
Abstract
The properties of mesenchymal stem cells (MSCs) have become better known over the past decade and show potentially attractive new capabilities in solid-organ transplantation. After systemic administration, MSCs migrate to the damaged tissues, engraft, and then display potent anti-inflammatory and immunomodulatory properties through cell-to-cell interactions and secretion of soluble factors. They are weakly immunogenic and influence the differentiation and function of both innate and adaptive immune cells, thus promoting a tolerogenic response. Moreover, the results of the preclinical studies and the initial clinical trials support the evidence that MSCs can, at least partially, induce allograft tolerance. This review describes the immunosuppressive properties of MSCs on cells involved in alloimmune response and the current understanding of their underlying mechanisms, which is a prerequisite for an optimal clinical use.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells (MSCs) are multipotent non-hematopoietic progenitor cells capable of self-renewing and differentiating into multiple mesodermal lineages, including bone, cartilage, fat, tendon, and muscle [1]. Human MSCs were originally identified in the 1960s as a subpopulation of bone-marrow stromal cells with the potential to regenerate a bone-marrow environment in vivo [2]. Indeed, MSCs secrete cytokines, growth factors, and matrix molecules that influence homing, proliferation, and maturation of hematopoietic progenitor cells [3, 4]. Since then, MSCs have been isolated from various adult and fetal tissues, such as adipose tissue, amniotic fluid, placenta, umbilical-cord blood, dental pulp, and fetal liver [5–8]. All these types of MSCs have equivalent capacities of regeneration and differentiation but express no specific markers. In order to compare the study outcomes, the International Society for Cellular Therapy (ISCT) proposed minimal criteria to define human MSCs, i.e., (i) plastic-adherence under standard culture conditions; (ii) expression of CD73, CD90, and CD105, and no expression of CD45, CD34, CD14, CD11b, CD79a, CD19, or major histocompatibility complex (MHC) class-II antigens; and (iii) an ability to differentiate into osteoblasts, adipocytes, and chondroblasts in vitro [9].
Over the past decade, the properties of MSCs have become better known and show potentially attractive new capabilities in solid-organ transplantation. Firstly, MSCs are easily expanded ex vivo without any loss of function. After systemic administration, they migrate to the damaged tissues, engraft, and then differentiate under the appropriate conditions [10]. Moreover, MSCs display potent anti-inflammatory and immunomodulatory properties in vitro and in vivo. They are poorly immunogenic and influence the differentiation and function of both innate and adaptive immune cells. Thus, MSCs are promising candidates for cell-based therapies in the field of transplantation and also for all immune-mediated disorders. This review describes the immunosuppressive properties of MSCs on both innate and adaptive immune cells, our current understanding of their underlying mechanisms, the results from preclinical animal studies, and the first clinical trials involving solid-organ transplantation.
MSCs and Adaptive Immunity
T Cells
Interactions between MSCs and T cells have been extensively studied over the past decade. T cells are the major cellular effectors of the adaptive immune response and play a central role in cellular-mediated immunity. Human MSCs share several adhesion molecules with thymic epithelium that are essential for interactions with T cells. They express constitutively vascular-cell adhesion molecule-1 (VCAM-1), leukocyte function-associated antigen-3 (LFA-3), and MHC class-I antigens. They can also express intercellular adhesion molecule-1 (ICAM-1) and MHC class-II antigens when exposed to interferon (IFN)-γ. However, they do not express the costimulation molecules CD80, CD86, CD40, or CD40L, even after IFN-γ stimulation [11]. Due to this peculiar immunophenotypic profile, MSCs are immunoprivileged and therefore fail to behave as antigen-presenting cells (APCs) or to elicit an allogeneic T cell proliferative response in vitro. Moreover, rodent, baboon, and human MSCs inhibit T cell proliferation triggered by allogeneic lymphocytes, non-specific mitogens, and antigenic peptides in vitro [12–15]. This suppressive effect is dose-dependent and concerns both naïve and memory CD4+ and CD8+ T cells. It is not MHC-restricted because it occurs regardless of the source of MSCs, including “third-party” MSCs. In vivo administration of MSCs prolongs MHC-mismatched skin-allograft survival in baboons [13] and reduces steroid-resistant acute graft-versus-host disease in humans [16, 17].
Three fundamental mechanisms account for the unresponsiveness of T cells: peripheral deletion, anergy, and suppression/regulation. Some reports suggest that inhibition of T cell proliferation is not caused by induction of apoptosis but rather by the anergy state, which is reversible after MSC removal or administration of exogenous interkeukin-2 (IL-2), both in vitro and in a mouse model of experimental autoimmune encephalomyelitis [12, 18]. This anergy state may not be secondary to the lack of a costimulatory signal because the addition of the anti-CD28 antibody fails to restore an allogeneic T cell response [11]. Expression and phosphorylation patterns of molecules involved in T cell signaling pathways are regulated differently in T cells that are activated or anergized. Thus, T cells that are stimulated in the presence of MSCs are arrested in the early G1 phase due to inhibition of cyclin D2 and upregulation of the inhibitory protein p27kip1, which is consistent with arrest anergy [19].
The mechanisms underlying the immunosuppressive effects of MSCs on T cells involve both soluble factors and cell-to-cell contact. Until now, many soluble factors have been reported, including hepatocyte growth factor, transforming growth factor β1 (TGF-β1), prostaglandin E2 (PGE2), human leukocyte antigen (HLA)-G, inducible nitric-oxide synthase (iNOS), heme oxygenase-1 (HO-1), and galectin-1 [12, 20–24]. MSCs also express membranous markers that can modulate the expression of cytokine receptors and signal transducers in T cells, such as the inhibitory molecule for programmed death 1 (PD-1) [25]. Finally, MSCs express indoleamine 2,3-dioxygenase (IDO), which catalyzes conversion from tryptophan to kynurenine. Intracellular tryptophan depletion prevents T cell entry into the S phase, and thus induces cell cycle arrest in the G1 phase [26]. Altogether, most of these mechanisms are probably redundant because blocking one of them does not completely abrogate the immunosuppressive functions of MSCs.
