
Abstract
Introduction
Assessment of KIT/PDGFRA mutations is essential for therapeutic decision making in patients with gastrointestinal stromal tumor (GIST). Blood-derived circulating tumor DNA can provide molecular information representative of the tumor tissue.
Methods
In this study, primary tumors and matched presurgical blood samples were collected from 25 patients with localized gastric GIST, and the DNAs were analyzed for KIT and PDGFRA mutations using a next-generation sequencing platform.
Results
Sequencing of the tumors identified mutations in KIT exon 11 in 18/25 cases (72 %). The mutations were detected in 13/18 (72 %) plasma samples from the patients harboring KIT mutation in the paired GIST tissue. Identical point mutations were found in three of the presurgical plasma samples, and insertion/deletions were detected as single-base substitutions in ten cases. No mutations were detected in plasma samples from the seven patients with KIT/PDGFRA wild-type GIST.
Conclusion
Our study demonstrates that primary KIT mutations can be detected in the presurgical plasma of patients with localized GIST; this would help clinicians reach proper diagnoses before surgery and assist them to make appropriate therapeutic decisions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
This is the first study demonstrating the possibility of using plasma DNA as a surrogate marker for the presence of KIT/PDGFRA mutations prior to resection of primary GIST. |
Given the frequent deletion and insertion mutations present in GISTs, we could detect these mutations in only half of the cases from plasma DNA. |
Although the analysis of plasma DNA is not enough to fully replace tissue sequencing, it can provide useful information for diagnostic purposes and therapeutic decisions. |
1 Introduction
In gastrointestinal stromal tumors (GISTs), 85–90 % of patients harbor activating mutations in the KIT or PDGFRA receptor tyrosine kinase genes with the most common mutations affecting the juxtamembrane domain encoded by KIT exon 11. Knowledge of the mutational status provides clinicians with a mandatory guide to therapeutic decision making concerning patients with unresectable or metastatic GIST [1]. Although a tumor itself is the major source of tumor DNA, acquiring DNA through biopsy or surgery is invasive and often not possible. Cell-free circulating tumor DNA (ctDNA) from a patient’s peripheral blood, known as liquid biopsy, can theoretically overcome the limitations of tissue biopsy and provide the same molecular information [2]. Detection of tumor-associated mutations in the blood can be used in the clinic, for purposes including the assessment of prognosis, early detection of disease recurrence, and as surrogates for traditional biopsies with the purpose of predicting response to treatments and the development of acquired resistance [3]. Recently, a few promising studies on the application of liquid biopsy in GISTs were carried out using the BEAMing (beads, emulsion, amplification, magnetics) technology and allele-specific ligation polymerase chain reaction (PCR) assays [4, 5]. As both are designed to detect specific, predefined mutations, negative test results cannot be interpreted as demonstrating the absence of mutations in the genes [6]. Thus, less targeted approaches may be better able to provide complete sequences of hotspot exons [2]. At present, several next-generation sequencing (NGS; or massive parallel sequencing) platforms are available, and they hold major advantages in terms of breadth of mutation coverage.
Prior studies in GISTs and other cancers have been conducted mostly in the metastatic settings, under which the amount of ctDNA tends to be greater [2, 7]. Previously, we reported that additional KIT and PDGFRA mutations were detected by ctDNA sequencing in two of the three GIST patients on tyrosine kinase inhibitor therapy [8]. To validate the use of ctDNA as a biomarker for determining KIT and PDGFRA mutations, we compared the mutation status between tumor tissue and presurgical plasma samples from patients with localized gastric GIST using an NGS platform.
2 Materials and Methods
Peripheral blood was drawn from 25 patients with a localized gastric mass before surgery. None of the patients was reported to receive neoadjuvant chemotherapy. The tumors, resected between 2009 and 2014, were diagnosed as GISTs based on histologic findings and immunohistochemical expression of KIT (CD117). No other primary tumor or metastasis was found at the time of diagnosis of gastric GIST. Formalin-fixed paraffin-embedded (FFPE) tumor and plasma samples with de-identified patient information were provided by the Korea Biobank Network. According to the SOPs of the Korea Biobank Network, blood collected in EDTA (ethylene diamine tetraacetic acid) or sodium citrate tube was centrifuged at 2500 rpm for 10 min and kept frozen at −20 or −70 °C. The frozen plasma samples were express shipped on dry ice and delivered to our diagnostic laboratory within 5 h. All samples derived from the biobanks were obtained with informed consent under institutional review board (IRB)-approved protocols, and this study was approved by the IRB of Inje University Sanggye Paik Hospital (SGPAIK 2014-07-023).
