
Abstract
Parkinson’s disease is a chronic, neurodegenerative disease, which manifests with a mixture of motor, cognitive and behavioural symptoms. Levodopa is the most effective antiparkinsonian treatment to date, although chronic use engenders a mixture of complications in a substantial proportion of patients. Amongst these is the occurrence of episodes of worsening symptoms—‘off’ phenomena. These episodes can manifest with either motor or non-motor symptoms or a combination of these features and have been found to have profound impacts on patients’ quality of life. Although preventative measures are poorly evidenced, avoiding excessive total daily levodopa intake in selected populations that are deemed to be of a higher risk for developing these episodes warrants further exploration. Methods to improve levodopa bioavailability and delivery to the brain are currently available and are of value in addressing these episodes once they have become established. These include modifications to levodopa formulations as well as the use of complimentary agents that improve levodopa bioavailability. The deployment of device-assisted approaches is a further dimension that can be considered in addressing these debilitating episodes. This review summarises the clinical manifestations of ‘off’ phenomena and the current approaches to treat them. Although we briefly discuss clinical advances on the horizon, the predominant focus is on existing, established treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
‘Off’ phenomena in Parkinson’s disease commonly occur as a result of long-term levodopa use and can have a profound impact on patients’ quality of life. |
Avoiding inappropriately high total levodopa daily dosage in high-risk cohorts could potentially mitigate their risk of occurrence. |
In patients suffering from ‘off’ phenomena, fractionating levodopa dosages, the addition of adjunctive agents (e.g. dopamine agonists, monoamine oxidase-B inhibitors, Catechol-O-methyltransferase inhibitors) or the introduction of device-assisted therapies (apomorphine infusion, deep brain stimulation, levodopa-carbidopa intestinal gel) can significantly improve symptom burden. |
1 Introduction
Parkinson’s disease (PD) is the second most highly prevalent neurodegenerative condition and results in substantial patient morbidity and care-giver burden [1]. Although motor features such as bradykinesia, rigidity, postural instability and rest tremor have become synonymous with the disease, patients with PD also experience a range of non-motor symptoms (NMS), which also impose negatively on quality of life (QOL) [2, 3].
Dopaminergic deficiency remains a key aspect of the pathogenesis and clinical manifestations of the disease [4], therefore treatment approaches targeting replacement of dopamine remain paramount. l-dopa (3, 4-dihydroxy-l-phenylalanine), was engineered to achieve this outcome over 50 years ago and still remains the gold standard for symptomatic management of PD today [5, 6]. This molecule is converted to dopamine by the enzyme aromatic amino acid-decarboxylase (AADC) largely upon crossing the blood–brain barrier. This increases presynaptic dopamine concentrations, synaptic release of dopamine and therefore post-synaptic dopamine receptor stimulation in the basal ganglia with resultant improvements to symptoms [7,8,9].
The beneficial effects of l-dopa replacement, although substantial, tend to wane as the disease progresses. The numerous explanations for this will be discussed in detail later. The net result is of an increasing frequency of rapid and, at times, unpredictable cycles between good therapeutic motor responses (‘on’ states), poorly controlled symptoms (‘off’ states)- known as motor fluctuations (MF) and involuntary movements (dyskinesia), collectively considered as motor complications. There is an increasing awareness that non-motor fluctuations (NMF) can also occur and mirror levels of dopamine release, which can be harder to detect because they are less obviously manifest to carers or clinicians [10, 11].
Fluctuations are a major source of disability and result in a substantial reduction to QoL. Modifying treatment approaches to prevent or improve fluctuations have therefore become a major focus of therapeutics in PD. Here, we aim to provide an overarching view of these occurrences and the principles and specific therapies currently utilised in their management. This review will focus on the ‘off’ state and does not encompass specific techniques employed for the management of dyskinesia although the majority of treatments described, potentially provide overlapping benefits for both ‘off’ periods and dyskinesia.
1.1 Clinical Characteristics
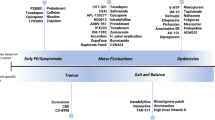
While subtle variations in PD motor severity are not uncommon, abrupt “On–Off” phases in the setting of chronic levodopa therapy are unique to patients with PD and were observed soon after the first introduction of l-dopa therapy into clinical practice [12]. A variety of terms have subsequently been coined to describe variations in their nature. These differences are depicted in Fig. 1 and predominant underlying mechanisms in Fig. 2.

