
Abstract
Purpose of Review
This review summarizes some of the most recent studies on single-cell resolution sequencing of the post-mortem human brain and the application of these techniques for the study of psychiatric and neurological disorders.
Recent Findings
Over the last several years, researchers have optimized single-cell transcriptome and genome sequencing in post-mortem human brain tissue. This has given us unprecedented access to cell-type specific gene expression profiles and somatic mutations unique to pathological states. Additionally, we can now measure epigenetic information from individual brain cells and with advanced statistical approaches, we can focus on the key cell-types underlying psychiatric and other brain phenotypes.
Summary
A new era of cell-type specific and single-cell resolution studies of the human brain is underway. With proper application of rapidly advancing laboratory and data analysis techniques, we should witness important advances in our understanding of molecular changes associated with psychiatric and neurological phenotypes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The extensive specialization and diversity of cell-types in the mammalian brain has been of great interest since the earliest days of neuroscience. Over the decades, technological advances have allowed us to interrogate various features of brain cell-types, including their morphology, electrophysiology, and molecular biology, in ever-increasing detail. Many of these techniques provide some insight into the functioning of single brain cells, such as patch-clamp recordings of individual neurons in brain slices or tracking of fluorescently labelled microglia in vivo. However, with the advent of single-cell next-generation sequencing (NGS) techniques, we are now able to study in minute detail the identity, function, and state of individual brain cells.
Studies of the human brain at single-cell resolution have become an invaluable resource for teasing apart the interactions between members of this complex cellular ecosystem. Cell-type specificity provided by single-cell approaches allows us to hone in on the individual components contributing to complex neurological and psychiatric disorders. In the context of psychiatry, there has been considerable interest in measuring cell-type specific gene expression changes associated with disease, detecting somatic mutations with single-cell genomics, and pin-pointing the cell-types which contribute most prominently to a given psychopathology.
Single-Cell Sequencing Technologies
Currently available techniques encompass single-cell level measurements of genomes, transcriptomes, proteomes, and epigenomes, potentially combined with morphological, spatiotemporal, and electrophysiological data (Fig. 1) [1, 2].

The human brain is composed of billions of individual cells which belong to an amazingly diverse array of cell-types. These cells and their mutual interactions give rise to the myriad complex functions of the brain, as well as dysfunctions such as psychiatric diseases. Each cell in turn is defined by its genome, epigenome, transcriptome, proteome, and other measurable properties. How do the specific states, functions, and interactions of a billion cells relate to the state and functioning of an entire human brain? A rapidly evolving suite of technologies which apply next-generation sequencing at the single-cell level may help us find the answer. We are now able to measure changes in the genomic sequences, gene expression, epigenetic modifications, chromatin states and sometimes combinations of these variables within individual cells of diverse cell-types in the brains of healthy and diseased individuals. The detailed picture of cell-types and states produced by these datasets will aid us in teasing apart the specific and complementary roles of diverse brain-cell types in psychiatric and neurological diseases
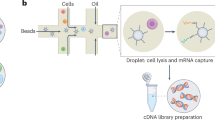
Some single-cell and single-nucleus RNA-seq (scRNA-seq and snRNA-seq) technologies rely on fluorescence assisted sorting of individual cells or nuclei into the wells of a microplate [3,4,5] followed by preparation of libraries from the RNA extracted in each well. Many flavours of microfluidics based technologies including Drop-seq [6], inDrops [7], DroNc-seq [8], sNucDrop-seq [9], and 10X Chromium [10], among others, have also been developed for scRNA-seq and snRNA-seq. These techniques rely on microfluidic devices which encapsulate individual cells or nuclei in oil droplets as the suspension is passed through the device. Each cell or nucleus is tagged with a unique barcode and mRNA molecules are captured by poly-A priming. Library preparation is performed in bulk and after sequencing, the reads are demultiplexed using barcodes, while unique molecular identifiers (UMIs) are used to generate counts of different RNA molecules originating from individual nuclei. Plate-based methods are generally low-throughput but allow for full-length cDNA sequencing whereas droplet-based methods can be ultra-high throughput (scalable up to millions of cells) but suffer from higher dropout rates and 3′ biased reads.
A suite of single-cell level NGS approaches have also been developed utilizing combinatorial indexing for measuring transcription [11, 12], DNA methylation [13], chromatin states [14,15,16], and even multiple modalities of information [17]. These approaches are based on split-and-pool strategies and involve sorting and tagging individual cells or nuclei with barcodes, followed by sequential pooling and attachment of additional barcodes. The numbers of barcodes used and cells pooled are adjusted such that the probability of multiple cells receiving the same combination of barcodes is very low. These methods are less expensive and may require less specialized equipment than droplet-based methods, but they can result in higher multiplet rates, when multiple cells or nuclei are tagged by the same barcode combination.
For profiling DNA sequence variations at the single-cell level, most studies thus far have relied on sorting of individual nuclei into the wells of a microplate. However, recently, commercial technology for droplet-based high-throughput single-nucleus genome sequencing has been created which holds promise for detecting both copy-number variations (CNVs) and single-nucleotide variations (SNVs) at single-cell resolution [18].
Droplet-based profiling of open chromatin in single-cells has also been developed [19]. Furthermore, several techniques for acquiring multimodal data at single-cell resolution exist, such as CITE-seq which measures cell-surface epitopes and transcriptomes [20] or Patch-seq which measures electrophysiological recordings and transcriptomes [21].