The diagnosis of each case was confirmed by a single pathologist (J. Pyo), and tumor-rich areas (40–80 %) were dissected from unstained FFPE sections. Genomic DNAs were then isolated from the tumor sections and matched frozen plasma aliquots using High Pure PCR Template Preparation Kit (Roche Diagnostics, Mannheim, Germany). First, all tissue samples were tested for mutations in exons 9, 11, 13, and 17 of KIT and in exons 12, 14, and 18 of PDGRFA by the Sanger sequencing method using an ABI 3700 automated sequencer (Applied Biosystems, Foster City, CA, USA). Next, NGS was performed in both tumor and plasma samples. Amplicon library preparation and pyrosequencing were carried out according to manufacturers’ protocols [8, 9]. The GIST panel used in this study (SeaSun Biomaterials, Daejeon, Korea) covers exons 9, 11, 13, and 17 of KIT and exon 18 of PDGFRA. Based on the literature and our prior experience, a variety of deletions and insertions (indels) that occur around codons 564–571 and a point mutation at codon 561 are the most common in PDGFRA exon 12, which comprised about 5 % of the total number of PDGFRA mutations listed in the COSMIC database and was not included in the NGS panel [10]. For negative control, we used DNA obtained from HeLa cells in each run. Emulsion PCR, breaking, and bead enrichment were performed using the GS Junior Titanium emPCR Kit (Lib-L), emPCR Reagents Lib-L Kit, Oil and Breaking Kit, and the Bead Recovery Reagents Kit (Roche Diagnostics, Mannheim, Germany). To increase the resolution of low-level somatic mutant molecules within a high background of wild-type molecules, the InsightTM Onco Panel for KIT and PDGFRA (SeaSun Biomaterials, Daejeon, Korea) was used for mutant enrichment PCR as described previously when analyzing the plasma samples. Peptide nucleic acid (PNA) probes were used as PCR blockers to suppress amplification of wild-type alleles [11]. To know the sensitivity of NGS only, the mutant enrichment method was not used in the first batch samples (case no. 4–10). Sequencing was performed using the GS Junior system (Roche Diagnostics, Mannheim, Germany), and the results were analyzed with the GS Amplicon Variant Analyzer (version 2.7; Roche Diagnostics, Mannheim, Germany). The analyses of ctDNA were carried out in a blind fashion with respect to mutation status of primary tumor.
In addition, qualitative analysis of plasma DNA was performed with microfluidic DNA chip gel electrophoresis using Agilent High Sensitivity DNA Kit and Agilent 2100 Bioanalyzer (Agilent Technology, Santa Clara, CA, USA). DNA chips were run with two internal DNA markers of known size (35 and 10387 bp, respectively), and a DNA ladder of standard concentrations that allowed automatic calculation of DNA fragment sizes and amounts. We determined the ratio of absorbance at 260 and 280 nm (A260/280) using a NanoDrop 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and obtained relatively low 260/280 nm ratios, suggesting contamination of protein or other blood-derived products. We also used an Agilent microfluidic DNA chip to determine the presence of nucleosome-sized DNA fragments (average of 160–180 bp), and Agilent software was used to calculate the concentrations of apoptotic DNA in each plasma sample (Table 1) [12].
3 Results
Twenty-five patients with gastric GIST were included in this study (13 men and 12 women), and their age ranged from 29 to 81 years (mean: 59.4 years). KIT exon 11 mutations were identified in 18/25 (72.0 %) tissue samples by both Sanger sequencing and NGS. The remaining seven cases, sequenced to a read depth of >1400, had no detectable mutation in KIT and PDGFRA. In these 18 cases, the tumor sizes ranged between 1.8 and 5.6 cm (mean: 3.4 cm), and the mitotic counts were less than 5 per 50 high-power fields (5 mm2). Two GISTs were classified as no risk and 16 as very low or low risk of progressive disease. Seventeen of them were spindle cell tumors, and one showed mixed spindle and epithelioid cells.
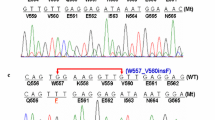
Matched presurgical plasma samples were analyzed for tumor-specific mutations. A summary of results comparing tumor tissue and ctDNA analysis is shown in Table 1. The amount of total plasma DNA varied among samples, ranging from 3.376 to 20.848 ng/μL, which is consistent with previous reports [7]. The most common type in tumor tissues was single-base substitutions (n = 8), followed by deletions (n = 7) and insertions (n = 3). Five of the deletions in exon 11 involved codons 557 and/or 558. Of the plasma samples from 18 patients with mutations detected in tumor tissues, 13 (72 %) were identified as harboring KIT exon 11 mutations with allele frequencies ranging from 0.19 to 21.96 %. Identical point mutations were found in three of the matched ctDNA samples (case No. 11, 12, and 13); whereas indels were detected as single-base substitutions in the other ten samples (case No. 1–10). None of the seven patients with wild-type GIST had detectable mutations in KIT (exons 9, 11, 13, and 17) and PDGFRA (exon 18) in plasma DNA.
