
Abstract
Cardiovascular disease (CVD) remains the leading cause of morbility and mortality worldwide. The identification of common cardiovascular risk factors has led to the development of effective treatments that enabled a significant reduction of the global cardiovascular disease burden. However, a significant proportion of cardiovascular risk remains unexplained by these risk factors leaving many individuals at risk of cardiovascular events despite good control of the risk factors. Recent randomized clinical trials and Mendelian randomization studies have suggested that inflammation explains a significant proportion of the residual cardiovascular risk in subjects with good control of risk factors. An accelerated process of vascular ageing is increasingly recognized as a potential mechanism by which inflammation might increase the risk of CVD. In turn, cellular ageing represents an important source of inflammation within the vascular wall, potentially creating a vicious cycle that might promote progression of atherosclerosis, independently from the individual cardiovascular risk factor burden. In this review, we summarise current evidence suggesting a role for biological ageing in CVD and how inflammation might act as a key mediator of this association.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Acute inflammatory responses evoked by tissue and cellular damage exert beneficial effects as they promote tissue repair and help to prevent colonization of the damaged tissues by opportunistic pathogens. Thus, a limited temporal evolution of the inflammatory processes is considered beneficial as it enables the elimination of the triggering insult and promotes tissue repair. Conversely, persistent inflammatory stimuli or dysregulation of mechanisms involved in the resolution of acute inflammatory responses result in a chronic inflammatory exposure, which is commonly observed in ageing and several age-related diseases, including atherosclerosis, type 2 diabetes mellitus, cancers and chronic neurodegenerative diseases, such as Alzheimer’s disease.
The reciprocal relationships between inflammation, ageing, and age-related diseases have led to the creation of the “inflammaging” theory that, since the early 2000s, refers to the reduced ability to cope with a variety of stressors accompanied by an increased pro-inflammatory status during unsuccessful ageing, namely ageing accompanied by diseases [1]. Thus, according to the “inflammaging” theory, the molecular and cellular mechanisms linking inflammation and ageing are also involved in the initiation and evolution of many chronic and degenerative conditions, including cardiovascular disease (CVD).
2 Telomere Length as a Potential Marker Underlying the Association Between Ageing and Inflammation
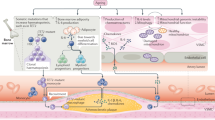
A 2–4 fold increase in serum levels of several pro-inflammatory cytokines, including IL-6 (also known as the “gerontologist cytokine”) [2] and tumor necrosis factor alpha (TNF-α), is commonly detected with advancing age [2,3,4,5]. Although this evidence was considered related to an increased burden of chronic inflammatory diseases commonly observed in the elderly, successful ageing (ageing without co-morbidity) is still associated with low-grade inflammatory activity in vivo. Numerous studies of older adults and centenarians indicate that, even in healthy individuals, levels of several cytokines (including IL-6 and TNF-α) increase with age, also in the absence of acute infections or associated diseases [2, 6,7,8]. Despite this observational evidence, the link between inflammation and ageing and the biological pathways potentially underlying this association have been difficult to identify, due to the lack of markers of cellular ageing that could also inform on the risk of unhealthy ageing. Ideally, this marker should be influenced by chronic inflammation, related to the risk of morbidity/mortality for age-related diseases and biologically involved in the mechanisms of cellular ageing (Fig. 1).
Telomeres are repeated DNA sequences located at the end of the chromosomes that maintain DNA stability during cellular division, also playing a role in regulating the replicative potential of the cell [9]. Their function is dependent on their length and is lost when they become critically short because of multiple cellular replications. The lack of adequate protection of the chromosomal ends by critically short telomeres triggers the constitutive activation of DNA damage response (DDR), ultimately resulting in a replicative arrest, acquisition of a dysfunctional cellular phenotype and cellular senescence or apoptosis [10] (Fig. 1). Cells with activated DDR do not contribute to the physiological tissue/organ function, leading to a decline in their biological reserves that is considered a hallmark of frailty and increased vulnerability to age-related disease. Thus, telomere length represents a replicative “clock” for human somatic cells and, as such, a good marker of biological ageing. Indeed, several observational studies have documented cross-sectional and prospective associations between short telomere length in peripheral leukocytes (leukocyte telomere length, LTL) and an increased risk of mortality [11,12,13,14] and age-related diseases [15,16,17].
The measure of LTL also provides a strong rationale for considering this marker as a reliable measure of the chronic inflammatory burden of an individual. Indeed, leukocytes represent inflammatory cells called into action in situations of acute or chronic inflammatory responses. Particularly, prolonged conditions of tissue stress or malfunction can alter tissue homeostasis and, consequently, induce production of pro-inflammatory cytokines by tissue resident macrophages [18]. These cytokines (directly or indirectly) stimulate proliferation and differentiation of peripheral lymphocytes and new inflammatory cells from the bone marrow, respectively. The increased replications stimulated by this adaptive response will be reflected in shorter telomere length measured in peripheral leukocytes [19].