Schematic depiction of different motor fluctuations and their relation to levodopa intake. EMO early morning ‘off’

Different ‘off’ phenomena and varying mechanisms resulting in their development. EMO = Early morning OFF
First, the more gradual transitions in clinical symptom control noted towards the end of doses are described as ‘wearing off’ phenomenon or end-of-dose deterioration [13].
The occurrence of slowness or immobility in a patient’s waking hours prior to the first medication dose is considered an early morning ‘off’ state (EMO). This is related to low plasma levodopa levels from a lack of overnight medication.
The term delayed ‘on’ is a further term often used to describe the lengthy latency for a levodopa dose to start working and can commonly occur with the first morning dose or after a meal [14].
In contrast, a dose failure or no ‘on’ refers to times when the medication fails to work.
When the symptoms experienced during ‘off’ states are of a non-motor nature, these can be referred to as a non-motor ‘off’ state, or non-motor fluctuation (NMF) [15].
The constellation of motor symptoms experienced by patients in the ‘off’ state is broad and variable. Although some report a mixture of slowness, stiffness, incoordination or reduced dexterity and muscle cramping, others experience difficulties in their ability to stand up, balance, or even swallow. Patients can also suffer changes in their voice and breathing while experiencing posturing (dystonia) in their hands, feet or legs. On occasion, a profound worsening of tremor and the emergence of gait freezing can be seen [16]. The non-motor symptoms commonly reported can be divided into neuropsychiatric, autonomic and sensory domains. Neuropsychiatric symptoms can manifest with fluctuations in cognition, attention, and mood (anxiety, depression, apathy). Autonomic symptoms on the other hand comprise altered sweating, light headedness, abdominal pain or bloating, and urinary urgency. Patients experiencing sensory symptoms report a mixture of visual disturbances, pain, dysaesthesia, akathisia and restless legs. A number of non-motor endo-phenotypes have also been described and comprise subtypes of either a depressed, anxious, or anxious-depressed variety. Though neurobehavioural syndromes [impulse control disorders (ICDs), punding and dopamine dysregulation syndrome] are not strictly considered NMFs, they can masquerade as such considering their varying occurrences with l-dopa intake. The phenomenon of anticipation of an impending ‘off’ period-metacognition is a further non-motor manifestation and can worsen the distress and severity of the fluctuation [17, 18].
Although fluctuations become more prevalent as the disease progresses, the frequency noted seems to vary between cohorts, presumably in view of discrepancies in ascertainment approaches. Broadly speaking however, 10% of patients will develop MF annually from the onset of initiation of levodopa [19]. Over time, this results in a compounded average of approximately 40% of patients reporting MF after 4–6 years [19] and an almost universal occurrence after 10 years [20, 21]. ‘Wearing off’ in isolation tends to predate dyskinesia and has been reported in up to 50% of patients within 2 years of starting levodopa, although it can be observed within months of initiation [22], indicating that the disease duration likely has the greater influence on MF development rather than the duration of l-dopa exposure. EMOs are the most common initial manifestation reported with a recent survey of 320 patients suggesting over 40% of early disease patients and 60% of patients overall suffer from it [23], although paradoxical transient improvement in symptom control following sleep is also well recognised. However, ‘end of dose wearing off’ is the most common l-dopa-related phenomenon overall with delays to ‘switching on’ from the following dose being strongly associated [24, 25].
Despite best current medical management, a large observational study suggested ‘off’ episodes continue to trouble patients on average 2–3 h per day stressing the need for more ambitious treatment approaches [26]. The prevalence of NMFs in conjunction with MF varies (17–100%) depending on evaluation tools utilised and populations tested [27]. While EMO was noted in approximately 60% of a cohort, almost 90% of patients reported severe NMS (such as urinary frequency, anxiety, depression, pain, dribbling of saliva) on awakening. Despite their commonly noted co-occurrence with MF, NMF can also occur in an isolated and heterogeneous manner and predate their motor counterpart. While anxiety, fatigue, pain, and paraesthesia commonly complicate motor fluctuations, their co-occurrence is also common with dyskinesia [28]. Psychiatric symptoms and pain appear to frequently fluctuate according to l-dopa replacement, while concentration difficulties, fatigue, depression and anxiety can persist in the on state while being worsened during ‘off’ states [29]. Apathy and panic attacks tend to characterise more severe NMFs and are amongst the most disabling presentations [27]. Although there is some suggestion of specific NMF endophenotypes occurring alongside MF, this finding is inconsistent [30].