Thus, a wide variety of sequencing technologies are available for single-cell resolution studies of complex tissues. However, not all technologies are equally applicable to human brain tissue, especially archived, frozen, post-mortem brain tissue which is of primary interest when studying psychopathology. For example, scRNA-seq is at best extremely challenging, and often impossible, for frozen post-mortem brain tissue because of the difficulties of extracting intact cells from such tissue, especially neurons which have extensive and fragile processes. Fortunately, several studies have indicated that identification of cell-types based on single-nucleus gene expression profiles is comparable to single-cell transcriptomic profiles, although the RNA content of the whole cell and the nucleus are not identical [22, 23]. In this review, we briefly touch upon single-cell sequencing studies of the mouse brain but focus mainly on studies that have harnessed NGS to examine gene expression (Table 1) and genomic variation (Table 2) at the single-cell level in the human brain.
Single-Cell and Single-Nucleus Sequencing of the Mouse Brain
Given that rodent models of psychopathology and neurological disease are a mainstay of modern neuroscience, in the past few years a plethora of studies have been published on single-cell and single-nucleus sequencing of the mouse brain. Many different brain regions have been profiled by scRNA-seq including cortical regions [8, 24,25,26], subcortical structures [27], and the hippocampus [8, 24, 28, 29]. Different stages in brain development have been investigated in detail [30,31,32] and specific cell-types have been targeted, such as oligodendrocytes [33, 34] or microglia [35,36,37]. In many experiments, cell-type specific signatures of a variety of experimental perturbations have been measured [26, 29, 38].
In fact, single-cell transcriptomics has recently produced several large scale atlases of mouse brain cellular diversity [12, 39,40,41]. Moreover, single-cell resolution ATAC-seq studies of several mouse brain regions have recently been published [14,15,16]. The explosion of single-cell and single-nucleus sequencing studies of the mouse brain has previously been reviewed in detail elsewhere [42,43,44,45].
Single-Cell and Single-Nucleus Transcriptomics of the Healthy Human Brain
Darmanis et al. (2015) performed one of the first scRNA-seq studies of human brain in surgically excised non-pathological tissue from the adult human temporal cortex of 8 epilepsy patients and on developing brain samples from 16 to 18-week old foetuses. They profiled gene expression in over 400 individual cells using Fluidigm C1 chips [5]. Unbiased (i.e. unsupervised) and biased (i.e. supervised) clustering approaches, now commonplace in scRNA-seq, and cell-type annotation, identified excitatory and inhibitory neurons as well as major glial cell-types. Soon after, Krishnaswami et al. (2016) established one of the first protocols for snRNA-seq on human post-mortem brain tissue. They used fluorescence assisted nuclei sorting (FANS) to place individual neuronal nuclei, selected based on NeuN expression, into the wells of a microplate and used a SMART-seq approach for generating libraries [3]. Their work demonstrated on a small-scale the feasibility of snRNA-seq in archived post-mortem tissues, such as those accessible through brain banks.
The same year, Lake et al. (2016) performed Fluidigm C1 chip capture of NeuN positive FAN sorted neuronal nuclei to profile more than 3000 neurons from various cortical regions of a single healthy subject. They identified numerous inhibitory and excitatory neuronal subtypes which were in broad agreement with the cell-types described by Darmanis et al. (2015), but revealed finer subtypes powered by the larger dataset, such as layer-specific and region-specific excitatory neuron subtypes [46].
Habib et al. (2017), created an snRNA-seq method they termed DroNc-seq and applied it to several post-mortem human prefrontal cortex (PFC) and hippocampus samples, in addition to mouse brain tissue. DroNc-seq incorporated several adjustments to the Drop-seq [6] protocol, including an alteration to the dimensions of the microfluidic device to allow for capture of nuclei, which are smaller than cells, and inclusion of intronic reads in analyses due to the preponderance of pre-mRNA in the nucleus. They were the first to demonstrate the feasibility of droplet-based high-throughput snRNA-seq in archived post-mortem human brain tissue [8]. Furthermore, there was good correspondence in cell-types between the mouse and human datasets and with the findings of Lake et al. (2016).
Lake et al. (2018) independently designed an adaptation of Drop-seq for snRNA-seq. Their modifications included heat-based lysis of nuclei and incorporation of intronic reads, similar to Habib et al. (2017). Moreover, they performed an assay for single-nucleus chromatin accessibility based on combinatorial barcoding, in addition to snRNA-seq, and used their snRNA-seq findings to refine clustering of single-nuclei based on chromatin accessibility [47••]. Although tissue was obtained from healthy subjects, the cell-type specific chromatin accessibility information generated was used to indirectly assess cell-type involvement in neurological and psychiatric diseases.
While most studies have focused on the cortex, Welch et al. (2019) performed high-throughput snRNA-seq on more than 40,000 nuclei derived from archived substantia nigra samples from 7 healthy donors. They developed and applied a single-cell data analysis tool called LIGER for aligning the data from multiple individuals into a consolidated dataset. Clustering driven by inter-individual variability is a recurring problem in snRNA-seq datasets, and LIGER was able to mitigate this effect. They identified the expected subtypes of glial and neuronal cells, including dopaminergic neurons. Moreover, they were able to pin-point subject specific effects: including activation of microglia in one subject who experienced traumatic brain injury (TBI) at the time of death, and distinct microglial and astrocytic signatures in another subject with histological signs of amyloid deposits upon post-mortem examination [48••]. Thus, not only will their dataset serve as a reference for future snRNA-seq studies of the substantia nigra, but their software, which can effectively combine results from multiple datasets for joint analysis without losing dataset-specific components of the information, will be widely applicable in future single-cell sequencing studies.