While the telomerase activity of bone marrow haematopoietic and progenitor cells (that support the leukocytosis associated with the inflammatory response) can compensate for the lost of telomere sequences related to acute inflammatory states, repeated or prolonged exposure to inflammatory triggers exhaust the compensatory capacities of this enzyme, ultimately resulting in shorter telomere length in peripheral leukocytes [20, 21]. Cell proliferation is therefore considered an important cause of telomere shortening in peripheral leukocytes, and the presence of short LTL in the elderly can be interpreted as the accumulation of cell divisions associated with their life time tissue renewal and, thus, inflammatory burden [22] (Fig. 1). Also, an elevated inflammatory exposure is commonly associated with increased oxidative stress, that, in turn, can cause oxidative damage to the telomere sequence leading to a greater loss of telomere repetitions for each cell replication [23]. The ability of LTL to inform on the individual cumulative exposure to inflammation and oxidative stress has been confirmed in multiple studies showing robust associations between short LTL and chronic inflammatory diseases [24, 25] or markers of oxidative stress [24,25,26,27].
Among the various sources of intracellular oxidative stress, a growing number of studies have identified mitochondrial dysfunction as a major source of free radicals, which might affect the control of telomere length through several mechanisms. The process of mitochondrial oxidative phosphorylation generates reactive oxygen species (mtROS) even in physiological conditions [28]. In the setting of inflammatory states, hypoxia or excessive availability of metabolic substrates, there is an increased mtROS production, that in turn leads to an hyperactivation of the inflammasome NLRP3, ultimately resulting in an increased production of the activated IL-1β, a proinflammatory cytokine that is centrally involved in the pathogenesis of CVD [29,30,31]. A novel and highly attractive pathway linking mtROS with CVD and ageing is represented by the protein p66Shc. Physiologically, this protein is contained in multimeric complexes in the cytoplasm and the mitochondrial matrix. Under cellular stress, it migrates in the intermembranous mitochondrial space, directly stimulating production of mtROS [32]. The consequent activation of the inflammatory response linked to mitochondrial dysfunction can accelerate telomere shortening [33]. Furthermore, the activation of the protein p66Shc is directly regulated by the activity of sirtuins, proteins with function of histone deacetylase that are sensitive to the energy status of the cells. An increased availability of substrates, as observed in obesity and diabetes mellitus, is accompanied by a down-regulation of the expression and activity of sirtuins which, in turn, results in an increased activation of p66Shc [34, 35], upregulation of the expression of several inflammatory pathways [36, 37] and faster telomere attrition [38]. In parallel, a deficit of sirtuin activity can activate several cardiac and vascular pathways leading to a faster progression of cardiovascular damage [39].
Although telomeres represent just one of the many connections between inflammation and cellular ageing, their ability to reflect the cumulative inflammatory exposure and inform on biological ageing make them an ideal marker to explore the inflammaging hypothesis in CVD.
3 Inflammation as a Driver of Ageing and CVD
3.1 Chronic Inflammatory Disease, LTL and CVD
Several human models of chronic inflammation have been used to study the association between CVD, telomeres length and inflammation. Among these, rheumatic diseases and, in particular, rheumatoid arthritis (RA) have been the most commonly adopted [40, 41]. Patients with RA have a 1.5–2.0 fold increased risk of developing coronary artery disease (CAD), similar in magnitude to the risk imparted by diabetes mellitus [42,43,44]. An expert committee of the European League Against Rheumatism has recommended that CV risk prediction models should be adapted for patients with RA by a 1.5 multiplication factor (if this is not already included in the risk algorithm) to reflect their increased risk of heart disease [45]. Inflammation and an increased exposure to oxidative stress have been considered the main factors accounting for the increased risk of CVD recorded in patients with RA. As the same factors have also an important impact on the rate of LTL shortening, it is not surprising that several studies conducted on subjects with RA have found evidence of reduced LTL accompanied to increased oxidative stress [46, 47]. Beyond the impact of inflammation and oxidative stress, altered telomere biology in patients with RA can also result from the defective activity of telomerase. Restoration of telomerase activity might improve the immune abnormalities in RA [48] highlighting the strong link between mechanism regulating cellular ageing and the inflammatory responses in this model.