4 Discussion
To our knowledge, this is the first study demonstrating the possibility of using ctDNA as a surrogate tissue to identify the presence of mutations in patients with GIST prior to resection of their primary tumors. This approach may hold the promise of detecting primary mutations when tissue samples are too small or unsuitable for standard mutation analysis, and may obviate the need for repeated biopsies. We also combined an enrichment-PCR method with next-generation pyrosequencing to enhance the detection of low frequency mutations in plasma [11].
BEAMing, one of the most advanced technologies for point-mutation analysis of ctDNA, previously detected secondary mutations more readily in plasma (47 %) than in tissue (12 %). However, the assay was less sensitive for identifying primary KIT exon 11 mutations in ctDNA (12 % of samples tested) [2, 5]. A recent report by Bauer et al. showed that primary and resistant mutations were found in 41 and 86 % of the plasma samples, respectively, from patients with metastatic GIST by using an NGS platform (Illumina’s MiSeq) and a cutoff of allele frequency of 0.5 % [13]. These discrepancies are partly attributable to the study design that specifically targeted secondary mutations, but mostly to the diversity of KIT mutations in GISTs [2].
Using a cutoff value of 0.5 %, the same as that used by Bauer et al., we were able to detect the presence of KIT mutations in 10 of the 18 cases (56 %) in plasma DNA. The present study provides a proof of concept of the feasibility of a noninvasive method for detecting primary mutations in GISTs; however, there were limitations in our study. First, the process of blood sample collection and plasma preparation, as well as the gap in time between blood draw and surgery, were not available to the authors, and might vary between patients and institutions. Circulating DNA in the blood is known to be highly fragmented, and the integrity and quality of ctDNA might be affected by several preanalytical factors, including clotting, delayed separation of blood cells from plasma, freeze-thaw, and storage [14]. To apply ctDNA sequencing in the clinic, acquisition of high-quality DNA is one of the most important prerequisites. In a previous study, we were able to detect identical primary KIT mutations (including deletion of K558_E562 in exon 11 and duplication of A502_Y503 in exon 9) in both tumor tissue and ctDNA using the same platform [8]. In that study, all the samples underwent the same pre-analytical processing steps; that is, blood was centrifuged immediately after collection, and the plasma was stored at −20 °C for no more than 3 days until DNA was extracted and analyzed. In addition, samples were from the patients with advanced GIST on tyrosine kinase inhibition therapy, in which the amount of ctDNA might be greater than those from the low-risk tumors of the present study. Second, some technical issues of NGS need to be considered when analyzing ctDNA. Detection of tumor-specific mutations in the blood using NGS technology may offer increased sensitivity. However, the liquid biopsy is not at a stage where it can fully replace sequencing of a tissue sample. Still, there are problems in identifying long indels using any current NGS platform, as well as the BEAMing technology. They are biased toward the shorter alterations, particularly for the analysis of DNAs that are degraded or highly fragmented. For the ten cases in which NGS failed to call the exact genotypes, five of the indel mutations in the tissue were detected as single-base substitutions within the deleted regions of KIT (indicated by asterisks in Table 1). Thus, for example, a PCR-based backup assay can be used to complement NGS and not to miss critical indels that occur in EGFR exon 19 and KIT exon 11 in cases of lung cancer and GIST, respectively. Third, a mutation could be detected in as low as 0.19 % of mutant allele by the combinatory use of PNA and NGS (case No. 13). In a previous study using a mixture of DNAs from HeLa and EGFR-mutant cells and the same analytical platform, the limit of detection was 0.5 % mutant allele by NGS alone, and the mutant-specific enrichment procedures using PNA probes resulted in an increase of detection limit and rate for mutations [11]. However, the diagnostic performance between the two methods could not be compared in this study, because only one of them was employed for analyzing each plasma sample. Finally, no mutations were detected in the ctDNA samples of five patients (case No. 14–18), but KIT mutations were detected in the tumor tissues. These five ctDNA samples remained negative even after using the PNA-NGS approach. The reason why a mutation was detected in tumor but not in synchronous plasma could be due to either the absence of the mutation in plasma DNA or to the presence of the mutation in allele frequency below the detection limit of our method [15]. There were no differences in tumor size and amount of ctDNA between the positive and the negative cases. We were also not able to establish a correlation between risk grade of GIST and amount of ctDNA due to the small sample size.