While the massive capacity of high-throughput snRNA-seq is enticing, some questions require a more targeted approach as exemplified by a recent human brain snRNA-seq study from the Allen Institute [49]. Boldog et al., (2018) identified a new subtype of inhibitory neuron, dubbed the rosehip neuron, which seems to be uniquely found in the human cortex. The information from snRNA-seq was complemented by morphological and electrophysiological data from surgical tissue as well as corroborated with fluorescent in situ hybridization (ISH). The data from this study is part of a larger human mid-temporal gyrus dataset [50], generated by the Allen Institute from both post-mortem samples and surgical tissue and it provides an excellent resource for benchmarking data produced by high-throughput platforms. Uniquely, this dataset accounted for cortical layer location during the dissection and extraction of nuclei.
Single-Nucleus Transcriptomic Studies of Human Brain Pathology
Over the past year, a slew of single-cell sequencing studies of the human brain has exploited the rapidly developing technology to ask questions about cell-type diversity and cell-type specific gene expression changes in pathological states, including autism spectrum disorders (ASD) [51••, 52•, 53], Alzheimer’s disease (AD) [54•], multiple sclerosis (MS) [55], and depression [56].
Among studies focusing on ASD, Renthal et al. (2018) performed snRNA-seq using the inDrops approach in a mouse model of Rett syndrome and in the post-mortem occipital cortex of Rett syndrome patients. Rett syndrome is an X-linked developmental disorder in the autism spectrum. Affected females are heterozygous carriers of causal mutations in the MECP2 gene. Since one X-chromosome carries the mutation and the other does not, random X-chromosome inactivation in individual cells results in mosaic expression of the mutant MECP2 allele in the brain. Using an innovative analytical approach, this group was able to utilize 3′ biased snRNA-seq reads to separate cells containing an active MECP2 mutation causal for Rett, from cells in which the mutant allele was not expressed [51••]. This was achieved by genotyping the Rett syndrome patients to identify SNPs in the 3′ region of genes near the MECP2 locus, in linkage disequilibrium with the MECP2 mutation carried by the patient. These SNPs could then be detected by 3’ snRNA-seq, thus allowing the authors to determine whether the mutant or healthy MECP2 allele was expressed in a particular cell [51••]. Their approach allowed direct comparison of gene expression within specific cell-types between cells expressing mutated versus normal MECP2, revealing similarities in the patterns of differential gene expression (DGE) in the human patients and the mouse model.
Further applying single-cell technology to study ASD, Velmeshev et al. (2019) performed high-throughput snRNA-seq of the PFC and anterior cingulate cortex (ACC) in individuals with ASD, epilepsy, or no pathology. Unbiased identification of cell-types across the brain regions identified the major cortical cell-types and revealed an over-representation of protoplasmic astrocytes in ASD subjects. Cell-type specific DGE analysis revealed over 500 differentially expressed genes (DEGs) which were in good agreement with previous literature on ASD-associated genes [52•]. The DEGs in upper layer neurons and in microglia were the best predictors for clinical autism severity. Since several of the ASD subjects also experienced seizures, the authors performed cell-type specific DGE analysis by snRNA-seq in the PFC of matched sporadic epilepsy patients, to tease apart the contributions of seizures and ASD. Only a small proportion of DEGs in epilepsy overlapped with the ASD findings, suggesting that most of the cell-type specific DEGs were specific to ASD.
Similar to the previous study, Sorrells et al. (2019) performed snRNA-seq post-mortem on the amygdala from 8 individuals between the ages of 4 and 15 years, both neurotypical controls and ASD patients. Their paper focused on identifying a subset of neurons within the paralaminar nucleus (PL) of the human amygdala which show protracted development and retain molecular and morphological features of immature neurons well into adulthood [53]. Their snRNA-seq experiment complemented the ISH and immunohistochemistry findings. Among more than 13,000 nuclei sequenced, they were in fact able to identify a small population marked by high expression of DCX, BCL2, NR2F2, and ROBO1, characteristic of the immature PL neurons identified using other techniques. Moreover, snRNA-seq allowed them to detect additional genes that were enriched in this immature neuronal population, namely ST8SIA2, SOX11, and MAP2. Finally, they were able to compare gene expression between ASD cases and controls in this immature PL neuron cluster and identify around 30 DEGs.
To explore Alzheimer’s disease at the single-cell level, Mathys et al. (2019) performed high-throughput snRNA-seq on the PFC of 48 individuals from the ROSMAP [57] cohort. They clustered cells into the major neuronal and non-neuronal cell-types of the PFC and measured DGE between individuals with detectable Alzheimer’s pathology and individuals without pathology. Over a thousand DEGs were identified, the majority of which were downregulated, with the largest contribution from excitatory neurons. A small subset of DEGs were validated in NeuN positive and negative populations separated by FANS and by ISH [54•]. They also examined progressive changes in gene expression with increase in pathological burden by grouping individuals according to clinico-pathological measures. Changes in gene expression were more cell-type specific between individuals with no pathology versus early pathology, compared to individuals with early pathology versus late pathology. Sub-clustering of the major cell-types identified specific sub-clusters with over-representation of cells from pathological or healthy states. Interestingly, some AD-associated sub-clusters had an over-representation of female cells. Further exploration of sex-differences revealed a global pathology-associated upregulation of genes in oligodendrocytes in males and a global pathology-associated downregulation of genes in neurons in females. An interesting feature of this dataset is the advanced age of the donors (> 70 years).