Human and murine MSCs can impair the activity of CD4+ and CD8+ effector T cells. After antigenic stimulation, T-helper cells are induced to differentiate into Th1, Th2, and Th17 or regulatory T cells (Tregs). MSCs cause effector T cells to decrease IFN-γ and tumor necrosis factor (TNF)-α production and to increase IL-4 both in vitro and in vivo, supporting the inhibition of the Th1 response [18, 20]. MSCs inhibit differentiation of naive T cells into Th17 cells in vitro and impair the production of the inflammatory cytokines IL-17 and IL-22 by differentiated Th17 cells. Moreover, MSCs induce the production of IL-10 and reciprocally modulate the expression of the transcription factors, retinoid-related orphan receptor (ROR)-C and forkhead box p3 (FOXP3), through epigenetic changes. Thus, Th17 cells acquire the capability of inhibiting in vitro proliferative responses of activated CD4+ T cells through the induction of a regulatory phenotype [27]. In vivo, MSCs inhibit naturally occurring Th17 cells derived from unilateral ureteral obstruction in mice [28]. These effects on Th17 cells seem to be mediated by both direct cellular contact and by the production of PGE2 and IL-10 [27, 29, 30].
Tregs play a major role in the maintenance of self-tolerance through the negative regulation of immune responses. In vitro, human MSCs increase the proportion of T cells with a regulatory phenotype that possesses a methylated FOXP3 gene with a Treg-specific demethylated region (TSDR) specific to induced Tregs [20, 31, 32]. More precisely, MSCs induce three major Treg subsets corresponding to IL-10+ T regulatory 1 (Tr1), TGF-β+ T helper 3 (Th3), and CD25+FOXP3+ natural Treg-like CD4+ cells [33]. These Tregs are functional and can efficiently suppress T-cell proliferation triggered by antigenic peptides [32, 34]. Activation of the Notch1 pathway in CD4+ T cells cocultured with MSCs could be responsible for Treg differentiation, especially as FOXP3 is a downstream target for Notch signaling [35]. In vivo, murine MSCs prevent autoimmune diabetes in NOD mice [36] and prolonged cardiac allograft survival in a semiallogeneic heart transplant mouse model [37] through the generation of Tregs. This effect requires both direct contact between MSCs and allogeneic T cells and soluble factors such as PGE2, TGF-β, and HO-1 [33, 34]. Taken together, these results suggest that MSCs induce a more anti-inflammatory or tolerant response after CD4+ T cell activation.
Finally, human MSCs inhibit the initial formation of cytotoxic T lymphocytes [38] and suppress proliferation of naive and memory T cells in response to allogeneic dendritic cells (DCs) or non-specific mitogens in vitro. In contrast, MSCs have little inhibitory effect on T cell responses to viruses, such as Epstein–Barr virus and cytomegalovirus, in vitro and in vivo [39]. Murine MSCs affect antigen-specific proliferation, IFN-γ production, and cytotoxic activity of naive and memory T cells [40]. In vivo, allogeneic MSCs have promoted tumor growth in a murine melanoma model, possibly through the generation of CD8+ regulatory T cells [14].
B Cells
Although the effects of MSCs on T cells have been extensively analyzed, the interactions between MSCs and B cells are less well documented and the results are controversial. B cells play a major role in humoral-mediated immunity. They differentiate into immunoglobulin [Ig]-secreting plasmablasts after antigenic stimulation and are potent APCs. Discrepancies found between published reports may be explained by differences between the B cell subset population studied (purified versus enriched B cells) and the stimuli used to trigger B cell proliferation and differentiation [41]. Using enriched B cells, Rasmusson et al. have shown that human MSCs do not improve B cell survival but can either stimulate or inhibit IgG secretion in peripheral and spleen-derived B cells, depending on the strength of stimulation with lipopolysaccharide (LPS) or virus antigens [42]. Comoli et al. consistently report that MSCs suppress antibody production after a strong stimulus such as alloantigens. Interestingly, this inhibition is abrogated when anti-CD40 is present, suggesting that MSC-mediated inhibition of B cell function is mainly indirect due to a suppressive effect on CD4+ T cells [43]. Both secreted factors (e.g., TGF-β, PGE2) and cell–cell contact seem to be involved in this process.
In contrast, some authors have focused on the direct effects of MSCs on B cells using a purified CD19+ population [44, 45]. Human MSCs increase B cell viability while also inhibiting proliferation after polyclonal stimulation mimicking the three signals of B cell activation (i.e., B cell receptor engagement, costimulation, and cytokine- or toll-like receptor [TLR]-activation). In the presence of MSCs, B cells are arrested in the G0/G1 phases of the cell cycle. MSCs also inhibit B cell differentiation, as shown by a decrease in CD38+/CD138+ expression after exposure to DCs, and impaired antibody production. Finally, MSCs downregulate CXCR4, CXCR5, and CCR7 expressions, as well as chemotaxis to their respective ligands, CXCL12 and CXCL13, which control homing to secondary lymphoid organs. These effects are associated with activation of the mitogen-activated protein kinase (MAPK) pathways, i.e., extracellular-response kinase (ERK) 1/2 and p38. In contrast, Traiggiai et al. found that MSCs supported both polyclonal expansion and differentiation of transitional, naive, and memory B cells isolated from healthy donors and total B cells from patients with systemic lupus erythematosus. In this particular case, B cells were stimulated with an agonist of TLR-9 without triggering the B cell receptor [46]. Underlying mechanisms appear to be primarily dependent on cell–cell contact despite that IL-6, a potent B cell growth factor, is produced by MSCs after stimulation with the TLR-9 agonist. These findings are in-line with the postulated role of MSCs in supporting stages of B-cell development in vivo.