Factors contributing to the development of fluctuations have been extensively studied and found to be similar for both MF and NMFs [31, 32]. These include a younger onset age, female gender, more severe disease, and higher levodopa dosages [33,34,35,36]. The practical implications of these identified factors are elaborated on in the discussion of preventative approaches. Although a number of genetic factors have been implicated in the development of dyskinesia [37,38,39,40], their role in the development of ‘off’ phenomena specifically is less clear.
Taken together, fluctuations of all types are undoubtedly highly disabling, although patients seem to be particularly troubled by ‘off’ phenomena in view of the functional disability, pain and distress associated with these periods [41, 42]. This assertion is further validated by consequent reductions in QoL scores and patients reporting a preference for the ON state with dyskinesia over the OFF condition [43]. Despite a traditional clinical bias for addressing MF, more recent demonstrations of relatively greater NMS and NMF impact on QoL (mood symptoms and pain in particular) have propelled these aspects into a more central consideration of patient care [27, 44]. Nevertheless, much more clinical education and better detection tools addressing these phenomena are still needed moving forward.
1.2 Pathogenic Mechanisms
‘Off’ phenomena need to be contextually understood in conjunction with how MFs develop as a whole. However, it is crucial to first understand that the development of fluctuations relies on the presence of dopaminergic therapy, in particular levodopa [45, 46]. Levodopa therapeutics consist of a short duration response (SDR) and long-duration response (LDR) [45]. The SDR impacts on symptoms over minutes to hours after an individual drug dose in accordance with the drug’s pharmacokinetic profile. In contrast, the LDR is a sustained antiparkinsonian effect resulting from prolonged levodopa treatment. Both these entities occur from initiation of levodopa therapy [47]. The predominant manifestation of the LDR at the beginning can mask the true clinical nature of SDRs despite fluctuations in plasma levodopa levels. A subsequent progressive decay of the LDR makes the magnitude of the SDR effect more apparent with the resultant clinical observation of fluctuations [48]. Change to the balance between LDR and SDR over time is a result of peripheral factors related to l-dopa pharmacokinetics, absorption, and transport, as well as central pharmacodynamic alterations [49] (Fig. 2).
Peripheral factors can make levodopa delivery to the brain less reliable because of their impact on the consistency of levodopa plasma levels. Clinically, they tend to be more responsible for the development of the delayed ‘on’ phenomena or dose failures, although they may also have some bearing on other MF [50]. Distinct abnormalities that contribute to PD include delayed gastric emptying and therefore delayed or absent proximal small intestine levodopa absorption as well as swallowing impairment. Oral cavity abnormalities (epiglottic valleculae, pyriform sinus) and oesophageal alterations (non-peristaltic swallows, belching, segmental spasms, dilatation, and gastro-oesophageal reflux) can also further compound the situation by impairing the physical transit of the drug or delaying its absorption [51,52,53,54].
A common issue relates to ingestion of high protein meals, which reduce the absorption of levodopa by virtue of competition for transfer across the intestinal mucosa and the blood–brain barrier [55]. The presence of Helicobacter pylori colonisation has been further demonstrated to partly contribute to the occurrence of ‘on–off’ phenomena by virtue of reducing levodopa absorption [56], although there is not yet unequivocal evidence that eradication of H. pylori can reliably improve MF. The impact of an individual’s unique gut microbiome architecture on PD therapeutics is also currently a topic of active exploration although evidence for specific causation of MF is far from conclusive [57].
Centrally, changes in the duration of the effects of levodopa occur partly as a result of the loss of presynaptic dopaminergic terminals due to the ongoing neurodegenerative processes of PD. This reduces the capacity of the striatum to store dopamine and buffer against the consequences of oscillations of plasma levodopa levels [58, 59]. Furthermore, postsynaptic changes occur as a result of chronic non-physiological dopaminergic stimulation via alterations in striatal genes and proteins mediating receptor expression and intracellular activity, as well as functional abnormalities in basal ganglia output pathways [60,61,62].