In a landmark study of oligodendroglial heterogeneity in the human brain, Jäkel et al. (2019) performed snRNA-seq on white matter obtained post-mortem from multiple sclerosis (MS) patients and controls. In addition to neurons and other glia, they identified many different oligodendrocyte and oligodendrocyte precursor (OPC) clusters, including an oligodendrocyte cluster with immune features. Characteristic gene expression features of these oligodendroglial clusters were verified by ISH and immunohistochemistry [55]. Pseudotime analysis indicated that end state oligodendrocytes most highly express genes involved in cell-signalling and adhesion, whereas myelinating oligodendrocytes most highly express genes involved in myelination. On combined clustering of control and MS datasets, there was a higher representation of immune cells, including macrophages, derived from MS tissue, indicating immune infiltration. Overall, the same cell-types were present in MS versus control white matter, but OPCs and intermediate oligodendrocytes were under-represented in MS and the distribution of nuclei among the mature oligodendrocyte clusters was also altered. DGE analysis revealed increased expression of genes for myelination in multiple oligodendrocytic clusters in MS, as well as distinct changes in gene expression between lesioned, non-lesioned, and remyelinating regions of white matter from patients.
In our lab, we sequenced around 80,000 nuclei from the PFC of 17 individuals who were depressed and died by suicide and 17 individuals who were psychiatrically healthy. The 26 quality-controlled cell-type clusters we identified were in good correspondence with the cell-types identified by Habib et al. (2017). We were able to detect cluster-specific DEGs associated with depression in more than half of these cell-types [56]. Many of these genes have previously been implicated in bulk gene expression studies of MDD.
Single-Cell Resolution Studies of Sequence Variation in the Human Brain
The potential impact of somatic mutations, including SNVs and CNVs, which may accumulate in the post mitotic cells of the central nervous system over the course of development and ageing, has long intrigued neuroscientists in the context of neurological and psychiatric conditions [58]. Experimental techniques exist for studying somatic mutations and their contributions to brain disorders using tissue homogenates, but single-nucleus genome sequencing has an undeniable advantage in this context. Although limitations, such as errors introduced during whole genome amplification (WGA) and the astronomical cost of whole genome sequencing (WGS) for large numbers of cells, still remain to be addressed, several groups have succeeded in sequencing the genomes of single-cells from post-mortem brain tissue and the results have yielded some intriguing insights.
McConnell et al. (2013) measured CNVs in single neurons from post-mortem human frontal cortex and induced pluripotent stem cell (iPSC) derived neurons using WGA followed by DNA microarrays or sequencing. Cultured neurons had higher incidence of CNVs compared to NPCs or fibroblasts, suggesting that accumulation of somatic mutations may be integral to the development of neuronal identity. Non-germline small and large CNVs distributed throughout the genome were detected in >40% of the brain-derived neurons, although only a small number of neurons showed extensive CNV burden [59].
Cai et al. (2014) measured CNVs in post-mortem brain tissue from 3 healthy individuals and 1 subject with hemimegaencephaly (HMG) and established that aneuploidy is rare but sub-chromosomal CNVs are common. They confirmed an expected CNV at chromosome 1q in the HMG brain, although they identified a tetrasomy rather than the predicted trisomy [60]. Some CNVs were shared by multiple neurons, providing evidence that they are not artefacts of the technology.
LINE1 (L1) retrotransposon insertion, a subtype of CNV, is of special interest in psychiatry. These mobile DNA elements are capable of “jumping” in the genome, i.e. inserting a copy of themselves into a new part of the genome via an RNA intermediate, and are thought to be especially active during neurogenesis. Most of the newly formed L1 insertions in the genome are not-capable of jumping but they can create variation in the genomes of individual neurons, even within the same individual, which may have gene regulatory consequences [58]. Moreover, studying somatic L1 retrotransposition with single-cell genomics is of interest in psychiatric research because changes in the rates of L1 retrotransposition have been linked to schizophrenia [61, 62], autism spectrum disorders [63], and major depressive disorder [64].
Evrony et al. (2012) examined the rates of L1 retrotransposition in 300 neuronal nuclei from the cortex and caudate nucleus in three neurologically healthy individuals by WGA, L1 insertion profiling (L1 IP), and sequencing. They detected hundreds of known and tens of novel L1 insertions in these single-nucleus genomes, but on average each neuron had less than one somatic L1 insertion, suggesting that such insertions are generally rare [65]. In contrast, Upton et al. (2015) reported much higher rates of somatic L1 insertions in hippocampal neurons (~13 on average) and glia (~6 on average) and cortical neurons (~16 on average) using single-cell retrotransposon capture sequencing (RC-seq). Somatic L1 insertions were identified based on their absence in bulk tissue RC-seq with brain and liver samples from the same individuals and seemed to be enriched in hippocampally transcribed genes in both neurons and glia from the hippocampus [66]. However, Evrony et al. (2016) later reanalysed these data and estimated that the true rates were closer to less than one L1 somatic insertion per cell [67], more consistent with their earlier paper.
In a 2015 study (Lodato et al.), 36 neurons from the cortex of three individuals were sequenced to detect somatic SNVs. On average, 1500 L1 variants were identified and a considerable proportion was found in transcriptionally active neuronal gene regions. Interestingly, certain SNVs seem to be caused by deamination of methyl cytosines to thymines, suggesting that they were produced post-mitotically rather than during DNA replication in development [68]. Using one subject to detect patterns of shared somatic mutations diverging over time, Lodato et al., were able to trace the developmental lineage of a subset of neurons, identifying clades of related neurons. Of note, some of the more frequently detected brain SNVs were present in non-brain tissue, indicating that they arose early in development.
