role in recovery from DILI, and, thus, interventions against the immune system may come with a potential cost. Hence, understanding the role of the immune system in the liver injury process after drug-induced liver injury remains an important and pressing issue for developing therapeutics and improving patient care.

Mechanisms of drug-induced liver injury through innate or adaptive immune mechanisms. (a) General scheme of the drug-induced formation of reactive metabolites and protein adducts, which trigger necrosis and initiate a sterile inflammatory response by release of damage-associated molecular patterns (DAMPs), the promotion of cytokine and chemokine formation by activating pattern-recognition receptors, such as toll-like receptors (TLRs), and the consequent activation and recruitment of inflammatory cells, which can aggravate the initial injury (see text for details). (b) Sterile inflammatory response after acetaminophen-induced liver injury. Part of the acetaminophen dose is metabolized to a reactive metabolite, which triggers mitochondrial dysfunction and eventually DNA fragmentation resulting in necrotic cell death. The release of DAMPs induces a sterile inflammatory response, which recruits neutrophils and monocyte-derived macrophages. The preponderance of evidence suggests that the inflammatory response does not aggravate the initial injury but removes necrotic cell debris and supports regeneration (see text for details). Abbreviations: HMGB1, high mobility group box 1 protein; HOCl, hypochlorous acid; ICAM-1, intercellular adhesion molecule-1; IL, interleukin; MHCII, major histocompatibility complex; MCP-1, monocyte chemoattractant protein-1; mtDNA, mitochondrial DNA; RAGE, receptor for advanced glycation end products; TLR, toll-like receptor
Kupffer cells, the resident macrophages in the liver, express a majority of the receptors necessary for detection of DAMPs and mediate the initial response to acute cell injury [1, 2]. Ligation of DAMP receptors, such as the purinergic receptor 2X7 (P2X7) or toll-like receptor-4 (TLR4) and -9 (TLR9), results in increased expression and release of a number of cytokines, including interleukins [10, 12]. These mediators then amplify the pro-inflammatory signal and recruit circulating immune cells into the liver to continue the inflammatory process. Furthermore, many DAMP receptors also serve as priming agents for the multimeric complex called the inflammasomes [1, 13•, 14•]. Activation of TLR4 and TLR9 by DAMPs, such as HMGB1 and DNA fragments, respectively, increases expression of pro-interleukin-1ß (pro-IL-1ß) [14•, 15]. Pro-IL-1ß is cleaved to generate the active cytokine IL-1β by caspase-1, which is activated through the inflammasome [15]. A number of different inflammasome complexes exist, but perhaps the most commonly studied one in the liver is dependent on the function of a protein called NACHT, LRR, and PYD domain-containing protein 3 (Nalp3) [15]. Nalp3 forms a complex with apoptosis-associated spec-like protein containing a CARD (ASC) and pro-caspase 1 [14•, 15]. The inflammasome activated through P2X7 ligation then serves to activate pro-caspase-1 into the active caspase-1, which processes pro-interleukin-1ß [13•, 16]. This occurs in the presence of simultaneous activation of multiple inflammatory signals, which trigger the inflammatory response serving as a sort of feed-forward loop that provides a redundant signal.
Upon activation by DAMPs or endotoxin, Kupffer cells produce also the pro-inflammatory and apoptosis-inducing cytokine tumor necrosis factor-α (TNF-α) and reactive oxygen species (ROS) when activated through the complement receptor [17]. Because both of these types of mediators can initiate cell death, the role of both endogenous Kupffer cells and recruited monocytes has been studied extensively [18]. In numerous models of liver injury, pretreatment with gadolinium chloride or clodronate liposomes yields a protective effect [18,19,20]. Similarly, plasma levels of glutathione disulfide (GSSG) rise acutely in diseases with a strong inflammatory component, especially Kupffer cell activation, indicating release of significant ROS levels in the vascular space by these inflammatory cells [20,21,22]. Elimination of TNF-α either genetically or through antibodies can also prevent injury in some models indicating TNF-α release from Kupffer cells may be a critical mediator of their ability to kill hepatocytes [22,23,24]. As such, it is clear that inflammatory signals released by Kupffer cells are capable of damaging hepatocytes directly and further provoking sterile inflammation. Their specific role in various pathological conditions will be discussed further.
After the initial cytokine wave is produced by Kupffer cells, both monocytes and neutrophils are recruited to the liver in large numbers (Fig. 1a). Neutrophils represent the most populous member of the innate immune cell classes and are noted to secrete cytokines to sustain immunological responses, produce ROS, and phagocytize cellular debris [25]. Notably though, due to their small size, neutrophils are largely incapable of directly phagocytizing epithelial cells. Instead, neutrophils can adhere to these cells and generate in an adherence-dependent process multiple ROS, some of which are highly toxic [25, 26]. Moreover, neutrophils can also secrete cytokines which can both serve to amplify or reduce the inflammatory response [27]. Similarly, monocytes can produce both pro-inflammatory and anti-inflammatory cytokines, and their role is likely highly context-dependent, similar to Kupffer cells [28, 29]. The capacity of neutrophils to produce ROS has led to their extensive study as potential mediators of injury [25, 30]. We will discuss this more specifically in the context of acetaminophen (APAP)-induced liver injury where substantial data exists both for and against their capacity to produce injury.
The sustained cytokine release from Kupffer cells also recruits other inflammatory cells, such as eosinophils, dendritic cells, and T cells, and more have also been implicated as pathogenic after DILI. While their specific roles have not been investigated as fully as in other disease, all of these inflammatory populations have been implicated in DILI [2, 6, 7, 31]. Many of these cells also have critical roles in immunity, and, thus, their overall contributions are not well understood.
Given the substantial differences present between different models of DILI, we will use APAP as a primary mechanism for describing understood mechanisms of inflammation after DILI.