In addition, two studies have used splenic B cells, purified by negative selection (CD43 depletion), to avoid inadvertent activation of B cells and have consistently reported that MSCs inhibit B cell proliferation and terminal differentiation into plasma cells [47, 48]. This suppressive effect is associated with downregulation of the transcription factor B-lymphocyte-induced maturation protein-1 (Blimp-1) and upregulation of the transcription factor PAX5 [47, 49]. Various mechanisms of action have been described, such as matrix metalloproteinase-processed CCL-2 or PD-1/PD-L1 interactions [48, 49].
In vivo, soluble factors released by MSCs reduce antigen-specific IgM and IgG1 secretion in mice immunized with T-independent or T-dependent antigens [47]. In murine models of systemic lupus erythematosus, a B cell-driven autoimmune disease, there are contradictory reports on the effect of MSCs. Youd et al. found that MSCs worsen disease-enhancing autoantibody production, the number of plasma cells, glomerular immune-complex deposition, and proteinuria [50]. In contrast, Schena et al. reported that MSCs do not affect autoantibody production but only reduce glomerular immune-complex deposition, lymphocytic infiltration, and glomerular proliferation [48]. Finally, Choi et al. showed that human adipose-derived MSCs (ADSCs) have a beneficial effect on systemic lupus erythematosus during the early stages of disease, by improving survival rate, histologic and serologic abnormalities, and immunologic function [51]. In solid-organ transplantation, infusion of MSCs reduces alloantigen-specific antibodies and improves long-term survival of heart and kidney allografts [52, 53••].
Regulatory B cells (Bregs) are a peculiar subset of B cells that express the surface markers CD24 and CD38, and produce IL-10 (CD19+CD24highCD38highIL-10+). Franquesa et al. recently reported that coculture of B cells with MSCs, anti-IgM, anti-CD40, and IL-2 significantly increased the percentage of Bregs secreting IL-10 [54]. Expansion of Bregs is an emerging approach in the treatment of autoimmune disorders; thus, the in vivo induction of Bregs secondary to MSC infusion seems an interesting option to consider.
Dendritic Cells
DCs are the most potent APCs that can initiate and regulate the adaptive immune responses by promoting antigen-specific T cell activation. Human MSCs strongly inhibit the initial differentiation of both CD34+ cells and monocytes into DCs. These cells, instead, develop macrophage morphology with numerous vacuoles; they retain high CD14+ expression and do not acquire CD1a expression [55–58]. Monocytes have been shown to enter into the cell cycle before differentiating into functional DCs. Similar to that observed in T cells, monocytes are arrested in the G0 phase of the cell cycle due to downregulation of cyclin D2 [59].
In addition, human MSCs impair DC maturation, as shown by the reduced expression of CD83, HLA-DR, and the costimulatory molecules CD80/CD86 and a decreased secretion of IL-12 [55–58]. MSCs also suppress the chemotactic activity of DCs in response to CCL21, an important chemokine that regulates DC migration into the T cell area of lymph nodes [60]. These suppressive effects are mediated via either soluble factors (M-CSF, IL-6, TGF-β, PGE2) or intercellular contact. In particular, MSCs have been shown to interfere with the DC-activation process by altering cytoskeleton organization, resulting in an inability to form active immune synapses with T cells [61]. DCs generated in the presence of MSCs fail to express proinflammatory cytokines (such as TNF-α and IL-12), class-II MHC, and costimulatory molecules but secrete large amounts of anti-inflammatory cytokine IL-10. Accordingly, DCs generated in coculture with MSCs fail to induce T cell activation or proliferation but promote alloantigen-specific Tregs that express both TGF-β and FOXP3 [62, 63].
In vivo, murine MSCs impair TLR4-induced activation of DCs, resulting in inhibition of cytokine secretion, downregulation of molecules involved in the migration to lymph nodes, antigen presentation to CD4+ T cells, and cross-presentation to CD8+ T cells. These effects are associated with inhibition of the MAPK pathways [64•]. Taking these findings together, the results indicate that human MSCs can inhibit T cell activation indirectly by inducing regulatory APCs.
MSCs and Innate Immunity
Macrophages
Macrophages are key effector cells in innate immunity and are involved in tissue defense, homeostasis, and repair. They can exhibit either a proinflammatory or an anti-inflammatory phenotype according to the microenvironment associated with the successive phases of the inflammatory response. In vitro, both human and murine MSCs can switch activated macrophages into a regulatory phenotype characterized by high expression of CD206 and IL-10, low expression of inflammatory cytokines (TNF-α, IL-6, IL-12p70, IFN-γ), and high phagocytic activity of apoptotic cells. MSCs also inhibit the upregulation of CD80 and CD86 costimulatory molecules and of MHC class-II molecules, while increasing the expression of inhibitory receptors ILT-3 and ILT-4 in macrophages. Thus, MSCs polarize proinflammatory M1 macrophages into anti-inflammatory M2 macrophages but also impair their capacity to activate antigen-specific CD4+ T cells [65–68].