In a follow-up study, Lodato et al. (2018) measured SNVs in the hippocampus and PFC of 24 individuals with ages spanning 4 months to 82 years. Nine subjects were diagnosed with either Cockayne syndrome (CS) or xeroderma pigmentosa (XP), neurodegenerative diseases caused by deficiencies in the DNA repair mechanism, and the remaining 15 were free of pathology. In line with previous studies, somatic SNVs were found to accumulate with age and to be enriched in neuronally expressed genes. However, SNVs seemed to accumulate at a higher rate in the hippocampus [69•]. As expected, somatic SNVs were more frequent in CS and XP subjects than in controls.
Most recently, a study looking at 1000 cells from brain-healthy individuals found, on average, that neurons harbour more CNVs than non-neuronal or non-neural cells (Chronister et al., 2019). Furthermore, these CNVs tended to affect a larger portion of the genomes [70••]. Neuronally expressed transcripts are generally longer and are reported to possess more somatic mutations within neurons [68]. However, in contrast to the reported neuronal increase of somatic SNVs overtime, this study [69•] found a decreased prevalence with age of neurons with CNVs in their genomes. The authors suggest that cells with more CNVs may be more susceptible to ageing-related loss.
Single-Nucleus Methylomics in the Post-Mortem Human Brain
Whole-genome bisulphite sequencing (WGBS) of single cells from archived tissue is extremely challenging as bisulphite-conversion leads to loss of material and is very limiting when starting with extremely small amounts of DNA derived from single cells. Furthermore, the process is expensive as each cell needs to be sequenced at sufficient coverage. Nevertheless, Luo et al. (2017) performed single-nucleus WGBS and produced a single-cell resolution map of DNA methylation in the human frontal cortex. FAN sorting into microplates, followed by bisulphite-conversion and sequencing, produced single-nucleus DNA methylation profiles for almost 3000 nuclei from the frontal cortex of a single subject. Despite data sparsity, clustering of cells using single-nucleus DNA-methylation signatures resulted in separation of the cortical excitatory and inhibitory neuronal subtypes with a resolution comparable to snRNA-seq [71]. Non-CG methylation was found to be more cell-type specific than CG methylation and overall patterns of cell-type specific methylation were highly conserved from mouse brain to human brain. This dataset is a valuable reference for cell-type specific DNA methylation in the human brain and enables deconvolution of bulk DNA methylation data to estimate constituent cell-types.
Insights, Challenges, and Future Applications of Human Brain Single-Cell Sequencing
Some limitations of snRNA-seq in the human brain may be inherent to the technology or the underlying biology, such as underrepresentation of glial cells [8, 47••, 52•] and consistently lower numbers of RNA molecules detected in glial cells compared to neurons [5, 8, 47••]. Other limitations may be overcome using computational methods such as imputation for addressing high gene dropout and sparse data [72,73,74] and dataset alignment algorithms for addressing inter-individual variability [48, 75].
As our knowledge of the strengths and limitations of these techniques increases, so does our ability to better design experiments. Using cryosections from histological dissections for extracting nuclei can ensure more even input from different microanatomical regions [47, 52]. Combining two subjects, differing in sex or in known SNVs, for nuclei capture on a microfluidic device, followed by deconvolution using sex-specific genes or based on known SNVs, can help account for technical variability between captures [76].
In addition to direct identification of cell-types and comparison of cell-type specific features between biological groups, different modalities of single-cell data can indirectly inform our understanding of disease states. Deconvolution algorithms [77, 78] can elucidate cell-type contributions to observed disease-related changes in gene expression or DNA methylation in bulk tissue studies. Findings from genome-wide association studies (GWAS) and bulk gene expression studies can in turn help pin-point disease-relevant cell-types from single-cell datasets [79,80,81]. Future work will likely involve integration of multimodal data [16, 48••, 75], and use of complementary approaches other than NGS, such as high-throughput ISH [82, 83], for single-cell resolution studies.
Conclusion
Promising initiatives such as the Human Cell Atlas [84] and the Brain Somatic Mosaicism Network [85] are currently underway, and should greatly enhance our understanding of diversity in the transcriptome and genome of individual cells in the brain. The findings will contribute to furthering our knowledge of complex diseases which affect the brain. The continued rapid advancement of single-cell technology is creating ample opportunities for applying these technologies to the study of the human brain in health and disease. Soon we can hope to unravel the intricate complexity of the multitude of cell-types that compose the human brain and to better explain how they contribute to the development of disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet. 2019;20:257–72. https://doi.org/10.1038/s41576-019-0093-7.
Kulkarni A, Anderson AG, Merullo DP, Konopka G. Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr Opin Biotechnol. 2019;58:129–36. https://doi.org/10.1016/j.copbio.2019.03.001.
Krishnaswami SR, Grindberg RV, Novotny M, Venepally P, Lacar B, Bhutani K, et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat Protoc. 2016;11(3):499–524. https://doi.org/10.1038/nprot.2016.015.
Johnson MB, Wang PP, Atabay KD, Murphy EA, Doan RN, Hecht JL, et al. Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat Neurosci. 2015;18:637–46. https://doi.org/10.1038/nn.3980. https://www.nature.com/articles/nn.3980#supplementary-information.
Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci. 2015;112(23):7285–90. https://doi.org/10.1073/pnas.1507125112.
Macosko Evan Z, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using Nanoliter droplets. Cell. 2015;161(5):1202–14. https://doi.org/10.1016/j.cell.2015.05.002.
Zilionis R, Nainys J, Veres A, Savova V, Zemmour D, Klein AM, et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat Protoc. 2016;12:44–73. https://doi.org/10.1038/nprot.2016.154. https://www.nature.com/articles/nprot.2016.154#supplementary-information.
Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods. 2017;14:955–8. https://doi.org/10.1038/nmeth.4407. https://www.nature.com/articles/nmeth.4407#supplementary-information.