Acetaminophen-Induced Liver Injury: A Clinically Relevant Model of Drug-Induced Liver Injury
APAP is a commonly used analgesic that is safe when used at therapeutic doses. However, an overdose of APAP remains the primary cause of drug-induced liver injury in Western societies [32]. The intracellular mechanisms of APAP-induced cell death have been examined extensively, and much of the molecular pathways are well understood. We will briefly discuss some of the relevant and well-established mechanisms that result in release of sterile inflammatory signals.
A majority of APAP is metabolized by glucuronidation and sulfation reactions and is then excreted through the urine [3]. Smaller amounts of APAP are metabolized by cytochrome P450 enzymes, mainly Cyp2E1, to the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) [33]. NAPQI is highly reactive, but it is largely captured by cellular glutathione (GSH) [34]. After an overdose, hepatic GSH levels are rapidly depleted and NAPQI begins to adduct cellular proteins [35]. In fasted mice, protein adduct formation occurs as early as 30 min and peaks at 2–3 h after the initial 300 mg/kg APAP overdose [35]. In humans, adduct formation is more delayed [36, 37]. Protein adduct formation, especially on mitochondrial proteins [38], is the primary initiating source of the injury process, and prevention of their formation is essentially completely protective against APAP-induced liver injury. Although inhibition of cytochrome P450 enzymes results in complete protection against APAP-induced liver injury [39], most patients seek medical attention after the metabolism phase, which makes cytochrome P450 enzymes not the most effective therapeutic targets in the clinic.
The mitochondrial protein adducts generate an initial oxidant stress, which activates c-Jun N-terminal kinase (JNK) [40•]. Phospho-JNK translocates to the mitochondria and amplifies the mitochondrial ROS and peroxynitrite formation [40•, 41•]. This amplification of ROS production in the mitochondria can be prevented through a number of different ROS quenching agents, all of which are highly protective, confirming the central role of the mitochondria [41•, 42]. The mitochondrial oxidant stress initiates cell death signaling outside the mitochondria as well, leading to translocation of proteins, such as Bax, to the mitochondria [43]. Bax forms pores in the outer mitochondria membrane, which results in the release of intermembrane proteins, such as endonuclease G and apoptosis-inducing factor (AIF) [43]. These proteins translocate to the nucleus and cause DNA fragmentation [44]. In addition, the mitochondrial oxidant stress and peroxynitrite cause the mitochondrial permeability transition pore (MPTP) opening [45]. Both DNA fragmentation and the MPTP formation are critical events in APAP-induced necrosis [46]. In addition to the intracellular signaling mechanisms of cell death, adaptive responses also have to be considered. Removal of damaged mitochondria and APAP protein adducts by autophagy [47] and induction of mitochondrial biogenesis [48•] are critical modulating events of APAP-induced liver injury and release of DAMPs.
During necrotic cell death, intracellular components are released into the serum [15, 49, 50]. This includes a number of the aforementioned DAMPs and initiates the sterile inflammatory response in APAP-induced liver injury [1, 13•] (Fig. 1b). The mechanisms that control inflammation at this point are fairly well-established as listed previously. The pathophysiological role of inflammation in APAP-induced liver injury has remained a controversial issue in the literature [1, 13•]. Many of the established mechanisms are derived from the studies attempting to determine the role of inflammation as an event that aggravates the initial injury, and thus, we will discuss these mechanisms in the context of the experimental evidence.
The Acetaminophen-Induced Sterile Inflammatory Response
Evidence exists both for and against the idea that activation of Kupffer cells and neutrophils can amplify the APAP-induced cellular injury. Data exists both with direct interventions against specific cell types and specific mediators of inflammation. We will go through these mediators individually to define their potential contribution (Fig. 1b).
Kupffer Cells and Monocytes
Because of the massive release of cellular components, it was hypothesized that activation of Kupffer cells would exacerbate APAP-induced liver injury [51] similar to other models of liver inflammation [26]. Surprisingly though, Kupffer cells are largely depleted during APAP-induced liver injury and monocyte-derived macrophages (MoMF) are recruited into the liver [52, 53]. As such, many experiments have focused on both the role of endogenous macrophages and recruited monocytes as potential mediators. Initial experiments using gadolinium chloride, a potent Kupffer cell inactivator, indicated a protective effect against APAP toxicity [51]. Later experiments using a strategy to destroy Kupffer cells prior to APAP administration demonstrated a beneficial effect of Kupffer cells rather than being the cause of the injury [18]. In addition, mice deficient in NADPH oxidase, which is required for Kupffer cell-mediated oxidative burst, did not show protection against APAP-induced liver injury suggesting that a Kupffer cell-derived oxidant stress is not involved in the injury process [18, 54, 55]. As such, it is highly unlikely that Kupffer cells directly promote APAP-induced hepatotoxicity. While livers injured by APAP produce large amounts of monocyte chemoattractant protein-1 (MCP-1), a primary monocyte recruitment chemokine and macrophage activator, mice deficient of MCP-1 were not protected against APAP toxicity [56]. Furthermore, animal deficient of the MCP-1 receptor CCR2 were not protected [52, 56]. In contrast, CCR2 mice showed reduced regeneration of the tissue damage suggesting that the recruited monocytes are important for the recovery by removing the cell debris [52, 56]. Despite the fact that no known function of macrophages is associated with APAP-induced cell death, a number of recent studies have again brought up the idea that monocytes or macrophages are capable of exacerbating inflammation [29, 57••, 58•]. These reports are in striking contrast to previous studies using the same interventions without explanation about the differences or attempts to address these controversies. Whether these differences are due to variations in experimental design, differences in the gut microbiome, mouse strain differences, or mismatched wild-type strains are not known. Unfortunately, contradicting results such as these can be difficult to explain. We can only emphasize that if investigators publish opposite results, they have the obligation to address the reasons for the differences to previous publications. In addition, an increased focus must be placed on the rigor of the experimental design and consideration of off-target effects of reagents and genetic interventions and their impact on the role of inflammation and inflammatory mediators in APAP-induced liver injury.