In vivo, MSCs have been shown to improve mouse survival and attenuate organ injuries in models of acute lung injury and peritonitis by specific reprogramming of IL-10-secreting macrophages [69–71]. Similarly, MSCs also promote repair and tissue remodeling, as demonstrated by the increased proliferation of tubular epithelial cells and a reduction in total collagen deposition in a mouse model of ischemia-reperfusion with acute kidney injury [72]. Furthermore, MSCs produce CCR2 ligands that are responsible for macrophage recruitment. This secretion was associated with accelerated wound closure in a mouse model of excisional skin healing but has also promoted tumorigenesis in mouse models of lymphoma, melanoma, and breast carcinoma [73, 74]. In addition, injection of MSC recruits alveolar macrophages, which led to decreased airway hyperresponsiveness, eosinophilic infiltration, and Th2 cytokine production in a mouse model of allergic asthma [75]. As engraftment of MSCs is limited in vivo, despite tissue-specific homing, macrophage polarization could be a key step in explaining their persistent effects after elimination. In addition, Melief et al. demonstrated that MSCs promote the generation of Tregs, both directly [see above] and indirectly, in skewing monocytes toward IL-10-secreting macrophages [76•]. In the same way, Akiyama et al. reported that infusion of allogeneic MSCs induced transient T cell apoptosis via the FAS pathway in mice with systemic sclerosis or experimental colitis. Apoptotic T cells trigger TGF-β production by macrophages loaded with apoptotic bodies, which in turn upregulated Tregs and led to immune tolerance in vivo [77••].
Contrary to what has been observed with other cell types, MSC immunoregulation of macrophages is mostly mediated by soluble factors. Two main mechanisms have been reported in mouse models of peritonitis [70, 71]. Nemeth et al. showed that inflammatory signals such as LPS or TNF-α activate MSCs that reprogram resident macrophages through the secretion of PGE2, which acts on EP2 and EP4 receptors on macrophages to induce secretion of IL-10. In addition, Choi et al. reported that activated MSCs secrete the anti-inflammatory protein TNF-α-stimulated gene-6 protein (TSG-6), which interacts through the CD44 receptor on resident macrophages. The CD44 molecule is dissociated from TLR-2, leading to impairment of TLR-2-induced NF-κB signaling [71]. Finally, IDO activity has also been implicated in the differentiation of monocytes into IL-10-secreting macrophages [78]. Altogether, MSCs induce an anti-inflammatory response and may involve macrophages in Treg expansion.
NK Cells
NK cells play a critical role in the defense against virus-infected cells and tumor cells. They are divided into two subtypes: CD56dim, which exerts cytolytic activity, and the CD56bright-producing cytokines, such as IFN-γ, TNF-α, IL-10, and GM-CSF. In vitro, human MSCs exert opposite effects on peripheral blood NK cells depending on the culture conditions. Interactions between fresh NK cells and MSCs lead to NK-cell activation, as shown by upregulation of CD69, whereas downregulation of CD69 is observed after interaction of IL-2-stimulated NK cells and MSCs [79, 80]. In addition, MSCs inhibit both IL-2- and IL-15-induced NK-cell proliferation without enhancing cell death [81–83]. This suppressive effect is dose-dependent and requires the presence of IFN-γ produced by activated NK cells, which in turn enhances the IDO activity by MSCs [82]. MSCs also influence NK-cell cytokine production. In standard media, NK cells release high amounts of IFN-γ and TNF-α upon binding with MSCs, via interactions with NKp30 and LFA1/ICAM1 [79]. Conversely, MSCs inhibit IFN-γ secretion by IL-2- or IL-15-activated NK cells [20, 81, 82] suggesting that MSC regulates NK cells in an inflammatory context.
In addition, MSCs have been shown to downregulate the natural cytotoxicity receptors NKp30 and NKp44 and the NK group 2D (NKG2D), which correlate with impaired cytotoxic activity [81, 84]. These effects are mediated by cell-to-cell contact and soluble factors, such as TGF-β, PGE2, IL-10, or HLA-G5 [81, 84, 85]. NK cells seem able to kill both autologous and allogeneic MSCs through LFA1/ICAM1 interaction and NKG2D engagement by MHC class I-related chain A/B (MICA/B) and UL16-binding proteins (ULBPs) expressed in MSCs [38, 79, 81]. However, in a proinflammatory environment, the cytotoxic effect of NK cells on MSCs may be partially neutralized in the presence of IFN-γ, which induces upregulation of HLA class-I molecules at the surface of MSCs, thus providing a strong inhibitory signal for NK cell activation [83]. Moreover, TLR3-primed MSCs are more resistant to IL-2-activated NK cells because of modulation of surface expression and secretion of MICA molecules [86]. Thus, MSCs could modulate their behavior in a proinflammatory environment to decrease their susceptibility to NK cell cytotoxicity.
iNKT and γδ T Cells
Invariant natural killer T (iNKT) and γδ T cells are two unconventional T cell populations involved in the defense against infections and cancers, autoimmune disease pathogenesis, and the maintenance of transplant tolerance. Similar to that observed with conventional αβ T cells, MSCs inhibit iNKT and γδ T cell proliferation from peripheral blood mononuclear cells in vitro via both cell-to-cell contact and soluble factors, such as PGE2. In contrast, MSCs only partially affect iNKT and γδ T cell cytokine production and cytotoxic activity and do not alter antigen presentation by activated γδ T cells to naive CD4+ T cells. Finally, activated γδ T cells can lyse MSCs through a TCR-dependent mechanism [87].