Hu P, Fabyanic E, Kwon DY, Tang S, Zhou Z, Wu H. Dissecting cell-type composition and activity-dependent transcriptional state in mammalian brains by massively parallel single-nucleus RNA-Seq. Mol Cell. 2017;68(5):1006–15.e7. https://doi.org/10.1016/j.molcel.2017.11.017.
Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R et al. Massively parallel digital transcriptional profiling of single cells. 2017;8:14049. doi:https://doi.org/10.1038/ncomms14049. https://www.nature.com/articles/ncomms14049#supplementary-information,
Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017;357(6352):661–7. https://doi.org/10.1126/science.aam8940.
Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 2018;360(6385):176–82. https://doi.org/10.1126/science.aam8999.
Mulqueen RM, Pokholok D, Norberg SJ, Torkenczy KA, Fields AJ, Sun D, et al. Highly scalable generation of DNA methylation profiles in single cells. Nat Biotechnol. 2018;36:428–31. https://doi.org/10.1038/nbt.4112. https://www.nature.com/articles/nbt.4112#supplementary-information.
Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. 2018;174(5):1309–24.e18. https://doi.org/10.1016/j.cell.2018.06.052.
Preissl S, Fang R, Huang H, Zhao Y, Raviram R, Gorkin DU, et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat Neurosci. 2018;21(3):432–9. https://doi.org/10.1038/s41593-018-0079-3.
Sinnamon JR, Torkenczy KA, Linhoff MW, Vitak SA, Mulqueen RM, Pliner HA, et al. The accessible chromatin landscape of the murine hippocampus at single-cell resolution. Genome Res. 2019;29(5):857–69. https://doi.org/10.1101/gr.243725.118.
Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361(6409):1380–5. https://doi.org/10.1126/science.aau0730.
Pellegrino M, Sciambi A, Treusch S, Durruthy-Durruthy R, Gokhale K, Jacob J, et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018;28(9):1345–52. https://doi.org/10.1101/gr.232272.117.
Lareau CA, Duarte FM, Chew JG, Kartha VK, Burkett ZD, Kohlway AS, et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol. 2019;37:916–24. https://doi.org/10.1038/s41587-019-0147-6.
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14:865–8. https://doi.org/10.1038/nmeth.4380. https://www.nature.com/articles/nmeth.4380#supplementary-information.
Cadwell CR, Palasantza A, Jiang X, Berens P, Deng Q, Yilmaz M, et al. Electrophysiological, transcriptomic and morphologic profiling of single neurons using patch-seq. Nat Biotechnol. 2015;34:199–203. https://doi.org/10.1038/nbt.3445. https://www.nature.com/articles/nbt.3445#supplementary-information.
Lake BB, Codeluppi S, Yung YC, Gao D, Chun J, Kharchenko PV, et al. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci Rep. 2017;7(1):6031. https://doi.org/10.1038/s41598-017-04426-w.
Grindberg RV, Yee-Greenbaum JL, McConnell MJ, Novotny M, O’Shaughnessy AL, Lambert GM, et al. RNA-sequencing from single nuclei. Proc Natl Acad Sci. 2013;110(49):19802–7. https://doi.org/10.1073/pnas.1319700110.
Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347(6226):1138–42. https://doi.org/10.1126/science.aaa1934.
Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. 2016;19:335–46. https://doi.org/10.1038/nn.4216. https://www.nature.com/articles/nn.4216#supplementary-information.
Hrvatin S, Hochbaum DR, Nagy MA, Cicconet M, Robertson K, Cheadle L, et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat Neurosci. 2018;21(1):120–9. https://doi.org/10.1038/s41593-017-0029-5.
Kalish BT, Cheadle L, Hrvatin S, Nagy MA, Rivera S, Crow M, et al. Single-cell transcriptomics of the developing lateral geniculate nucleus reveals insights into circuit assembly and refinement. Proc Natl Acad Sci. 2018;115(5):E1051–60. https://doi.org/10.1073/pnas.1717871115.
Habib N, Li Y, Heidenreich M, Swiech L, Avraham-Davidi I, Trombetta JJ, et al. Div-Seq: single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 2016;353(6302):925–8. https://doi.org/10.1126/science.aad7038.
Arneson D, Zhang G, Ying Z, Zhuang Y, Byun HR, Ahn IS, et al. Single cell molecular alterations reveal target cells and pathways of concussive brain injury. Nat Commun. 2018;9(1):3894. https://doi.org/10.1038/s41467-018-06222-0.
Hochgerner H, Zeisel A, Lönnerberg P, Linnarsson S. Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat Neurosci. 2018;21(2):290–9. https://doi.org/10.1038/s41593-017-0056-2.
Tiklová K, Björklund ÅK, Lahti L, Fiorenzano A, Nolbrant S, Gillberg L, et al. Single-cell RNA sequencing reveals midbrain dopamine neuron diversity emerging during mouse brain development. Nat Commun. 2019;10(1):581. https://doi.org/10.1038/s41467-019-08453-1.
La Manno G, Gyllborg D, Codeluppi S, Nishimura K, Salto C, Zeisel A, et al. Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell. 2016;167(2):566–80.e19. https://doi.org/10.1016/j.cell.2016.09.027.
Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcao A, Xiao L, et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science. 2016;352(6291):1326–9. https://doi.org/10.1126/science.aaf6463.
Marques S, Vanichkina D, van Bruggen D, Floriddia E, Munguba H, Varemo L et al. Single-cell transcriptomic profiling of progenitors of the oligodendrocyte lineage reveals transcriptional convergence during development. bioRxiv. 2017.
Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017;21(2):366–80. https://doi.org/10.1016/j.celrep.2017.09.039.
Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50(1):253–71.e6. https://doi.org/10.1016/j.immuni.2018.11.004.
Sousa C, Golebiewska A, Poovathingal SK, Kaoma T, Pires-Afonso Y, Martina S, et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018;19(11):e46171. https://doi.org/10.15252/embr.201846171.
Lacar B, Linker SB, Jaeger BN, Krishnaswami SR, Barron JJ, Kelder MJE, et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat Commun. 2016;7:11022. https://doi.org/10.1038/ncomms11022 https://www.nature.com/articles/ncomms11022#supplementary-information.
Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, et al. Molecular architecture of the mouse nervous system. Cell. 2018;174(4):999–1014.e22. https://doi.org/10.1016/j.cell.2018.06.021.
Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 2018;174(4):1015–30.e16. https://doi.org/10.1016/j.cell.2018.07.028.
Häring M, Zeisel A, Hochgerner H, Rinwa P, Jakobsson JET, Lönnerberg P, et al. Neuronal atlas of the dorsal horn defines its architecture and links sensory input to transcriptional cell types. Nat Neurosci. 2018;21(6):869–80. https://doi.org/10.1038/s41593-018-0141-1.
Cuevas-Diaz Duran R, Wei H, Wu JQ. Single-cell RNA-sequencing of the brain. Clin Transl Med. 2017;6(1):20. https://doi.org/10.1186/s40169-017-0150-9.
Ofengeim D, Giagtzoglou N, Huh D, Zou C, Yuan J. Single-cell RNA sequencing: unraveling the brain one cell at a time. Trends Mol Med. 2017;23(6):563–76. https://doi.org/10.1016/j.molmed.2017.04.006.
Guillaumet-Adkins A, Heyn H. Single-cell genomics unravels brain cell-type complexity. In: Delgado-Morales R, editor. Neuroepigenomics in aging and disease. Cham: Springer International Publishing; 2017. p. 393–407.
Cembrowski MS. Single-cell transcriptomics as a framework and roadmap for understanding the brain. J Neurosci Methods. 2019;326:108353. https://doi.org/10.1016/j.jneumeth.2019.108353.
Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352(6293):1586–90. https://doi.org/10.1126/science.aaf1204.
•• Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol. 2018;36(1):70–80. https://doi.org/10.1038/nbt.4038This study measured both gene expression and chromatin accessibility in individual cells at high-throughput in mulitple regions of the post-mortem human brain. Single-nucleus gene expression was used to refine cell-type classfication based on single-nucleus chromatin accessibility, and cell-type specific chromatin accessibility was used to assess cell-type contributions to neurological and psychiatric diseases.
•• Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell. 2019;177(7):1873–87.e17. https://doi.org/10.1016/j.cell.2019.05.006This study introduces LIGER, a computational tool for analysis of single-cell sequencing data which allows aligment of multiple datasets into a single consolidated dataset while preserving dataset specific information and allows for integration of multi-modal data.
Boldog E, Bakken TE, Hodge RD, Novotny M, Aevermann BD, Baka J, et al. Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat Neurosci. 2018;21(9):1185–95. https://doi.org/10.1038/s41593-018-0205-2.
Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, et al. Conserved cell types with divergent features between human and mouse cortex. bioRxiv. 2018:384826. https://doi.org/10.1101/384826.
•• Renthal W, Boxer LD, Hrvatin S, Li E, Silberfeld A, Nagy MA, et al. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat Neurosci. 2018;21(12):1670–9. https://doi.org/10.1038/s41593-018-0270-6This study demonstrates the possibility of studying somatic mutations using high-throughput 3'-biased single-nucleus RNA-seq and the technique is widely applicable for studying cell-type specific contributions to X-linked neurological conditions.
• Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, et al. Single-cell genomics identifies cell type–specific molecular changes in autism. Science. 2019;364(6441):685. https://doi.org/10.1126/science.aav8130This study reports for the first time the cell-type specific contributions to autism spectrum disorders in the prefrontal and anterior cingulate cortex at single-cell resolution.
Sorrells SF, Paredes MF, Velmeshev D, Herranz-Pérez V, Sandoval K, Mayer S, et al. Immature excitatory neurons develop during adolescence in the human amygdala. Nat Commun. 2019;10(1):2748. https://doi.org/10.1038/s41467-019-10765-1.
• Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570(7761):332–7. https://doi.org/10.1038/s41586-019-1195-2This study reports for the first time the cell-type specific contributions to Alzheimer's disease in the human prefrontal cortex at single-cell resolution.
Jäkel S, Agirre E, Mendanha Falcão A, van Bruggen D, Lee KW, Knuesel I, et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. 2019;566(7745):543–7. https://doi.org/10.1038/s41586-019-0903-2.
Nagy C, Maitra M, Suderman M, Theroux J-F, Mechawar N, Ragoussis J, et al. Single-nucleus RNA sequencing shows convergent evidence from different cell types for altered synaptic plasticity in major depressive disorder. bioRxiv. 2018. https://doi.org/10.1101/384479.
Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimer's Dis : JAD. 2018;64(s1):S161–s89. https://doi.org/10.3233/jad-179939.
Singer T, McConnell MJ, Marchetto MCN, Coufal NG, Gage FH. LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci. 2010;33(8):345–54. https://doi.org/10.1016/j.tins.2010.04.001.
McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, et al. Mosaic copy number variation in human neurons. Science. 2013;342(6158):632–7. https://doi.org/10.1126/science.1243472.