In contrast, a number of studies have suggested a potential immune-regulatory role of macrophages after APAP overdose. In patients with APAP toxicity, monocyte-derived macrophages are recruited in significant numbers into the liver [28, 53, 59]. These cells have a largely anti-inflammatory phenotype indicative of a pro-regenerative, pro-wound resolution phenotype [28, 53, 59]. This may be mediated by secretory leukocyte protease inhibitor (SLPI), which has a potent anti-inflammatory effect after APAP-induced liver injury [28, 59]. Murine studies largely confirm this as recruited monocytes regulate both neutrophil survival and neutrophil clearance [60••]. The effects of SLPI are apparently mediated by Mer tyrosine kinase, providing a potential therapeutic target or biomarker for understanding macrophage function during APAP overdose [61••]. Furthermore, restoration of innate immune activity in vivo in patients using colony-stimulating factor-1 improves outcome in patients with acute liver failure, including patients with APAP overdose [62]. Similarly, monocytopenia is associated with far worse outcomes after APAP-induced liver injury that is not found in other models of acute liver failure [63]. As such, interventions designed at limiting the effects of Kupffer cells and monocyte-derived macrophages may be highly inadvisable given the role of these phagocytes in regeneration and the prominent role of the liver in innate immunity and the pressing need for avoiding sepsis in patients.
Neutrophils
The role of neutrophils remains the most controversial issue in APAP-induced liver injury, while also being a major area of research [1, 13•]. It has been proposed routinely over the previous decade that neutrophils may play a significant role as mediators of either Kupffer cell-derived [50] or NK cell-derived inflammation [64]. A number of papers have also directly looked at the role of neutrophils and determined that inhibition of inflammation or direct interventions against neutrophils themselves can be protective against APAP-induced liver injury [15, 64, 65]. A large number of these papers have focused on an axis wherein DAMPs, such as ATP, HMGB1, formyl peptides, and mitochondrial DNA, are released, Kupffer cells are primed and activated to generate mediators, such as IL-1ß, via the inflammasome, neutrophils are recruited, and then neutrophils further provoke inflammation and hepatic injury through hepatocyte killing [13•]. Neutrophil recruitment then occurs through release of chemokines and cytokines, such as keratinocyte factor (KC), macrophage inflammatory proteins 1 and 2 (MIP-1/MIP-2), and interleukin-6 (IL-6), in addition to release of IL-1ß and IL-1α in the mouse [15, 57••, 66]. Knockout of DAMPs and their receptors ameliorate inflammation and simultaneously reduce APAP-induced liver injury [15, 65]. Similarly, augmentation of cytokine levels or cytokine receptor levels has been proposed to reduce injury after APAP overdose [15, 57••]. Recruited neutrophils have been proposed to kill cells through a variety of mechanisms after APAP-induced liver injury including release of ROS [25] and release of proteases, such as elastase [65]. Thus, there is a growing list of studies that suggest a role of neutrophils in aggravation of APAP-induced liver injury [1, 13•].
However, in stark contrast to these data, more specific interventions against neutrophils were found to be ineffective and classical indicators of neutrophil-mediated tissue injury were not present after APAP-induced liver injury [54, 55, 66,67,68]. Similarly, when markers of neutrophil activation, such as CD11b expression and priming for ROS, were measured in circulating and in liver-infiltrating neutrophils, they were largely not elevated during the period of injury [55, 68]. Instead, increased CD11b expression, ROS priming, and enhanced phagocytosis capacity occurred after the injury and during regeneration suggesting a pro-wound resolution role [55]. Furthermore, it is well-established that neutrophils kill target cells by ROS formation, especially hypochlorite, in different models of acute liver injury [25, 30, 69]. However, there is no evidence for a direct neutrophil-induced oxidant stress during APAP toxicity [67]. Consistent with these observations is that animals deficient in NADPH oxidase (NOX2) activity, the key enzyme responsible for ROS formation in these phagocytes, show similar oxidant stress and injury as wild-type animals [54, 55]. Given the established mechanisms of neutrophil-induced cell killing in the liver [25, 70], these data make it highly unlikely that neutrophils aggravate APAP-induced liver injury [13•, 31].
Attempts to repeat some of the previous results using animals deficient in mediators of the inflammasome failed to yield repeatable results [57••, 68, 71]. Administering high doses of IL-1ß directly during APAP-induced liver injury did not further enhance liver injury despite the fact that it potently enhanced neutrophil recruitment [72]. Other groups have proposed that the alarmin IL-1α and not IL-1β may actually be responsible for the injury [57••]. In both cases, the injury would depend on the IL-1 receptor although its role has been questioned as IL-1R-deficient mice were found not to be protected [72]. Similarly, mice deficient in CD18, a primary neutrophil adhesion molecule critically involved in neutrophil extravasation and ROS formation in the liver [26, 70], also displayed no protection, and there is no established role for CD18 or its binding partner intracellular adhesion molecule (ICAM-1) [66,67,68]. Most importantly, neutrophil depletion directly has been shown to yield a protective response when given 24 h prior to APAP; however, this protection is caused by off-target pre-conditioning effects independently of neutrophil cytotoxicity [73]. When neutrophil-depleting agents are given before neutrophil recruitment, but after APAP metabolism, there is no protection [67]. Moreover, global knockout of elastase, a key protease secreted by neutrophils did not protect against APAP-induced liver injury (Woolbright and Jaeschke unpublished).
Thus, despite that a number of studies appear to support a role of neutrophils in APAP-induced liver injury, many investigations using specific interventions against neutrophil cytotoxicity failed to show an impact on the pathophysiology. Most importantly, there is no evidence for neutrophil activation during the injury phase in APAP overdose patients suggesting that neutrophils are not involved in the injury but contribute to the recovery [55].