A state of chronic inflammation with elevated levels of TNF- α and IL-1 can also be found in diabetes in which inflammation may also be one of the causative mechanisms that lead to insulin resistance and metabolism dysregulation [49]. Vascular damage mediated by inflammation in diabetes is mainly linked to the hyperglycemic state that increases cytokines production, favours rolling, adhesion and migration of leukocytes in post-capillary venules and increases expression of P-selectin on endothelial cells [50, 51]. Several studies have also demonstrated an increase in oxidative stress in diabetic patients, in particular of mitochondrial origin. Oxidative stress is also correlated to the risk of developing macro- and microvascular diabetic complications [52]. This was specifically demonstrated by Hinokio et al. in a study that correlated levels of urinary 8-hydroxy-2-deoxyguanosine (8-OHdG) to the development of diabetic nephropathy [53]. 8-OHdG is a marker of oxidative DNA damage that increases in the presence of oxidative stress and that can also promote apoptosis through the activation of caspases [54]. Sampson et al. found greater LTL attrition in patients with type 2 diabetes compared to controls, an alteration that was accompanied by a concomitant increase in urinary levels of 8-OHdG [55]. These findings suggest that the increased susceptibility of DNA to oxidative damage in type 2 diabetes patients translates into accelerated telomeres shortening and a progression to cellular senescence. This is further supported by a study from our group that in patients with type 1 and 2 diabetes mellitus documented shorter LTL in subjects with a lower plasma total antioxidant capacity. Remarkably, patients with short LTL and low plasma antioxidant activity were also those with a higher risk of ischaemic heart disease at the 10 years follow up [56]. This evidence highlights the strong relationships between oxidative stress, LTL biology and risk of CVD in patients with diabetes mellitus. Mitochondria might represent a major cause of oxidative stress-induced telomere shortening in patients with diabetes mellitus. This is supported by the study from Salpea et al., who described shorter LTL in patients with diabetes carrying a mutation of the uncoupling protein 2 (UCP2), a key regulator of mtROS production [57].
Periodontitis (and its treatment) represents another model of human systemic inflammation that has been commonly used to study the effect of acute and chronic inflammatory responses and its resolution on the cardiovascular system and markers of ageing. Poor periodontal health has been positively correlated with CVD in numerous observational studies [58,59,60,61]. This relationship is thought to be related to the migration in the bloodstream of microorganisms present in dental plaque, giving raise of a systemic inflammatory response [62]. Although no randomised controlled trials have assessed the impact of periodontal treatment on the risk of CVD, there are several clinical trials showing that the same treatment has a beneficial effect on CVD risk factors as it can reduce systemic levels of IL-6, CRP, blood pressure, total cholesterol, and E-selectin. An improvement of endothelial function, renal function and glycemic control in individuals with and without diabetes have also been described [63,64,65,66,67,68].
Several reports suggest that the chronic inflammatory and oxidative stress exposure related to periodontitis might contribute to a greater shortening of LTL. In a case–control study, we documented that patients affected by severe periodontitis had shorter LTL than healthy controls. LTL was inversely related with both the total amount of plasma oxidant metabolites and measures reflecting the chronic inflammatory exposure of the gum tissue [25]. Further support to the hypothesis that a chronic inflammatory exposure might account for the shorter LTL detected in patients with periodontitis was provided in a subsequent report, including patients with type 2 diabetes. In this population, patients with periodontitis had shorter LTL than controls and LTL was inversely related to the circulating levels of lipopolysaccharides, which might reflect the chronic trigger of the inflammatory response [24]. Despite these convincing findings, the ultimate cause accounting for the inverse relationship between periodontitis and LTL remains to be determined. Indeed, in the Atherosclerosis Risk in Communities (ARIC) study patients with periodontitis had shorter LTL than unaffected controls but the rate of LTL over a period of 6 years did not differ between groups [69]. This suggests that LTL shortening due to periodontitis may require a much longer observational period to become evident. Another intriguing hypothesis is that short LTL might underpin the immune-inflammatory deregulation that is thought to contribute to the periodontal disease, thus preceding its clinical diagnosis.