Cai X, Evrony Gilad D, Lehmann Hillel S, Elhosary Princess C, Mehta Bhaven K, Poduri A, et al. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 2014;8(5):1280–9. https://doi.org/10.1016/j.celrep.2014.07.043.
Doyle GA, Crist RC, Karatas ET, Hammond MJ, Ewing AD, Ferraro TN, et al. Analysis of LINE-1 elements in DNA from postmortem brains of individuals with schizophrenia. Neuropsychopharmacology. 2017;42:2602–11. https://doi.org/10.1038/npp.2017.115. https://www.nature.com/articles/npp2017115#supplementary-information.
Guffanti G, Gaudi S, Klengel T, Fallon JH, Mangalam H, Madduri R, et al. LINE1 insertions as a genomic risk factor for schizophrenia: preliminary evidence from an affected family. Am J Med Genet B Neuropsychiatr Genet. 2016;171(4):534–45. https://doi.org/10.1002/ajmg.b.32437.
Shpyleva S, Melnyk S, Pavliv O, Pogribny I, Jill James S. Overexpression of LINE-1 retrotransposons in autism brain. Mol Neurobiol. 2018;55(2):1740–9. https://doi.org/10.1007/s12035-017-0421-x.
Liu S, Du T, Liu Z, Shen Y, Xiu J, Xu Q. Inverse changes in L1 retrotransposons between blood and brain in major depressive disorder. Sci Rep. 2016;6:37530. https://doi.org/10.1038/srep37530.
Evrony Gilad D, Cai X, Lee E, Hills LB, Elhosary PC, Lehmann Hillel S, et al. Single-neuron sequencing analysis of L1 Retrotransposition and somatic mutation in the human brain. Cell. 2012;151(3):483–96. https://doi.org/10.1016/j.cell.2012.09.035.
Upton Kyle R, Gerhardt Daniel J, Jesuadian JS, Richardson Sandra R, Sánchez-Luque Francisco J, Bodea Gabriela O, et al. Ubiquitous L1 Mosaicism in Hippocampal Neurons. Cell. 2015;161(2):228–39. https://doi.org/10.1016/j.cell.2015.03.026.
Evrony GD, Lee E, Park PJ, Walsh CA. Resolving rates of mutation in the brain using single-neuron genomics. eLife. 2016;5:e12966. https://doi.org/10.7554/eLife.12966.
Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350(6256):94–8. https://doi.org/10.1126/science.aab1785.
• Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018;359(6375):555. https://doi.org/10.1126/science.aao4426This study explores somatic single-nucleotide variations in individual neurons of the human brain in health and disease.
•• Chronister WD, Burbulis IE, Wierman MB, Wolpert MJ, Haakenson MF, Smith ACB, et al. Neurons with Complex Karyotypes Are Rare in Aged Human Neocortex. Cell Rep. 2019;26(4):825–35.e7. https://doi.org/10.1016/j.celrep.2018.12.107This study investigates the relationship between aging and somatic copy-number variations in neruons of the human brain.
Luo C, Keown CL, Kurihara L, Zhou J, He Y, Li J, et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science. 2017;357(6351):600–4. https://doi.org/10.1126/science.aan3351.
van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell. 2018;174(3):716–29.e27. https://doi.org/10.1016/j.cell.2018.05.061.
Li WV, Li JJ. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat Commun. 2018;9(1):997. https://doi.org/10.1038/s41467-018-03405-7.
Huang M, Wang J, Torre E, Dueck H, Shaffer S, Bonasio R, et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods. 2018;15(7):539–42. https://doi.org/10.1038/s41592-018-0033-z.
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–902.e21. https://doi.org/10.1016/j.cell.2019.05.031.
Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, et al. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 2018;22(3):600–10. https://doi.org/10.1016/j.celrep.2017.12.072.
Gong T, Szustakowski JD. DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-Seq data. Bioinformatics (Oxford, England). 2013;29(8):1083–5. https://doi.org/10.1093/bioinformatics/btt090.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7. https://doi.org/10.1038/nmeth.3337. https://www.nature.com/articles/nmeth.3337#supplementary-information.
Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. Genetic identification of brain cell types underlying schizophrenia. Nat Genet. 2018;50(6):825–33. https://doi.org/10.1038/s41588-018-0129-5.
Skene NG, Grant SGN. Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Front Neurosci. 2016;10:16. https://doi.org/10.3389/fnins.2016.00016.
Calderon D, Bhaskar A, Knowles DA, Golan D, Raj T, Fu AQ, et al. Inferring relevant cell types for complex traits by using single-cell gene expression. Am J Hum Genet. 2017;101(5):686–99. https://doi.org/10.1016/j.ajhg.2017.09.009.
Codeluppi S, Borm LE, Zeisel A, La Manno G, van Lunteren JA, Svensson CI, et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods. 2018;15(11):932–5. https://doi.org/10.1038/s41592-018-0175-z.
Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, Perez JD, et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science. 2018;362(6416):eaau5324. https://doi.org/10.1126/science.aau5324.
Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. The human cell atlas. eLife. 2017;6:e27041. https://doi.org/10.7554/eLife.27041.
McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, Cortes-Ciriano I, et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: the brain somatic mosaicism network. Science. 2017;356(6336):eaal1641. https://doi.org/10.1126/science.aal1641.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Malosree Maitra, Corina Nagy and Gustavo Turecki declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Genetics and Neuroscience
Rights and permissions
About this article

Cite this article
Maitra, M., Nagy, C. & Turecki, G. Sequencing the Human Brain at Single-Cell Resolution. Curr Behav Neurosci Rep 6, 197–208 (2019). https://doi.org/10.1007/s40473-019-00192-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40473-019-00192-3