5 Conclusion
Along with rigorous concordance studies, investigations to assess the applicability of monitoring tumor-associated genetic aberrations using ctDNA analysis as the primary endpoint are needed [3]. In parallel, it is also important to implement optimal standardization of preanalytical steps to adopt ctDNA into routine clinical practice. Despite the limitations, this noninvasive approach may provide information for diagnostic purposes and therapeutic decisions for patients with GIST. This can be further used to assess microscopic residual disease that could lead to recurrence, and guide adjuvant therapy recommendations based on the presence of mutation-positive ctDNA after surgery.
References
Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–9. doi:10.1200/JCO.2003.04.190.
Nannini M, Astolfi A, Urbini M, Biasco G, Pantaleo MA. Liquid biopsy in gastrointestinal stromal tumors: a novel approach. J Transl Med. 2014;12:210. doi:10.1186/1479-5876-12-210.
Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10(8):472–84. doi:10.1038/nrclinonc.2013.110.
Maier J, Lange T, Kerle I, Specht K, Bruegel M, Wickenhauser C, et al. Detection of mutant free circulating tumor DNA in the plasma of patients with gastrointestinal stromal tumor harboring activating mutations of CKIT or PDGFRA. Clin Cancer Res. 2013;19(17):4854–67. doi:10.1158/1078-0432.CCR-13-0765.
Demetri GD, Jeffers M, Reichardt P, Kang Y-K, Blay J-Y, Rutkowski P et al., editors. Mutational analysis of plasma DNA from patients (pts) in the phase III GRID study of regorafenib (REG) versus placebo (PL) in tyrosine kinase inhibitor (TKI)-refractory GIST: Correlating genotype with clinical outcomes. ASCO Annual Meeting Proceedings; 2013.
Richardson AL, Iglehart JD. BEAMing up personalized medicine: mutation detection in blood. Clin Cancer Res. 2012;18(12):3209–11. doi:10.1158/1078-0432.CCR-12-0871.
Beaver JA, Jelovac D, Balukrishna S, Cochran RL, Croessmann S, Zabransky DJ, et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res. 2014;20(10):2643–50. doi:10.1158/1078-0432.CCR-13-2933.
Kang G, Bae BN, Sohn BS, Pyo JS, Kang GH, Kim KM. Detection of KIT and PDGFRA mutations in the plasma of patients with gastrointestinal stromal tumor. Target Oncol. 2015;. doi:10.1007/s11523-015-0361-1.
Lee B, Han G, Kwon MJ, Han J, Choi YL. KRAS mutation detection in non-small cell lung cancer using a peptide nucleic acid-mediated polymerase chain reaction clamping method and comparative validation with next-generation sequencing. Korean J Pathol. 2014;48(2):100–7. doi:10.4132/KoreanJPathol.48.2.100.
Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23(23):5357–64. doi:10.1200/JCO.2005.14.068.
Kang S, Kim BG, Han HH, Lee JH, Kim JE, Shim HS, et al. Targeted sequencing with enrichment PCR: a novel diagnostic method for the detection of EGFR mutations. Oncotarget. 2015;6(15):13742–9.
Urosevic N, Inglis T, Grasko J, Lim E. Evaluation of clinical laboratory methods for plasma cell-free DNA analysis in suspected septicaemia. J Med Diagn Methods. 2013;2:123. doi:10.4172/2168-9784.1000123.
Bauer S, Herold T, Muhlenberg T, Reis A-C, Falkenhorst J, Backs M et al., editors. Plasma sequencing to detect a multitude of secondary KIT resistance mutations in metastatic gastrointestinal stromal tumors (GIST). ASCO Annual Meeting Proceedings; 2015.
Chan KC, Yeung SW, Lui WB, Rainer TH, Lo YM. Effects of preanalytical factors on the molecular size of cell-free DNA in blood. Clin Chem. 2005;51(4):781–4. doi:10.1373/clinchem.2004.046219.
Rothe F, Laes J-F, Lambrechts D, Smeets D, Vincent D, Maetens M, et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol. 2014;25(10):1959–65.
Acknowledgments
The biospecimens and data for this study were provided by the Inje Biobank of Inje University Busan Paik Hospital and the Keimyung Human Bio-Resource Bank, members of the Korea Biobank Network. This work was conducted during a research year supported by Inje University in 2015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors (GK, BSS, J-S P, JYK, BL, K-M K) declare that they have no conflict of interest.
Funding
The authors have no funding to declare.
Ethical approval and informed content
All samples were obtained with informed consent under IRB-approved protocols, and this study was approved by the IRB of Inje University Sanggye Paik Hospital (SGPAIK 2014-07-023).
Rights and permissions
About this article

Cite this article
Kang, G., Sohn, B.S., Pyo, JS. et al. Detecting Primary KIT Mutations in Presurgical Plasma of Patients with Gastrointestinal Stromal Tumor. Mol Diagn Ther 20, 347–351 (2016). https://doi.org/10.1007/s40291-016-0203-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-016-0203-6