DAMPs, Cytokines, and Other Inflammatory Mediators in APAP-Induced Inflammation
Regardless of the role of individual cell types, a number of mediators have established roles in the actual inflammatory process as discussed previously. Removal of their cellular receptors or direct removal of the component reduces inflammation after APAP-induced liver injury largely confirming the role of DAMPs as initial mediators of inflammation. Some roles have been established for other cytokines that mediate subsequent effects as well.
Individual cytokines have also been observed to have direct effects. IL-10 levels are substantially elevated in mice and in patients with APAP overdose [74], notably though, while IL-10 levels are associated with non-survival in patients [75], knockout of IL-10 in mice substantially increases lethality of APAP [76]. As studies with IL-10-deficient mice showed, IL-10 is limiting pro-inflammatory cytokine formation and as a consequence, the induction of inducible nitric oxide synthase, which promotes cell death through peroxynitrite formation [76]. The role of IL-22 is currently being studied with mixed results in APAP hepatotoxicity. Knockout of IL-22 binding protein (IL-22BP) results in increased inflammation and increased liver injury in an IL-22/CXCL10-dependent manner [77]. However, pretreatment with IL-22 reduced injury through STAT3 activation [78]. In contrast, chronic IL-22 overexpression in transgenic mice caused increased APAP-induced injury, but this is likely due to constitutive overexpression of CYP2E1 and increased metabolic activation in these animals [78]. IL-6 is also elevated in mice after APAP and is linked to regeneration [79]. IL-6 KO mice showed delayed regeneration after APAP, which was corrected by treatment with recombinant IL-6 [79]. However, the reduced APAP-induced liver injury enhanced regeneration and increased survival in animals treated with GSH correlated with lower plasma IL-6 levels [80]. Similarly, reduced mortality of APAP overdose patients also correlated with lower plasma IL-6 levels [81]. In addition, IL-10 KO mice [76] and IL-10/IL-4 double KO mice [82], which are both more susceptible to APAP-induced liver injury, had higher plasma levels of IL-6 compared to wild-type animals. Treatment with an IL-6 antibody protected the IL-10/IL-4 double KO mice, and IL-10/IL-4/IL-6 triple KO mice were also protected [82]. These data suggest that IL-6 can promote regeneration, but under conditions, such as low expression of anti-inflammatory cytokines, it can also enhance liver injury. IL-4 has been proposed to reduce APAP-induced liver injury [83]. The protective effect of IL-4 may be related to enhancing hepatic GSH synthesis and protection against oxidant stress [83]. However, IL-4 can also regulate CYP2E1 [84]. IL-13 is another endogenous cytokine that appears to attenuate pro-inflammatory cytokine and chemokine formation and reduce APAP-induced liver injury [85]. Although pretreatment with a neutropenia-induced antibody attenuated liver injury in IL-13 KO mice [85], the role of neutrophils in this context is still unclear because the animals were pretreated with the antibody raising the previously discussed concern of preconditioning [73]. Another cytokine, which is implicated in neutrophil recruitment, is IL-17. IL-17 KO showed partially reduced APAP hepatotoxicity, which is correlated with lower neutrophil and macrophage accumulation in the liver [86]. Although it was concluded that IL-17 promotes liver injury through neutrophil recruitment [86], the fact that the mitogen-activated protein kinase ERK activation was attenuated in the IL-17 KO mice raises the possibility that the pro-inflammatory effect may be a consequence rather than the cause of the reduced injury. Given immune differences between humans and mice, the immune response that occurs in humans during APAP overdose may be deleterious to survival or may be linked to concurrent infection due to reduced liver function. Notably, mice given colony-stimulating factor (CSF) have increased innate immune function and increased macrophage function in the liver, yet they do not have increased liver injury [87]. Instead, these mice are protected against APAP, which correlates with results found in patients where those with high serum levels of CSF had better outcomes [87].
As such, the role and consequences of inflammation after APAP overdose are still very much under debate. What is widely conserved between studies is the idea that the initial cell death response provokes sterile inflammatory signals that then provoke further inflammation. Although DAMP-mediated inflammation is well-established, the default assumption is mostly that this will trigger an aggravation of the injury through cytotoxic neutrophils and monocytes. However, given the strong data against the involvement of neutrophils in the injury, alternative explanations need to be considered. Clearly, IL-10 with its effect on suppressing iNOS and peroxynitrite formation [76] and IL-4 with its effect on hepatic GSH recovery [83] are examples of inflammatory mediators affecting the intracellular mechanisms of cell death. More of these types of connections between inflammatory mediators and their impact on cell death signaling mechanisms need to be studied.
Idiosyncratic Drug-Induced Liver Injury
APAP-induced liver injury is a well-characterized type of drug-induced liver injury wherein a primary necrotic stimulus triggers secondary inflammation. While APAP remains the most common cause of drug-induced liver injury, idiosyncratic drug-induced liver injury (IDILI) ranks second in overall causes of acute liver failure [88]. A number of drugs from diverse classes of therapeutics have been associated with IDILI (Table 1). This is in spite of the fact that IDILI is difficult to diagnose due to issues with reporting and problems with establishing a drug as the root cause of the injury through exclusion. While IDILI may be caused by different mechanisms depending on the drug, one commonly noted mechanism is the potential for an innate immune response as an initiating event in IDILI. Classically, it is believed that some drugs cause formation of drug–protein conjugates (haptens), which then illicit an immune response from the adaptive immune system over time [96••, 97] (Fig. 1a). These data are strongly supported by previous measurements of haptens after exposure to drugs, such as halothane, and the fact that removal of the drug usually results in complete recovery if done in time [96••]. As to why these haptens form is still under debate. What is fairly well understood is that the hapten molecules result in activation of antigen-presenting cells (APCs) [98]. These cells then activate T cells, which can be cytotoxic to other cells, including hepatocytes. Halothane-induced hepatitis is a classic example of this type of injury [89]. Recent advances indicate halothane-induced hepatitis may be partially mediated by eosinophils, which likely act as co-stimulatory units for T cell-mediated cell death or may be able to kill hepatocytes themselves through release of major basic proteins [89]. This links them more directly to the adaptive immune system, which has long been believed to be a player in the allergen-like hepatitis associated with halothane and many other types of IDILI.