The normal functioning of the nigrostriatal dopaminergic system depends on tonic dopaminergic activity and phasic bursts of dopamine release during times of activity. Initially, dopaminergic terminal loss can be ‘passively stabilised’ by the endogenous tonic input [63]. Over time however, exogenous levodopa becomes necessary to improve symptoms of PD and during the first years can effectively reduce the severity of the symptoms of most patients with PD. However, exogenous levodopa replacement does not restore normal dopaminergic physiology [64]. The administration of repeated doses of short-acting levodopa results in alternating high and low concentrations and therefore “pulsatile” receptor stimulation [49, 65, 66]. This pulsatile pattern of dopaminergic release is thought, at least in part, to be responsible for MF and wearing ‘off’ [67].
Although specific underlying mechanisms contributing to NMFs are far more varied and often less clear, the higher incidence of non-motor symptoms in the motor ‘off’ state and their partial responsiveness to dopaminergic therapy point to some partial role for dopaminergic denervation [68] at least to some of the non-motor symptoms. Furthermore, evidence from electrophysiological studies suggest links between MFs and NMFs have a physiological basis. In particular, while abnormal synchronisation of neuronal activity in the primary motor cortex is implicated in akinesia, a similar mechanism in the prefrontal areas potentially results in some non-motor symptoms [69]. Further parallels related to excessive synchronisation in motor areas as a result of chronic non-physiological stimulation may underpin the onset of dyskinesia, while excessive gamma synchronisation in the cognitive and limbic basal ganglia loops can conceivably lead to psychosis as well as cognitive and affective abnormalities [18, 70, 71]. However, these concepts are not an all-encompassing explanation for NMF and co-occurring mechanisms are likely to be involved. These include further involvement of serotonergic, noradrenergic and cholinergic pathways and aberrant release of dopamine as a false transmitter from these neuronal populations. Dysfunction in these systems potentially explains some of the diversity in the presentation and treatment responses noted between MF and NMF. Also, the variable degree of degeneration of these neurotransmitter systems between different people with PD results in interactions between both the dopaminergic and non-dopaminergic factors that ultimately underlie NMF [70,71,72].
2 Prevention Strategies
Community-based studies performed over several decades have suggested that younger age, greater disease severity, and higher levodopa doses are consistent risk factors for developing motor complications [36]. The ELLDOPA study compared the effect of initiating treatment with different l-dopa doses. Although higher doses provided increased clinical benefits at 9 months, patients were at an increased risk of developing both ‘off’ periods and dyskinesia [33]. The longer (approximately 4 years) STRIDE-PD study specifically compared the risk of developing dyskinesia and wearing ‘off’ in patients randomised to l-dopa/carbidopa or l-dopa/carbidopa plus the Catechol-O-methyltransferase (COMT) inhibitor, entacapone [34]. Although the addition of entacapone did not modify the risk of developing ‘off’ time, a subsequent multivariate analysis suggested that younger age and higher l-dopa doses (> 600 mg per day) were critical risk factors for the development of dyskinesia. A further crucial observation was that this risk also applied to patients developing ‘off’ phenomena, which to some would seem counterintuitive given the presumption that higher l-dopa doses would reduce ‘off’ time.
These observations implicate levodopa pharmacodynamic and pharmacokinetic factors in the development of not only dyskinesia but also wearing ‘off’. The possibility of these findings reflecting that these patients might simply have had worse disease and by virtue of this, higher dose requirements, was not supported by close scrutiny of the ELLDOPA study. In this study, patients were not only randomised to fixed levodopa doses, but there were also statistical sub-analyses suggesting that higher levodopa doses increased the risk of developing wearing ‘off’ independently of the UPDRS motor score and disease severity [35].