MSCs in Solid-Organ Transplantation
Transplantation still remains the only therapeutic solution for end-stage failure of several organs. However, the long-term use of immunosuppressive drugs can cause life-threatening infections, malignancies, and metabolic side effects and cannot prevent chronic allograft injury, which limits the survival of transplanted organs and patients. MSCs have already been tested in various preclinical studies and some clinical studies to assess their ability to prevent antibody-mediated and cellular acute rejection after solid-organ transplantation (Table 1). In rodent models, most studies show that MSC infusion prolongs allograft survival [23, 37, 52, 88–91]. Both recipient and donor-derived MSCs are able to prevent acute rejection after heart or kidney transplantation [37, 90••, 91], but the possibility of using third-party MSCs is still unclear [52, 89]. The timing of a MSC injection (pretransplant vs. posttransplant) appears to be crucial. In vivo distribution of infused MSCs and their consequences on MSC-induced immunomodulation are mainly influenced by tissue injury or inflammatory signals. Pretransplant-infused MSCs preferentially migrate into the recipient’s spleen and lymph nodes, where they interact with immune cells at sites of initial T cell priming, thus promoting a tolerogenic response. In contrast, posttransplant-infused MSCs migrate into allografts where they can induce early graft dysfunction. This “engraftment syndrome” has been documented in both rats and humans [90••, 92]. Eventually, MSCs are cleared from the recipient; therefore, the risk of side effects should be low. Although MSC infusion alone prolongs allograft survival, some studies have reported a synergistic effect between MSCs and mycophenolate mofetil or sirolimus, thereby inducing a donor-specific tolerance [52, 89]. Because MSCs modulate immune cells, they need time to initiate their immunosuppressive properties in vivo. Their infusion with immunosuppressive drugs appears necessary to blunt the allogeneic immune response and, thus, to enable successful MSC engraftment. The discrepancies between outcomes in the different rodent transplantation models can be partially explained by the fact that immune responses in kidney allografts are weaker than in cardiac allografts.
In humans, syngeneic MSC infusion has been evaluated as a replacement for basiliximab induction therapy in living-related donor kidney transplantation for patients with low immunological risk [93••]. An induction therapy with MSCs, compared to basiliximab, resulted in a lower rate of acute rejection episodes at 6 months, less opportunistic infections, improved renal outcomes at 1 year, and no adverse events. However, the benefit of MSCs to reduce acute rejection at 1 year is less pronounced. This suggests that additional injection of these cells have to be evaluated to prevent acute rejection.
Due to their regenerative properties, MSCs may also help prevent and treat chronic allograft dysfunction. Thus, Franquesa et al. assessed the effect of a single delayed infusion of MSCs in a rat-kidney transplantation model of chronic allograft dysfunction. Interestingly, this treatment was associated with stabilization of renal function and a decreased proteinuria rate, as well as reduced interstitial fibrosis and tubular atrophy at 24 weeks [53••]. These results suggest that MSCs may modulate the mechanisms involved in chronic allograft dysfunction, thereby opening up new opportunities to treat patients with a chronic rejection.
Conclusion
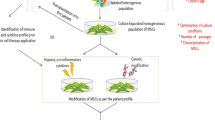
The induction of allograft tolerance defined as drug-free acceptance with preserved immunocompetence has long been a dream in solid-organ transplantation. MSCs are multipotent non-hematopoietic progenitor cells capable of self-renewing and differentiating into multiple mesodermal lineages. They inhibit the activation and function of both adaptive and innate immune cells involved in allogeneic rejection (Fig. 1). In rodent models of heart and kidney transplantation, MSCs induce donor-specific tolerance in combination with immunosuppressive drugs, and they allow drug minimization in human renal transplantation indicating their immunosuppressive properties. However, the adequacy between immunosuppressive therapies and MSCs still need to be determined. Finally, the recent reports evaluating the use of MSCs in tissue repair and/or treatment of chronic rejection open new perspectives for the long-term benefits of MSCs on allograft function and survival but safety also has to be ascertained.

Immunosuppressive properties of MSCs. When tissue is damaged, MSCs migrate into the injury site and are activated by inflammatory stimuli. Then, MSCs influence the differentiation and function of both innate and adaptive immune cells, and promote tolerogenic immune response. IDO indoleamine-2,3-dioxygenase, IFN-γ interferon-γ, IL interleukin; Ig immunoglobulin, PAMPs pathogen-associated molecular patterns, TGF-β transforming growth factor-β, TNF-α tumor necrosis factor-α
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–7.
Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6(2):230–47.
Majumdar MK, Thiede MA, Haynesworth SE, Bruder SP, Gerson SL. Human marrow-derived mesenchymal stem cells [MSCs] express hematopoietic cytokines and support long-term hematopoiesis when differentiated toward stromal and osteogenic lineages. J Hematother Stem Cell Res. 2000;9(6):841–8.
Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–34.
Gronthos S, Franklin DM, Leddy HA, Robey PG, Storms RW, Gimble JM. Surface protein characterization of human adipose tissue-derived stromal cells. J Cell Physiol. 2001;189(1):54–63.
In ‘t Anker PS, Scherjon SA, Kleijburg-van der Keur C, Noort WA, Claas FHJ, Willemze R, et al. Amniotic fluid as a novel source of mesenchymal stem cells for therapeutic transplantation. Blood. 2003;102(4):1548–9.
In‘t Anker PS, Scherjon SA, Kleijburg-van der Keur C, DeGroot-swings GM, Claas FH, Fibbe WE, et al. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells. 2004;22(7):1338–45.
Campagnoli C, Roberts IA, Kumar S, Bennett PR, Bellantuono I, Fisk NM. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood. 2001;98(8):2396–402.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. Cytotherapy. 2006;8(4):315–7.
Mackenzie TC, Flake AW. Human mesenchymal stem cells persist, demonstrate site-specific multipotential differentiation, and are present in sites of wound healing and tissue regeneration after transplantation into fetal sheep. Blood Cells Mol Dis. 2001;27(3):601–4.
Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 2003;75(3):389–97.
Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99(10):3838–43.
Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30(1):42–8.
Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J, et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102(10):3837–44.
Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringdén O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand J Immunol. 2003;57(1):11–20.
Le Blanc K, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, et al. Treatment of severe acute graft-versus-host disease with third party haplo identical mesenchymal stem cells. Lancet. 2004;363(9419):1439–41.
Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371(9624):1579–86.
Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood. 2005;106(5):1755–61.
Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. 2005;105(7):2821–7.
Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105(4):1815–22.
Nasef A, Mathieu N, Chapel A, Frick J, François S, Mazurier C, et al. Immunosuppressive effects of mesenchymal stem cells: involvement of HLA-G. Transplantation. 2007;84(2):231–7.
Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T, et al. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood. 2007;109(1):228–34.
Chabannes D, Hill M, Merieau E, Rossignol J, Brion R, Soulillou JP, et al. A role for heme oxygenase-1 in the immunosuppressive effect of adult rat and human mesenchymal stem cells. Blood. 2007;110(10):3691–4.
Gieseke F, Böhringer J, Bussolari R, Dominici M, Handgretinger R, Müller I. Human multipotent mesenchymal stromal cells use galectin-1 to inhibit immune effector cells. Blood. 2010;116(19):3770–9.
Augello A, Tasso R, Negrini SM, Amateis A, Indiveri F, Cancedda R, et al. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol. 2005;35(5):1482–90.
Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103(12):4619–21.
Ghannam S, Pène J, Moquet-Torcy G, Torcy-Moquet G, Jorgensen C, Yssel H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J Immunol. 2010;185(1):302–12.
Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res Ther. 2011;2(4):34.
Duffy MM, Pindjakova J, Hanley SA, McCarthy C, Weidhofer GA, Sweeney EM, et al. Mesenchymal stem cell inhibition of T-helper 17 cell-differentiation is triggered by cell-cell contact and mediated by prostaglandin E2 via the EP4 receptor. Eur J Immunol. 2011;41(10):2840–51.
Qu X, Liu X, Cheng K, Yang R, Zhao RCH. Mesenchymal stem cells inhibit Th17 cell differentiation by IL-10 secretion. Exp Hematol. 2012;40(9):761–70.
Maccario R, Podestà M, Moretta A, Cometa A, Comoli P, Montagna D, et al. Interaction of human mesenchymal stem cells with cells involved in alloantigen-specific immune response favors the differentiation of CD4+ T-cell subsets expressing a regulatory/suppressive phenotype. Haematologica. 2005;90(4):516–25.
Engela AU, Hoogduijn MJ, Boer K, Litjens NHR, Betjes MGH, Weimar W, et al. Human adipose-tissue derived mesenchymal stem cells induce functional de-novo regulatory T cells with methylated FOXP3 gene DNA. Clin Exp Immunol. 2013;173(2):343–54.
Mougiakakos D, Jitschin R, Johansson CC, Okita R, Kiessling R, Le Blanc K. The impact of inflammatory licensing on heme oxygenase-1-mediated induction of regulatory T cells by human mesenchymal stem cells. Blood. 2011;117(18):4826–35.
English K, Ryan JM, Tobin L, Murphy MJ, Barry FP, Mahon BP. Cell contact, prostaglandin E[2] and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4 + CD25[high] forkhead box P3+ regulatory T cells. Clin Exp Immunol. 2009;156(1):149–60.
Del Papa B, Sportoletti P, Cecchini D, Rosati E, Balucani C, Baldoni S, et al. Notch1 modulates mesenchymal stem cells mediated regulatory T-cell induction. Eur J Immunol. 2013;43(1):182–7.
Madec AM, Mallone R, Afonso G, Abou Mrad E, Mesnier A, Eljaafari A, et al. Mesenchymal stem cells protect NOD mice from diabetes by inducing regulatory T cells. Diabetologia. 2009;52(7):1391–9.
Casiraghi F, Azzollini N, Cassis P, Imberti B, Morigi M, Cugini D, et al. Pretransplant infusion of mesenchymal stem cells prolongs the survival of a semiallogeneic heart transplant through the generation of regulatory T cells. J Immunol. 2008;181(6):3933–46.
Rasmusson I, Ringdén O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit the formation of cytotoxic T lymphocytes, but not activated cytotoxic T lymphocytes or natural killer cells. Transplantation. 2003;76(8):1208–13.
Karlsson H, Samarasinghe S, Ball LM, Sundberg B, Lankester AC, Dazzi F, et al. Mesenchymal stem cells exert differential effects on alloantigen and virus-specific T-cell responses. Blood. 2008;112(3):532–41.
Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101(9):3722–9.
Franquesa M, Hoogduijn MJ, Bestard O, Grinyó JM. Immunomodulatory effect of mesenchymal stem cells on B cells. Front Immunol. 2012;3:212.
Rasmusson I, Le Blanc K, Sundberg B, Ringdén O. Mesenchymal stem cells stimulate antibody secretion in human B cells. Scand J Immunol. 2007;65(4):336–43.
Comoli P, Ginevri F, Maccario R, Avanzini MA, Marconi M, Groff A, et al. Human mesenchymal stem cells inhibit antibody production induced in vitro by allostimulation. Nephrol Dial Transplant. 2008;23(4):1196–202.
Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, et al. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006;107(1):367–72.
Tabera S, Pérez-Simón JA, Díez-Campelo M, Sánchez-Abarca LI, Blanco B, López A, et al. The effect of mesenchymal stem cells on the viability, proliferation and differentiation of B-lymphocytes. Haematologica. 2008;93(9):1301–9.