The other predominant hypothesis is the “danger hypothesis” wherein the presence of co-stimulatory signals by DAMPs further amplifies the immunogenic signal from the hapten signal and initiates the actual immune intolerance and the subsequent T cell response and cell death [96••, 97] (Fig. 1a). A recent paper examined this potential and found that mice deficient in programmed cell death protein 1 (PD1) displayed the characteristic cell death associated with troglitazone, but not its analogue pioglitazone [4••]. Importantly, this is the first animal model that has demonstrated a fundamental difference between a compound that is known to produce IDILI (troglitazone) and one that does not produce DILI but is analogous in its pharmacological mechanism (pioglitazone). Notably microsomal fractions of cells incubated with troglitazone activated the inflammasome in macrophages [4••]. As such, the drug–antibody conjugates produced by troglitazone metabolism may activate the inflammasome and thus serve as DAMPs themselves. Why this occurs in PD-1−/− mice, but not normal mice, is not yet well understood, but may have to do with immune tolerance in T cells given the prominent role of PD-1 in immune tolerance and the recent advent of successful treatment of patients with checkpoint inhibitors. Similar results were previously obtained with isoniazid and nevirapine, also characteristic DILI inducers [94]. This is an exciting and novel area that may have serious therapeutic benefit both for drug development and for patients.
A third model of IDILI has also been proposed, called the p-I Concept [99]. The fundamental concept of this hypothesis is that the agent can directly interact with the immune system. This hypothesis supposes that drugs bind to highly variable antigen specific regions directly instead of covalently modifying peptides as in the hapten hypothesis. The primary evidence for this hypothesis are the findings that only a portion of T cells react with a drug and that many drugs are capable of causing T cell activation when T cell clones are used instead of T cells [100]. This reaction occurs very rapidly (< 1 min) which eliminates the potential for antibody processing [99]. These T cells require co-stimulation in the sulfamethoxazole model, which may recapitulate the human condition [101]. Activation of these T cells results in a prolonged and potent immune response that dramatically damages the liver. The primary difference between this model and the aforementioned models is the direct interaction of the drug with the antigen receptors.
The presence of auto-antibodies against proteins, such as CYP2E1 or liver endoplasmic reticulum proteins, is another noted feature of IDILI [102]. These autoantibodies are present in halothane hepatitis, isoniazid, as well as human autoimmune hepatitis that is not associated with DILI [103, 104]. A number of different drugs result in development of these autoantibodies and a subsequent immune response [102]. CYP2E1 autoantibodies in particular have been noted in other diseases as well and thus may be a source of self-immune rejection common in the liver [102]. Notably, a recent large study in human patients indicates that autoantibodies, such as antinuclear antibodies (ANA), are present in a diverse array of injuries associated with DILI, and their levels decrease with both treatment and recovery, indicating a potentially causative role [105]. These autoantibodies may function highly similarly to hapten molecules and functionally serve the same role as to provide a source of autoimmunity that sparks T cell-mediated rejection. It remains undetermined whether antibody recognition by drugs can elicit the same response as covalent binding or hapten formation, although these studies have laid the groundwork for future efforts aimed at defining this mechanism in multiple models.
Conclusions
Xenobiotic metabolism can result in the formation of reactive metabolites that bind to proteins and damage cells. Therefore, it is imperative to understand the interaction between cell death with release of DAMPs and the resulting induction of a sterile inflammatory response (innate immunity) and potentially also an adaptive immune response. Although the main purpose of the immune response after cell death in the liver is the removal of necrotic cell debris and initiation of repair, it also has the potential to aggravate the existing injury through multiple pathways. However, it is incompletely understood when protein adducts act as haptens and when DAMP release triggers a detrimental inflammatory response versus promoting repair. The large number of studies published with controversial results indicates the need for more detailed mechanistic studies that specifically address these controversies and identify clinically relevant therapeutic targets.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major Importance
Woolbright BL, Jaeschke H. The impact of sterile inflammation in acute liver injury. J Clin Transl Res. 2017;3(Suppl 1):170–88.
Jaeschke H, Woolbright BL. Current strategies to minimize hepatic ischemia–reperfusion injury by targeting reactive oxygen species. Transplant Rev (Orlando). 2012;26(2):103–14.
McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013;30(9):2174–87.
•• Mak A, Kato R, Weston K, Hayes A, Uetrecht J. Editor’s highlight: an impaired immune tolerance animal model distinguishes the potential of troglitazone/pioglitazone and tolcapone/entacapone to cause IDILI. Toxicol Sci. 2018;161(2):412–20. First demonstration that immune tolerance impairment might mediate differences in DILI in similar drug types.
Jaeschke H. Acetaminophen: dose-dependent drug hepatotoxicity and acute liver failure in patients. Dig Dis. 2015;33(4):464–71.
Jaeschke H, Naisbitt DJ. Immune mechanisms in drug-induced liver injury. In: Drug-induced liver toxicity, Chen, M. and Will, Y. (eds.), 1st edn. Humana Press, New York, pp. 511–531, 2018.
Kim SH, Naisbitt DJ. Update on advances in research on idiosyncratic drug-induced liver injury. Allergy Asthma Immunol Res. 2016;8(1):3–11.
Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. 2003;4(2):95–104.
Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81.
Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012 Nov;143(5):1158–72.
Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3(9):e955691.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20.
• Woolbright BL, Jaeschke H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol. 2017;66(4):836–48. Recent review fully examining conflicts in the role of the inflammasome in APAP induced liver injury.
• Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–20. Landmark review on the inflammasome from leading authors.
Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119(2):305–14.
Vince JE, Silke J. The intersection of cell death and inflammasome activation. Cell Mol Life Sci. 2016;73(11–12):2349–67.
Jaeschke H, Farhood A, Bautista AP, Spolarics Z, Spitzer JJ. Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. Am J Phys. 1993;264(4 Pt 1):G801–9.
Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, et al. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. 2002;15(12):1504–13.
Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. 1994;20(2):453–60.
Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Phys. 1991;260(3 Pt 1):G355–62.
Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic Res Commun. 1991;15(5):277–84.
Witthaut R, Farhood A, Smith CW, Jaeschke H. Complement and tumor necrosis factor-alpha contribute to Mac-1 (CD11b/CD18) up-regulation and systemic neutrophil activation during endotoxemia in vivo. J Leukoc Biol. 1994;55(1):105–11.
Pfeffer K, Matsuyama T, Kündig TM, Wakeham A, Kishihara K, Shahinian A, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73(3):457–67.
Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA Jr. Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest. 1990;85(6):1936–43.
Jaeschke H. Mechanisms of liver injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia–reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006;290(6):G1083–8.
Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. J Gastroenterol Hepatol. 2011;26(Suppl 1):173–9.
Taïeb J, Mathurin P, Elbim C, Cluzel P, Arce-Vicioso M, Bernard B, et al. Blood neutrophil functions and cytokine release in severe alcoholic hepatitis: effect of corticosteroids. J Hepatol. 2000;32(4):579–86.
Antoniades CG, Quaglia A, Taams LS, Mitry RR, Hussain M, Abeles R, et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology. 2012;56(2):735–46.
Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. 2016;64(5):1667–82.
Hasegawa T, Malle E, Farhood A, Jaeschke H. Generation of hypochlorite-modified proteins by neutrophils during ischemia–reperfusion injury in rat liver: attenuation by ischemic preconditioning. Am J Physiol Gastrointest Liver Physiol. 2005;289(4):G760–7.
Jaeschke H, Williams CD, Ramachandran A, Bajt ML. Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity. Liver Int. 2012;32(1):8–20.
Lee WM. Drug-induced acute liver failure. Clin Liver Dis. 2013;17(4):575–86.
Corcoran GB, Mitchell JR, Vaishnav YN, Horning EC. Evidence that acetaminophen and N-hydroxyacetaminophen form a common arylating intermediate, N-acetyl-p-benzoquinoneimine. Mol Pharmacol. 1980;18(3):536–42.
Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther. 1973;187(1):211–7.
McGill MR, Lebofsky M, Norris HR, Slawson MH, Bajt ML, Xie Y, et al. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose–response, mechanisms, and clinical implications. Toxicol Appl Pharmacol. 2013;269(3):240–9.
Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, et al. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol. 2014;279(3):266–74.
Xie Y, McGill MR, Cook SF, Sharpe MR, Winefield RD, Wilkins DG, et al. Time course of acetaminophen-protein adducts and acetaminophen metabolites in circulation of overdose patients and in HepaRG cells. Xenobiotica. 2015;45(10):921–9.
Xie Y, McGill MR, Du K, Dorko K, Kumer SC, Schmitt TM, et al. Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol Appl Pharmacol. 2015;289(2):213–22.
Akakpo JY, Ramachandran A, Kandel SE, Ni HM, Kumer SC, Rumack BH, et al. 4-Methylpyrazole protects against acetaminophen hepatotoxicity in mice and in primary human hepatocytes. Human Exp Toxicol. 2018; in press
• Kaplowitz N, Win S, Than TA, Liu ZX, Dara L. Targeting signal transduction pathways which regulate necrosis in acetaminophen hepatotoxicity. J Hepatol. 2015;63(1):5–7. Signature review on mechanisms of regulated necrosis in APAP induced liver injury.
• Du K, Ramachandran A, Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol. 2016;10:148–56. Authoritative review on the role of oxidative stress in APAP overdose.
Du K, Farhood A, Jaeschke H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch Toxicol. 2017;91(2):761–73.
Bajt ML, Farhood A, Lemasters JJ, Jaeschke H. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2008;324(1):8–14.
Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci. 2006;94(1):217–25.
Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40(5):1170–9.
Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci. 2002;67(2):322–8.
Chao X, Wang H, Jaeschke H, Ding WX. Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int. 2018 Apr 23; https://doi.org/10.1111/liv.13866.
• Du K, Ramachandran A, MR MG, Mansouri A, Asselah T, Farhood A, et al. Induction of mitochondrial biogenesis protects against acetaminophen hepatotoxicity. Food Chem Toxicol. 2017 Oct;108(Pt A):339–50. First demonstration of the role of mitochondrial biogenesis in APAP overdose.
McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122(4):1574–83.
Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, et al. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol. 2012;56(5):1070–9.
Laskin DL, Gardner CR, Price VF, Jollow DJ. Modulation of macrophage functioning abrogates the acute hepatotoxicity of acetaminophen. Hepatology. 1995;21(4):1045–50.
Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol. 2008;84(6):1410–21.
Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident Kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. 2014;193(1):344–53.
James LP, McCullough SS, Knight TR, Jaeschke H, Hinson JA. Acetaminophen toxicity in mice lacking NADPH oxidase activity: role of peroxynitrite formation and mitochondrial oxidant stress. Free Radic Res. 2003;37(12):1289–97.
Williams CD, Bajt ML, Sharpe MR, McGill MR, Farhood A, Jaeschke H. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol Appl Pharmacol. 2014;275(2):122–33.