3.2 The Relationship of Short LTL with Clinical and Subclinical Measures of Atherosclerosis
Several large epidemiological studies have confirmed the association between short LTL and increased risk of CVD morbidity and mortality [13, 15, 70,71,72,73,74]. Strong associations have been also described between short LTL and exposure to CVD risk factors, including indices of obesity/insulin resistance [16, 55, 72, 73, 75,76,77], dyslipidemia, smoking habit [76] and hypertension [26]. Cross-sectional and prospective associations between LTL and subclinical measures of vascular remodeling/damage, including the carotid intima-media thickness (cIMT) and pulse wave velocity (PWV), have been reported as well [70, 71, 78,79,80]. The evidence produced in these reports, however, is in contrast with other studies that could not find associations between short LTL and cardiovascular mortality in elderly populations [81,82,83,84]. Factors that could account for these conflicting results includes the different methods used to assess LTL, the low sample size of some studies, the different age of the samples used for the statistical analyses and the lack of accuracy in the measure of LTL in older ages. In addition, all these studies did not report longitudinal assessment of LTL, limiting the degree to which causal inferences could be made and leaving an open question onto whether the risk of subjects with short LTL was related to a heritable trait or secondary to an increased rate of LTL attrition during the lifespan due to higher inflammatory exposure. Such a gap of knowledge was filled by our group that documented strong associations between the rate of LTL shortening over 10 years and cIMT, while weaker associations were detected with cross-sectional LTL measures [85]. Following this publication, a role for LTL in CVD was confirmed in a large metanalysis including 43,725 participants and 8400 patients that documented clear associations between short LTL and the risk of coronary heart disease [86]. The final prove of a causal role of LTL in the evolution of CVD was provided, however, by Codd et al. who used the complex statistical approach of Mendelian randomization to document an association of alleles related with shorter LTL with an increased risk of coronary artery disease [87]. These findings have been confirmed by another Mendelian randomization study that documented a potentially causal role of short LTL in the risk of coronary heart disease and abdominal aortic aneurysm [86], two conditions on which the interleukin-6 (IL-6) inflammatory pathway has also shown a causal impact [88,89,90,91]. Comprehensively, these results further support the idea that a process of accelerated biological ageing induced by inflammation might underpin the risk of CVD.
Based on this bulk of evidence, LTL has gained a role as a marker of vascular ageing, possibly representing an increased exposure to CV risk factor and inflammation.
4 Reverse Relationship: Ageing as a Driver of Inflammation and CVD
Although there is extensive evidence of inflammation promoting vascular ageing and CVD [24, 92, 93], a reverse relationship in which ageing itself (represented by telomeres shortening) could promote inflammation and CVD has also been described. Indeed, many cells of the vascular wall can produce inflammatory cytokines under conditions of replicative senescence. This was reported, for the first time, by Rodier et al. who demonstrated that constitutive activation of the DDR due to critically short telomeres induces activation of a complex senescence-associated secretory phenotype (SASP) in human fibroblasts, characterised by several changes in the gene expression, including the activation of IL-6 and interleukin-8 (IL-8) production. In turn, a reduction of the DDR obtained by transfection of exogenous telomerase as well as loss of ATM or other factors responding to DNA damage dumped the IL-6 and IL-8 production, confirming the hypothesis that an altered cellular phenotype acquired by cells with critically short telomeres and consequent activation of the DDR was the main mechanisms leading to the IL-6 production [94]. In older subjects and patients with atherosclerotic plaques, it is possible to observe an increased activation of markers of DDR compared to control subjects in both the atherosclerotic tissue and circulating cells [95,96,97,98]. Notably, Morgan et al. documented that this age-related increase in markers of DNA damage response is accompanied by greater expression of P21, interleukin 8, and monocyte chemotactic protein 1 mRNA within skeletal muscle small resistance arteries [99]. A more comprehensive assessment of the potential mechanisms by which senescent cells within the vascular wall might contribute to establishing an inflammatory environment was provided by Clarke et al., who documented that senescent human smooth muscle cells undergo activation of SASP and release high levels of IL-6, promoting chemotaxis of inflammatory cells and perpetuating the inflammatory state that leads to vascular damage [100]. Thus, growing evidence suggests the presence of a vicious cycle, where inflammation might act as a main cause and consequence of vascular ageing (Fig. 1).

Inflammation as a cause and consequence of biological ageing. Lifetime exposure to cardiovascular risk factors leads to a chronic, low grade, systemic inflammatory exposure which is sustained by recruitment and proliferation of inflammatory cells. This results in telomere shortening that can be assessed in peripheral leukocytes. When the size of telomeres become critically short, the cells undergo senescence and activation of the DNA damage response (DDR) which, in turn, leads to the development of a senescence-associated secretory phenotype (SASP). After the acquisition of such a phenotype, cells can produce inflammatory cytokines, perpetuating the chronic inflammatory exposure. Following this hypothesis, a proportion of the inflammatory burden commonly detected in the elderly or subjects with advanced cardiovascular ageing might be independent of the exposure to cardiovascular risk factors, as sustained by the senescent cells of the vascular wall
The hypothesis that vascular ageing might promote inflammation and perpetuate a chronic inflammatory status could provide a novel and intriguing interpretation of the Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS). This was the first randomised, double-blinded, placebo-controlled trial that tested the inflammatory hypothesis of atherothrombosis. The monoclonal antibody Canakinumab that blocks the IL-1β signaling was used to reduce systemic levels of inflammation in high-risk patients with established atherosclerosis that, despite the use of aggressive secondary prevention strategies, presented with persistent activation of systemic inflammation. At baseline, participants displayed an optimal control of cardiovascular risk factors, with values of LDL cholesterol of approximately 82 mg/dl. However, the average levels of hsCRP were 4.2 mg/dl [101]. All patients recruited in the trial had a previous cardiovascular event, suggesting that the long term exposure to CVD risk factors before recruitment might have promoted the evolution of vascular ageing up to its final clinical manifestation. In this context, it is possible that a constitutional activation of DDR in immune-inflammatory cells and/or cells residing within the vascular wall could represent a major driver of the persistent inflammatory activation, despite optimal control of cardiovascular risk factors. Therefore, it is not surprising that using a monoclonal antibody that specifically targets the inflammatory axis activated by the DDR resulted in an effective reduction of the risk of subsequent cardiovascular events, while did not modify the lipid profile.