Traggiai E, Volpi S, Schena F, Gattorno M, Ferlito F, Moretta L, et al. Bone marrow-derived mesenchymal stem cells induce both polyclonal expansion and differentiation of B cells isolated from healthy donors and systemic lupus erythematosus patients. Stem Cells. 2008;26(2):562–9.
Asari S, Itakura S, Ferreri K, Liu C-P, Kuroda Y, Kandeel F, et al. Mesenchymal stem cells suppress B-cell terminal differentiation. Exp Hematol. 2009;37(5):604–15.
Schena F, Gambini C, Gregorio A, Mosconi M, Reverberi D, Gattorno M, et al. Interferon-γ-dependent inhibition of B cell activation by bone marrow-derived mesenchymal stem cells in a murine model of systemic lupus erythematosus. Arthritis Rheum. 2010;62(9):2776–86.
Rafei M, Hsieh J, Fortier S, Li M, Yuan S, Birman E, et al. Mesenchymal stromal cell-derived CCL2 suppresses plasma cell immunoglobulin production via STAT3 inactivation and PAX5 induction. Blood. 2008;112(13):4991–8.
Youd M, Blickarz C, Woodworth L, Touzjian T, Edling A, Tedstone J, et al. Allogeneic mesenchymal stem cells do not protect NZBxNZW F1 mice from developing lupus disease. Clin Exp Immunol. 2010;161(1):176–86.
Choi EW, Shin IS, Park SY, Park JH, Kim JS, Yoon EJ, et al. Reversal of serologic, immunologic, and histologic dysfunction in mice with systemic lupus erythematosus by long-term serial adipose tissue-derived mesenchymal stem cell transplantation. Arthritis Rheum. 2012;64(1):243–53.
Ge W, Jiang J, Baroja ML, Arp J, Zassoko R, Liu W, et al. Infusion of mesenchymal stem cells and rapamycin synergize to attenuate alloimmune responses and promote cardiac allograft tolerance. Am J Transplant. 2009;9(8):1760–72.
Franquesa M, Herrero E, Torras J, Ripoll E, Flaquer M, Gomà M, et al. Mesenchymal stem cell therapy prevents interstitial fibrosis and tubular atrophy in a rat kidney allograft model. Stem Cells Dev. 2012;21(17):3125–35 This study is the first that evaluates the long-term beneficial effect of MSCs in a chronic allograft dysfunction model.
Franquesa M, Mensah FK, Huizinga R, Strini T, Boon L, Lombardo E, et al. Human adipose tissue-derived mesenchymal stem cells abrogate plasmablast formation and induce regulatory B cells independently of T helper cells. Stem Cells. 2015;33(3):880–91.
Jiang X-X, Zhang Y, Liu B, Zhang S-X, Wu Y, X-D Y, et al. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. 2005;105(10):4120–6.
Nauta AJ, Kruisselbrink AB, Lurvink E, Willemze R, Fibbe WE. Mesenchymal stem cells inhibit generation and function of both CD34+-derived and monocyte-derived dendritic cells. J Immunol. 2006;177(4):2080–7.
Djouad F, Charbonnier L-M, Bouffi C, Louis-Plence P, Bony C, Apparailly F, et al. Mesenchymal stem cells inhibit the differentiation of dendritic cells through an interleukin-6-dependent mechanism. Stem Cells. 2007;25(8):2025–32.
Spaggiari GM, Abdelrazik H, Becchetti F, Moretta L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: central role of MSC-derived prostaglandin E2. Blood. 2009;113(26):6576–83.
Ramasamy R, Fazekasova H, Lam EW-F, Soeiro I, Lombardi G, Dazzi F. Mesenchymal stem cells inhibit dendritic cell differentiation and function by preventing entry into the cell cycle. Transplantation. 2007;83(1):71–6.
Jung Y-J, S-Y J, Yoo E-S, Cho SJ, Cho K-A, Woo S-Y, et al. MSC-DC interactions: MSC inhibit maturation and migration of BM-derived DC. Cytotherapy. 2007;9(5):451–8.
Aldinucci A, Rizzetto L, Pieri L, Nosi D, Romagnoli P, Biagioli T, et al. Inhibition of immune synapse by altered dendritic cell actin distribution: a new pathway of mesenchymal stem cell immune regulation. J Immunol. 2010;185(9):5102–10.
Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105(5):2214–9.
Li Y-P, Paczesny S, Lauret E, Poirault S, Bordigoni P, Mekhloufi F, et al. Human mesenchymal stem cells license adult CD34+ hemopoietic progenitor cells to differentiate into regulatory dendritic cells through activation of the notch pathway. J Immunol. 2008;180(3):1598–608.
Chiesa S, Morbelli S, Morando S, Massollo M, Marini C, Bertoni A, et al. Mesenchymal stem cells impair in vivo T-cell priming by dendritic cells. Proc Natl Acad Sci U S A. 2011;108(42):17384–9 This study provides a detailed analysis of MSC effects on DC functions in vivo and links this impairment to the inability of priming T cells and mounting an efficient antigen-specific immune response in the secondary lymphoid organs.
Kim J, Hematti P. Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol. 2009;37(12):1445–53.
Maggini J, Mirkin G, Bognanni I, Holmberg J, Piazzón IM, Nepomnaschy I, et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS One. 2010;5(2):e9252.
Ylöstalo JH, Bartosh TJ, Coble K, Prockop DJ. Human mesenchymal stem/stromal cells cultured as spheroids are self-activated to produce prostaglandin E2 that directs stimulated macrophages into an anti-inflammatory phenotype. Stem Cells. 2012;30(10):2283–96.