Dambach DM, Watson LM, Gray KR, Durham SK, Laskin DL. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology. 2002;35(5):1093–103.
•• Zhang C, Feng J, Du J, Zhuo Z, Yang S, Zhang W, et al. Macrophage-derived IL-1α promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell Mol Immunol. 2017; https://doi.org/10.1038/cmi.2017.22. Important paper discussing the implications of inflammatory cell death and alarmin release.
• Perugorria MJ, Esparza-Baquer A, Oakley F, Labiano I, Korosec A, Jais A, Mann J, Tiniakos D, Santos-Laso A, Arbelaiz A, Gawish R, Sampedro A, Fontanellas A, Hijona E, Jimenez-Agüero R, Esterbauer H, Stoiber D, Bujanda L, Banales JM, Knapp S, Sharif O, Mann DA. Non-parenchymal TREM-2 protects the liver from immune-mediated hepatocellular damage. Gut 2018. Recent publication on a potential role for TREM2 in the control macrophages of macrophage-mediated liver injury.
Antoniades CG, Khamri W, Abeles RD, Taams LS, Triantafyllou E, Possamai LA, et al. Secretory leukocyte protease inhibitor: a pivotal mediator of anti-inflammatory responses in acetaminophen-induced acute liver failure. Hepatology. 2014;59(4):1564–76.
•• Graubardt N, Vugman M, Mouhadeb O, Caliari G, Pasmanik-Chor M, Reuveni D, et al. Ly6Chi monocytes and their macrophage descendants regulate neutrophil function and clearance in acetaminophen-induced liver injury. Front Immunol. 2017;8:626. Recent publication emphasizing a bi-phasic role for macrophages in neutrophil activation and wound healing after APAP overdose.
•• Triantafyllou E, Pop OT, Possamai LA, Wilhelm A, Liaskou E, Singanayagam A, et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut. 2018;67(2):333–47. Signature paper on a role for macrophage mediated wound resolution after APAP overdose in patients.
Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, et al. CSF1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology. 2015;149(7):1896–909.e14.
Moore JK, MacKinnon AC, Man TY, Manning JR, Forbes SJ, Simpson KJ. Patients with the worst outcomes after paracetamol (acetaminophen)-induced liver failure have an early monocytopenia. Aliment Pharmacol Ther. 2017;45(3):443–54.
Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology. 2004;127(6):1760–74.
Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2015;125(2):539–50.
Lawson JA, Farhood A, Hopper RD, Bajt ML, Jaeschke H. The hepatic inflammatory response after acetaminophen overdose: role of neutrophils. Toxicol Sci. 2000;54(2):509–16.
Cover C, Liu J, Farhood A, Malle E, Waalkes MP, Bajt ML, et al. Pathophysiological role of the acute inflammatory response during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2006;216(1):98–107.
Williams CD, Bajt ML, Farhood A, Jaeschke H. Acetaminophen-induced hepatic neutrophil accumulation and inflammatory liver injury in CD18-deficient mice. Liver Int. 2010;30(9):1280–92.
Gujral JS, Hinson JA, Farhood A, Jaeschke H. NADPH oxidase-derived oxidant stress is critical for neutrophil cytotoxicity during endotoxemia. Am J Physiol Gastrointest Liver Physiol. 2004;287(1):G243–52.
Jaeschke H. Molecular mechanisms of hepatic ischemia–reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284(1):G15–26.
Williams CD, Antoine DJ, Shaw PJ, Benson C, Farhood A, Williams DP, et al. Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol Appl Pharmacol. 2011;252(3):289–97.
Williams CD, Farhood A, Jaeschke H. Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol Appl Pharmacol. 2010;247(3):169–78.
Jaeschke H, Liu J. Neutrophil depletion protects against murine acetaminophen hepatotoxicity: another perspective. Hepatology. 2007;45(6):1588–9.
James LP, Simpson PM, Farrar HC, Kearns GL, Wasserman GS, Blumer JL, et al. Cytokines and toxicity in acetaminophen overdose. J Clin Pharmacol. 2005;45(10):1165–71.
Berry PA, Antoniades CG, Hussain MJ, McPhail MJ, Bernal W, Vergani D, et al. Admission levels and early changes in serum interleukin-10 are predictive of poor outcome in acute liver failure and decompensated cirrhosis. Liver Int. 2010;30(5):733–40.
Bourdi M, Masubuchi Y, Reilly TP, Amouzadeh HR, Martin JL, George JW, et al. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. 2002;35(2):289–98.
Kleinschmidt D, Giannou AD, McGee HM, Kempski J, Steglich B, Huber FJ, et al. A protective function of IL-22BP in ischemia reperfusion and acetaminophen-induced liver injury. J Immunol. 2017;199(12):4078–90.
Feng D, Wang Y, Wang H, Weng H, Kong X, Martin-Murphy BV, et al. Acute and chronic effects of IL-22 on acetaminophen-induced liver injury. J Immunol. 2014;193(5):2512–8.
James LP, Lamps LW, McCullough S, Hinson JA. Interleukin 6 and hepatocyte regeneration in acetaminophen toxicity in the mouse. Biochem Biophys Res Commun. 2003;309(4):857–63.
Bajt ML, Knight TR, Farhood A, Jaeschke H. Scavenging peroxynitrite with glutathione promotes regeneration and enhances survival during acetaminophen-induced liver injury in mice. J Pharmacol Exp Ther. 2003;307(1):67–73.
Moore JK, Craig DG, Pryde EA, Walker SW, Beckett GJ, Hayes PC, et al. Persistently elevated troponin I in paracetamol hepatotoxicity: association with liver injury, organ failure, and outcome. Clin Toxicol (Phila). 2013;51(7):532–9.