5 Clonal Hematopoiesis of Indeterminate Potential (CHIP): A Novel Mechanisms Linking Ageing, Inflammation and CVD
Besides the influence of the telomere-associated cellular ageing, recent research has identified somatic mutations that further emphasise the strong link between inflammation, ageing and CVD [102]. Acquisition of somatic mutation is mostly significant in highly proliferative tissues like the hematopoietic system. Hematopoietic stem/progenitor cells (HSPCs) accumulate somatic mutations with age, some of which confer a competitive advantage and leads to an expansion of their clonal progeny in the peripheral blood. The most common of these mutations are located in genes that are also involved in the development of haematological malignancies, including Tet2, DNMT2A and Jak2. Thus, the identification of such clones in the peripheral blood confers to the carrier an elevated risk of hematologic malignancies [103]. However, only ≈ 0.5% of subjects per year will progress towards an overt malignancy [104], leaving many of these subjects in a premalignancy state. When the clones exceed 2% of the peripheral leukocyte count, this premalignant condition is identified with the term “clonal hematopoiesis of indeterminate potential (CHIP)” [102]. The prevalence of CHIP is remarkably high, considering that by the age of 70 years, individuals harboring these clones amount to > 10% of the general population. People with CHIP show an increased risk of all-cause mortality that is conferred by an excess of CVD, including coronary heart disease and stroke [104]. From its seminal work, the group of Jaiswal et al. has confirmed that CHIP has a direct role in the initiation and evolution of atherosclerosis and that this is due to an upregulated activity of several inflammatory signals in the causal pathway for CVD, including IL-1β, IL-8 and IL-6 [105]. Indeed, hypercholesterolaemia-prone mice engrafted with bone marrow of Tet2 knockout mice develop more severe atherosclerosis than controls, and this is accompanied to an elevated expression of proinflammatory genes such as IL-6. In humans, carriers of CHIP showed a greater extension of coronary calcifications and a greater level of circulating IL-8 [105].
Intriguingly, recent evidence suggests that CHIP might be involved in the whole cardiovascular continuum, extending from the development of atherosclerosis to its late complications, such as heart failure. Sano et al. documented that, after induction of myocardial infraction or pressure overload, mice with partial (10%) bone marrow reconstitution with Tet2-deficient cells (10% Tet2 KO-BMT) undergo greater left ventricular remodeling, characterized by the development of larger left ventricular systolic and diastolic volumes as well as lower ejection fraction compared to their while type littermates (10% WT-BMT). The histological analysis of the myocardial tissue revealed an increased collagen deposition and myocardiocyte hypertrophy in the 10% Tet2 KO-BMT mice than in the 10% WT-BMT littermates. These alterations were accompanied by an increased expression of IL-1β and IL-6 in macrophages of 10% Tet2 KO-BMT mice and were prevented by infusion of an inflammasome NLRP3 inhibitor, highlighting the importance of the IL-1β inflammatory pathway in the development of the cardiac remodeling post myocardial infraction or pressure overload [106]. The confirmation that these mechanisms are likely to act also in humans was provided by Dorsheimer et al. In a population of 200 patients with chronic heart failure owing to ischemic origin, the authors found that: (1) CHIP was more common than in the general population, (2) the two most commonly mutated genes were Tet2 and DNMT3a, (3) patients harboring either DNMT3A or TET2 mutations in their bone marrow cells had significantly worse long-term clinical outcome for both death and death combined with rehospitalization for heart failure compared with non-CHIP carriers, and (4) there was a statistically significant dose-response association between clone size and clinical outcome [107].
Thus, while factors affecting the risk of developing somatic mutations remains to be fully elucidated, ageing is currently considered the greater risk for the development of CHIP, which therefore represents a novel potential mechanism explaining the high level of proinflammatory cytokines detected in the peripheral blood of older people and their association with an increased risk of CVD.