Hof-Nahor I, Leshansky L, Shivtiel S, Eldor L, Aberdam D, Itskovitz-Eldor J, et al. Human mesenchymal stem cells shift CD8+ T cells towards a suppressive phenotype by inducing tolerogenic monocytes. J Cell Sci. 2012;125(Pt 19):4640–50.
Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007;179(3):1855–63.
Németh K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E[2]-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15(1):42–9.
Choi H, Lee RH, Bazhanov N, Oh JY, Prockop DJ. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-κB signaling in resident macrophages. Blood. 2011;118(2):330–8.
Wise AF, Williams TM, Kiewiet MBG, Payne NL, Siatskas C, Samuel CS, et al. Human mesenchymal stem cells alter macrophage phenotype and promote regeneration via homing to the kidney following ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2014;306(10):F1222–35.
Chen L, Tredget EE, Wu PYG, Wu Y. Paracrine factors of mesenchymal stem cells recruit macrophages and endothelial lineage cells and enhance wound healing. PLoS One. 2008;3(4):e1886.
Ren G, Zhao X, Wang Y, Zhang X, Chen X, Xu C, et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell. 2012;11(6):812–24.
Mathias LJ, Khong SML, Spyroglou L, Payne NL, Siatskas C, Thorburn AN, et al. Alveolar macrophages are critical for the inhibition of allergic asthma by mesenchymal stromal cells. J Immunol. 2013;191(12):5914–24.
Melief SM, Schrama E, Brugman MH, Tiemessen MM, Hoogduijn MJ, Fibbe WE, et al. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells. 2013;31(9):1980–91 This study underlies the cellular networks involved in MSC-mediated Treg formation and shows a crucial role for macrophages in MSC-induced immunomodulation.
Akiyama K, Chen C, Wang D, Xu X, Qu C, Yamaza T, et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand−/FAS-mediated T cell apoptosis. Cell Stem Cell. 2012;10(5):544–55 This study provides evidence that MSC-mediated immunoregulation involves FAS/FASL to induce T cell apoptosis that induces anti-inflammatory macrophages and Tregs in vivo.
François M, Romieu-Mourez R, Li M, Galipeau J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther. 2012;20(1):187–95.
Poggi A, Prevosto C, Massaro A-M, Negrini S, Urbani S, Pierri I, et al. Interaction between human NK cells and bone marrow stromal cells induces NK cell triggering: role of NKp30 and NKG2D receptors. J Immunol. 2005;175(10):6352–60.
Giuliani M, Oudrhiri N, Noman ZM, Vernochet A, Chouaib S, Azzarone B, et al. Human mesenchymal stem cells derived from induced pluripotent stem cells down-regulate NK-cell cytolytic machinery. Blood. 2011;118(12):3254–62.
Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells. 2006;24(1):74–85.
Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A, et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells. 2006;24(2):386–98.
Spaggiari GM, Capobianco A, Becchetti S, Mingari MC, Moretta L. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood. 2006;107(4):1484–90.
Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood. 2008;111(3):1327–33.
Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4 + CD25highFOXP3+ regulatory T cells. Stem Cells. 2008;26(1):212–22.
Giuliani M, Bennaceur-Griscelli A, Nanbakhsh A, Oudrhiri N, Chouaib S, Azzarone B, et al. TLR ligands stimulation protects MSC from NK killing. Stem Cells. 2014;32(1):290–300.
Prigione I, Benvenuto F, Bocca P, Battistini L, Uccelli A, Pistoia V. Reciprocal interactions between human mesenchymal stem cells and gammadelta T cells or invariant natural killer T cells. Stem Cells. 2009;27(3):693–702.
Zhou HP, Yi DH, Yu SQ, Sun GC, Cui Q, Zhu HL, et al. Administration of donor-derived mesenchymal stem cells can Prolong the survival of rat cardiac allograft. Transplant Proc. 2006;38(9):3046–51.
Popp FC, Eggenhofer E, Renner P, Slowik P, Lang SA, Kaspar H, et al. Mesenchymal stem cells can induce long-term acceptance of solid organ allografts in synergy with low-dose mycophenolate. Transpl Immunol. 2008;20(1–2):55–60.
Casiraghi F, Azzollini N, Todeschini M, Cavinato RA, Cassis P, Solini S, et al. Localization of mesenchymal stromal cells dictates their immune or proinflammatory effects in kidney transplantation. Am J Transplant. 2012;12(9):2373–83 This study evaluates the optimal conditions and settings for fully harnessing MSC tolerogenic properties in a mouse kidney transplantation model.
Ge W, Jiang J, Arp J, Liu W, Garcia B, Wang H. Regulatory T-cell generation and kidney allograft tolerance induced by mesenchymal stem cells associated with indoleamine 2,3-dioxygenase expression. Transplantation. 2010;90(12):1312–20.
Inoue S, Popp FC, Koehl GE, Piso P, Schlitt HJ, Geissler EK, et al. Immunomodulatory effects of mesenchymal stem cells in a rat organ transplant model. Transplantation. 2006;81(11):1589–95.
Tan J, Wu W, Xu X, Liao L, Zheng F, Messinger S, et al. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. JAMA. 2012;307(11):1169–77 This study is the first prospective randomized clinical trial showing the interest of MSCs in solid organ transplantation. MSCs can safely replace anti-IL-2 receptor induction therapy in kidney-transplanted patients with a low immunological risk.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Cellular Transplants
Rights and permissions
About this article

Cite this article
Brunel, M., Herr, F. & Durrbach, A. Immunosuppressive Properties of Mesenchymal Stem Cells. Curr Transpl Rep 3, 348–357 (2016). https://doi.org/10.1007/s40472-016-0120-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40472-016-0120-y