Bourdi M, Eiras DP, Holt MP, Webster MR, Reilly TP, Welch KD, et al. Role of IL-6 in an IL-10 and IL-4 double knockout mouse model uniquely susceptible to acetaminophen-induced liver injury. Chem Res Toxicol. 2007;20(2):208–16.
Ryan PM, Bourdi M, Korrapati MC, Proctor WR, Vasquez RA, Yee SB, et al. Endogenous interleukin-4 regulates glutathione synthesis following acetaminophen-induced liver injury in mice. Chem Res Toxicol. 2012;25(1):83–93.
Lagadic-Gossmann D, Lerche C, Rissel M, Joannard F, Galisteo M, Guillouzo A, et al. The induction of the human hepatic CYP2E1 gene by interleukin 4 is transcriptional and regulated by protein kinase C. Cell Biol Toxicol. 2000;16(4):221–33.
Yee SB, Bourdi M, Masson MJ, Pohl LR. Hepatoprotective role of endogenous interleukin-13 in a murine model of acetaminophen-induced liver disease. Chem Res Toxicol. 2007;20(5):734–44.
Lee HC, Liao CC, Day YJ, Liou JT, Li AH, Liu FC. IL-17 deficiency attenuates acetaminophen-induced hepatotoxicity in mice. Toxicol Lett. 2018;292:20–30.
Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, et al. CSF1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology. 2015;149(7):1896–909.
Tujios SR, Lee WM. Acute liver failure induced by idiosyncratic reaction to drugs: challenges in diagnosis and therapy. Liver Int. 2018;38(1):6–14.
Proctor WR, Chakraborty M, Chea LS, Morrison JC, Berkson JD, Semple K, et al. Eosinophils mediate the pathogenesis of halothane-induced liver injury in mice. Hepatology. 2013;57(5):2026–36.
Yano A, Higuchi S, Tsuneyama K, Fukami T, Nakajima M, Yokoi T. Involvement of immune-related factors in diclofenac-induced acute liver injury in mice. Toxicology. 2012;293(1–3):107–14.
Oker-Blom C, Mäkinen J, Gothoni G. Toxicological studies on tienilic acid in rats. Toxicol Lett. 1980;6(2):93–9.
Pariente EA, Pessayre D, Bernuau J, Degott C, Benhamou JP. Dihydralazine hepatitis: report of a case and review of the literature. Digestion. 1983;27(1):47–52.
Li F, Lu J, Cheng J, Wang L, Matsubara T, Csanaky IL, et al. Human PXR modulates hepatotoxicity associated with rifampicin and isoniazid co-therapy. Nat Med. 2013;19(4):418–20.
Metushi IG, Hayes MA, Uetrecht J. Treatment of PD-1(−/−) mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology. 2015;61(4):1332–42.
Wuillemin N, Terracciano L, Beltraminelli H, Schlapbach C, Fontana S, Krähenbühl S, et al. T cells infiltrate the liver and kill hepatocytes in HLA-B(∗)57:01-associated floxacillin-induced liver injury. Am J Pathol. 2014;184(6):1677–82.
•• Cho T, Uetrecht J. How reactive metabolites induce an immune response that sometimes leads to an idiosyncratic drug reaction. Chem Res Toxicol. 2017;30(1):295–314. Recent review on mechanisms of idiosyncratice liver disease initiated by reactive metabolite formation.
Uetrecht J. Idiosyncratic drug reactions: past, present, and future. Chem Res Toxicol. 2008;21(1):84–92.
Pohl LR, Kenna JG, Satoh H, Christ D, Martin JL. Neoantigens associated with halothane hepatitis. Drug Metab Rev. 1989;20(2–4):203–17.
Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med. 2003;1:683–93.
Naisbitt DJ, Farrell J, Wong G, Depta JP, Dodd CC, Hopkins JE, et al. Characterization of drug-specific T cells in lamotrigine hypersensitivity. J Allergy Clin Immunol. 2003;1:1393–403.
Pichler WJ, Adam J, Watkins S, Wuillemin N, Yun J, Yerly D. Drug hypersensitivity: how drugs stimulate T cells via pharmacological interaction with immune receptors. Int Arch Allergy Immunol. 2015;168(1):13–24.
deLemos AS, Foureau DM, Jacobs C, Ahrens W, Russo MW, Bonkovsky HL. Drug-induced liver injury with autoimmune features. Semin Liver Dis. 2014;34(2):194–204.
Hintermann E, Holdener M, Bayer M, Loges S, Pfeilschifter JM, Granier C, et al. Epitope spreading of the anti-CYP2D6 antibody response in patients with autoimmune hepatitis and in the CYP2D6 mouse model. J Autoimmun. 2011;37(3):242–53.
Kitteringham NR, Kenna JG, Park BK. Detection of autoantibodies directed against human hepatic endoplasmic reticulum in sera from patients with halothane-associated hepatitis. Br J Clin Pharmacol. 1995;40(4):379–86.
de Boer YS, Kosinski AS, Urban TJ, Zhao Z, Long N, Chalasani N, et al. Features of autoimmune hepatitis in patients with drug-induced liver injury. Clin Gastroenterol Hepatol. 2017;15(1):103–112.e2.
Acknowledgments
Work in the authors’ laboratory was supported in part by the National Institutes of Health grants R01 DK102142 and R01 AA12916, the Robert Hanlon Trust, and by grants from the National Institute of General Medical Sciences (P20 GM103549 and P30 GM118247) of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no conflict of interest to disclose.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
This article is part of the Topical Collection on Drug-induced Liver Injury
Rights and permissions
About this article

Cite this article
Woolbright, B.L., Jaeschke, H. Mechanisms of Inflammatory Liver Injury and Drug-Induced Hepatotoxicity. Curr Pharmacol Rep 4, 346–357 (2018). https://doi.org/10.1007/s40495-018-0147-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-018-0147-0