6 Conclusions
The understanding of the relationship between inflammation, ageing and CVD is still incomplete, but increasing evidence is being collected that suggests a close and possibly mutual connection between inflammation and ageing in determining the development of CVD. The CANTOS trial has provided evidence that selective inhibition of the IL-1β/IL-6/CRP pathway can improve the outcome of patients affected by age-related diseases, including CVD and cancer. Further studies are needed to assess the impact of these treatments on markers of biological ageing.
References
Franceschi C, et al. Inflammaging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54.
Ershler WB. Interleukin-6: a cytokine for gerontologists. J Am Geriatr Soc. 1993;41(2):176–81.
Bruunsgaard H, et al. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J Gerontol A Biol Sci Med Sci. 1999;54(7):M357–64.
Hager K, et al. Interleukin-6 and selected plasma proteins in healthy persons of different ages. Neurobiol Aging. 1994;15(6):771–2.
Paolisso G, et al. Advancing age and insulin resistance: role of plasma tumor necrosis factor-alpha. Am J Physiol. 1998;275(2 Pt 1):E294–9.
Cohen HJ, et al. The association of plasma IL-6 levels with functional disability in community-dwelling elderly. J Gerontol A Biol Sci Med Sci. 1997;52(4):M201–8.
Fagiolo U, et al. Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol. 1993;23(9):2375–8.
Ferrucci L, et al. The origins of age-related proinflammatory state. Blood. 2005;105(6):2294–9.
Blackburn EH. Structure and function of telomeres. Nature. 1991;350(6319):569–73.
Rossiello F, et al. DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat Commun. 2017;8:13980.
Astrup AS, et al. Telomere length predicts all-cause mortality in patients with type 1 diabetes. Diabetologia. 2010;53(1):45–8.
Bakaysa SL, et al. Telomere length predicts survival independent of genetic influences. Aging Cell. 2007;6(6):769–74.
Cawthon RM, et al. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet. 2003;361(9355):393–5.
Deelen J, et al. Leukocyte telomere length associates with prospective mortality independent of immune-related parameters and known genetic markers. Int J Epidemiol. 2014;43(3):878–86.
Brouilette SW, et al. Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case–control study. Lancet. 2007;369(9556):107–14.
Gardner JP, et al. Rise in insulin resistance is associated with escalated telomere attrition. Circulation. 2005;111(17):2171–7.
Zhan Y, et al. Telomere length shortening and alzheimer disease—a Mendelian randomization study. JAMA Neurol. 2015;72(10):1202–3.
Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–64.
Aviv A. Leukocyte telomere length, hypertension, and atherosclerosis: are there potential mechanistic explanations? Hypertension. 2009;53(4):590–1.
Flores I, et al. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 2008;22(5):654–67.
Yui J, Chiu CP, Lansdorp PM. Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood. 1998;91(9):3255–62.
Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol. 2007;3(10):640–9.
von Zglinicki T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann N Y Acad Sci. 2000;908:99–110.
Masi S, et al. Association between short leukocyte telomere length, endotoxemia, and severe periodontitis in people with diabetes: a cross-sectional survey. Diabetes Care. 2014;37(4):1140–7.
Masi S, et al. Oxidative stress, chronic inflammation, and telomere length in patients with periodontitis. Free Radic Biol Med. 2011;50(6):730–5.
Bekaert S, et al. Telomere length and cardiovascular risk factors in a middle-aged population free of overt cardiovascular disease. Aging Cell. 2007;6(5):639–47.
Fouquerel E, et al. Oxidative guanine base damage regulates human telomerase activity. Nat Struct Mol Biol. 2016;23(12):1092–100.
Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013;112(8):1171–88.
Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208(3):417–20.
Usui F, et al. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35(1):127–36.
Masi S, et al. Mitochondrial oxidative stress, endothelial function and metabolic control in patients with type II diabetes and periodontitis: a randomised controlled clinical trial. Int J Cardiol. 2018;271:263–8.
Giorgio M, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122(2):221–33.
Guha M, et al. hnRNPA2 mediated acetylation reduces telomere length in response to mitochondrial dysfunction. PLoS One. 2018;13(11):e0206897.
Kumar S, et al. Sirtuin1-regulated lysine acetylation of p66Shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction. Proc Natl Acad Sci USA. 2017;114(7):1714–9.
Zhou S, et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res. 2011;109(6):639–48.
Vachharajani VT, et al. Sirtuins link inflammation and metabolism. J Immunol Res. 2016;2016:8167273.
Xie J, Zhang X, Zhang L. Negative regulation of inflammation by SIRT1. Pharmacol Res. 2013;67(1):60–7.
De Bonis ML, Ortega S, Blasco MA. SIRT1 is necessary for proficient telomere elongation and genomic stability of induced pluripotent stem cells. Stem Cell Rep. 2014;2(5):690–706.
Xu S, Bai P, Jin ZG. Sirtuins in cardiovascular health and diseases. Trends Endocrinol Metab. 2016;27(10):677–8.
Goronzy JJ, Shao L, Weyand CM. Immune aging and rheumatoid arthritis. Rheum Dis Clin N Am. 2010;36(2):297–310.
Nurmohamed MT, Heslinga M, Kitas GD. Cardiovascular comorbidity in rheumatic diseases. Nat Rev Rheumatol. 2015;11(12):693–704.
Hippisley-Cox J, et al. Predicting cardiovascular risk in England and Wales: prospective derivation and validation of QRISK2. BMJ. 2008;336(7659):1475–82.
Lindhardsen J, et al. The risk of myocardial infarction in rheumatoid arthritis and diabetes mellitus: a Danish nationwide cohort study. Ann Rheum Dis. 2011;70(6):929–34.
van Halm VP, et al. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: a cross-sectional study, the CARRE Investigation. Ann Rheum Dis. 2009;68(9):1395–400.
Agca R, et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis. 2017;76(1):17–28.
Steer SE, et al. Reduced telomere length in rheumatoid arthritis is independent of disease activity and duration. Ann Rheum Dis. 2007;66(4):476–80.
Gamal RM, et al. Telomere dysfunction-related serological markers and oxidative stress markers in rheumatoid arthritis patients: correlation with diseases activity. Clin Rheumatol. 2018;37(12):3239–46.
Fujii H, et al. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci USA. 2009;106(11):4360–5.
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Investig. 2005;115(5):1111–9.
Morigi M, et al. Leukocyte–endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-kB-dependent fashion. J Clin Investig. 1998;101(9):1905–15.
Singer G, Granger DN. Inflammatory responses underlying the microvascular dysfunction associated with obesity and insulin resistance. Microcirculation. 2007;14(4–5):375–87.
Friederich M, Hansell P, Palm F. Diabetes, oxidative stress, nitric oxide and mitochondria function. Curr Diabetes Rev. 2009;5(2):120–44.
Hinokio Y, et al. Urinary excretion of 8-oxo-7, 8-dihydro-2′-deoxyguanosine as a predictor of the development of diabetic nephropathy. Diabetologia. 2002;45(6):877–82.
Loft S, et al. Oxidative DNA damage estimated by 8-hydroxydeoxyguanosine excretion in humans: influence of smoking, gender and body mass index. Carcinogenesis. 1992;13(12):2241–7.
Sampson MJ, et al. Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care. 2006;29(2):283–9.
Masi S, et al. Telomere length, antioxidant status and incidence of ischaemic heart disease in type 2 diabetes. Int J Cardiol. 2016;216:159–64.
Salpea KD, et al. Association of telomere length with type 2 diabetes, oxidative stress and UCP2 gene variation. Atherosclerosis. 2010;209(1):42–50.
Humphrey LL, et al. Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J Gen Intern Med. 2008;23(12):2079–86.
Janket SJ, et al. Meta-analysis of periodontal disease and risk of coronary heart disease and stroke. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95(5):559–69.
Lafon A, et al. Periodontal disease and stroke: a meta-analysis of cohort studies. Eur J Neurol. 2014;21(9):1155–61, e66–7.
Mustapha IZ, et al. Markers of systemic bacterial exposure in periodontal disease and cardiovascular disease risk: a systematic review and meta-analysis. J Periodontol. 2007;78(12):2289–302.
Tonetti MS, Van Dyke TE, E.F.P.A.A.P.W. Working group 1 of the joint. Periodontitis and atherosclerotic cardiovascular disease: consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Clin Periodontol. 2013;40(Suppl 14):S24–9.
D’Aiuto F, et al. Systemic effects of periodontitis treatment in patients with type 2 diabetes: a 12 month, single-centre, investigator-masked, randomised trial. Lancet Diabetes Endocrinol. 2018;6(12):954–65.
D’Aiuto F, et al. Short-term effects of intensive periodontal therapy on serum inflammatory markers and cholesterol. J Dent Res. 2005;84(3):269–73.
D’Aiuto F, Orlandi M, Gunsolley JC. Evidence that periodontal treatment improves biomarkers and CVD outcomes. J Clin Periodontol. 2013;40(Suppl 14):S85–105.
Teeuw WJ, et al. Treatment of periodontitis improves the atherosclerotic profile: a systematic review and meta-analysis. J Clin Periodontol. 2014;41(1):70–9.
Tonetti MS, et al. Treatment of periodontitis and endothelial function. N Engl J Med. 2007;356(9):911–20.
Wang X, et al. The effect of periodontal treatment on hemoglobin a1c levels of diabetic patients: a systematic review and meta-analysis. PLoS One. 2014;9(9):e108412.
Sanders AE, et al. Telomere length attrition and chronic periodontitis: an ARIC Study nested case–control study. J Clin Periodontol. 2015;42(1):12–20.
Benetos A, et al. Telomere length as an indicator of biological aging: the gender effect and relation with pulse pressure and pulse wave velocity. Hypertension. 2001;37(2 Pt 2):381–5.
Benetos A, et al. Short telomeres are associated with increased carotid atherosclerosis in hypertensive subjects. Hypertension. 2004;43(2):182–5.
Demissie S, et al. Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell. 2006;5(4):325–30.
Fitzpatrick AL, et al. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am J Epidemiol. 2007;165(1):14–21.
Samani NJ, et al. Telomere shortening in atherosclerosis. Lancet. 2001;358(9280):472–3.
Aviv A, et al. Menopause modifies the association of leukocyte telomere length with insulin resistance and inflammation. J Clin Endocrinol Metab. 2006;91(2):635–40.
Valdes AM, et al. Obesity, cigarette smoking, and telomere length in women. Lancet. 2005;366(9486):662–4.
Al-Attas OS, et al. Telomere length in relation to insulin resistance, inflammation and obesity among Arab youth. Acta Paediatr. 2010;99(6):896–9.
O’Donnell CJ, et al. Leukocyte telomere length and carotid artery intimal medial thickness: the Framingham Heart Study. Arterioscler Thromb Vasc Biol. 2008;28(6):1165–71.
Panayiotou AG, et al. Leukocyte telomere length is associated with measures of subclinical atherosclerosis. Atherosclerosis. 2010;211(1):176–81.
Willeit P, et al. Cellular aging reflected by leukocyte telomere length predicts advanced atherosclerosis and cardiovascular disease risk. Arterioscler Thromb Vasc Biol. 2010;30(8):1649–56.
Bischoff C, et al. No association between telomere length and survival among the elderly and oldest old. Epidemiology. 2006;17(2):190–4.
Houben JM, et al. Telomere length and mortality in elderly men: the Zutphen Elderly Study. J Gerontol A Biol Sci Med Sci. 2011;66(1):38–44.
Martin-Ruiz CM, et al. Telomere length in white blood cells is not associated with morbidity or mortality in the oldest old: a population-based study. Aging Cell. 2005;4(6):287–90.
Strandberg TE, et al. Association of telomere length in older men with mortality and midlife body mass index and smoking. J Gerontol A Biol Sci Med Sci. 2011;66(7):815–20.
Masi S, et al. Rate of telomere shortening and cardiovascular damage: a longitudinal study in the 1946 British Birth Cohort. Eur Heart J. 2014;35(46):3296–303.
Haycock PC, et al. Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. Br Med J. 2014;349:g4227.
Codd V, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45(4):422–7, 427e1–2.
I.R.G.C.E.R.F. Collaboration, et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 2012;379(9822):1205–13.
Harrison SC, et al. Interleukin-6 receptor pathways in abdominal aortic aneurysm. Eur Heart J. 2013;34(48):3707–16.
Interleukin-6 Receptor Mendelian Randomisation Analysis Consortium, et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a Mendelian randomisation analysis. Lancet. 2012;379(9822):1214–24.
Paige E, et al. Interleukin-6 receptor signaling and abdominal aortic aneurysm growth rates. Circ Genomic Precis Med. 2019;12(2):e002413.
Kranzhofer R, et al. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19(7):1623–9.
Wu J, et al. The role of oxidative stress and inflammation in cardiovascular aging. BioMed Res Int. 2014;2014:615312.
Rodier F, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973–9.
Botto N, et al. Evidence for DNA damage in patients with coronary artery disease. Mutat Res. 2001;493(1–2):23–30.
Mahmoudi M, et al. Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ Res. 2008;103(7):717–25.
Martinet W, et al. Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res. 2001;88(7):733–9.
Martinet W, et al. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002;106(8):927–32.
Morgan RG, et al. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol. 2013;305(2):H251–8.
Gardner SE, et al. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1alpha-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol. 2015;35(9):1963–74.
Ridker PM, et al. antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–31.
Libby P, Ebert BL. CHIP (clonal hematopoiesis of indeterminate potential). Circulation. 2018;138(7):666–8.
Steensma DP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16.
Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98.
Jaiswal S, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111–21.
Sano S, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J Am Coll Cardiol. 2018;71(8):875–86.
Dorsheimer L, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019;4(1):25–33.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
This article is part of the topical collection on Vascular Aging and Arterial Stiffness.
Rights and permissions
About this article

Cite this article
Chiriacò, M., Georgiopoulos, G., Duranti, E. et al. Inflammation and Vascular Ageing: From Telomeres to Novel Emerging Mechanisms. High Blood Press Cardiovasc Prev 26, 321–329 (2019). https://doi.org/10.1007/s40292-019-00331-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40292-019-00331-7