This study of risk factors for MF is useful in potentially tailoring preventative management of MF in early PD patients. Considering that strategies utilising higher levodopa doses are associated with an increased risk of developing ‘off’ time, it may be that in higher-risk people, e.g. younger women with relatively low weight, prescribing l-dopa on a mg/kg basis may be a worthwhile tactic. Based on this premise, we might also suggest that levodopa be used in the lowest dose that provides a satisfactory clinical response, and that using low doses of multiple drugs in combination may minimise the need of raising levodopa doses to higher doses until clinically necessary. Anecdotally, this potentially delays the onset of MF although specific studies examining this are lacking. Arguments for this approach are that first, crossing a certain dopaminergic threshold (and by extrapolation dosage) seems to be necessary for fluctuations to develop. Also, levodopa-sparing agents (e.g. dopamine agonists, monoamine oxidase inhibitors) tend not to lead to the pulsatile dopamine receptor stimulation like levodopa, and therefore are less likely to evoke fluctuations. Dopamine receptors can be divided into D1 (D1 and D5) and D2 (D2–D4) families. The D1 receptor family is more involved in the nigro-striatal pathway and agents acting on them such as levodopa tend to be more efficacious in managing motor symptoms in contrast to agents predominantly acting on D2 receptors (e.g. ropinirole, pramipexole). This lack of D1 ‘efficacy’ could partly explain the lower tendency for fluctuations to develop with these agents. However, these potential merits, have to be weighed against the substantial down sides of these agents compared to levodopa, e.g. impulsive compulsive behaviours and lower efficacy in motor symptom relief. Although previously argued, the approach of delaying levodopa in favour of levodopa sparing agents initially to delay the onset of fluctuations has now been fairly conclusively dispelled. The recent LEAP trial was conducted in early PD patients (up to 2 years since diagnosis, treatment-naïve) with the aim of distinguishing between disease-modifying and symptomatic effects of levodopa [73]. The lack of difference in motor progression as well as rates of motor fluctuations between immediately treated and delayed start patients in the study provided clear evidence that delaying levodopa does not prevent MF. Therefore, l-dopa should be initiated when the clinician deems it to be clinically appropriate with subsequent consideration to a balanced multi-agent approach (bearing in mind the potential risks and benefits of each medication type) in order to achieve the minimum necessary dosages of medication to ameliorate symptoms while avoid accelerating the onset of fluctuations.
3 Treatment Strategies
Treatment approaches to deal with fluctuations have evolved over several decades, although the targets for intervention have broadly speaking remained unchanged. The main approaches capitalise first on strategies to bypass some of the peripheral impediments to levodopa absorption described. This has resulted in an expansion in the number of levodopa formulations being developed with the goal of extending the duration of action of each dose. Also, a number of alternative therapeutic targets have been formulated to complement levodopa while potentially minimising the dosage required for optimal treatment. The modes of action of these agents are summarised in Fig. 3. Therapies that achieve improvements with greater consistency often require the assistance of devices for administration and the use of these has revolutionised the effectiveness of levodopa treatment for a sub-group of PD patients. Figure 4 presents a recommended approach towards addressing ‘off’ symptoms in a stepwise manner.

Summary of modes of action of different medication groups at the synaptic level in improving dopaminergic signalling. A2A adenosine 2A, COMT catechol-O-methyltransferase, DAT dopamine transporter, DDC dopa decarboxylase, MAO-B monoamine oxidase type B, NMDA N-methyl-d-aspartate, TH tyrosine hydroxylase, VMAT vesicular monoamine transporter

Summary of proposed step-wise approach towards managing ‘off’ symptoms. Each column addresses individual ‘off’ symptoms and specific intervention steps to improve them
3.1 l-Dopa Preparations
Immediate-release (IR) l-dopa is a short acting agent with pharmacokinetics resulting in unstable plasma l-dopa concentrations. Peak plasma levels occur within an hour of oral administration dropping to less than 10% within 5 h in healthy adults [74]. This is further compounded by potential variability in bioavailability that is influenced by gastrointestinal hypo-motility [75]. Strategies to improve oral levodopa pharmacokinetics (adjusting timing/interval and/or improving absorption) are the mainstay of initial management of fluctuations. Dividing the levodopa dosage into smaller but more frequent doses (levodopa dose fractionation) is a practical first step in attempting to address fluctuations. This approach has been demonstrated in an open-label trial to be as effective at reducing ‘off’ time as adding an adjunctive agent to achieve this outcome [76]. Dose reduction with fractionation may not result in the full ‘on’ response patients tend to be accustomed to. Shortening of inter-dose intervals while maintaining an individually optimal and often increased dose may therefore be required. Clinical experience suggests that these benefits tend to be short lived with eventual alternative approaches becoming necessary.
Meals have been established to affect levodopa absorption with consequent fluctuations in plasma peak concentrations [77]. This is particularly the case with high-protein meals or large oral intake of neutral amino acids [50]. A competition between levodopa and other amino acids seems to occur at the absorption stage as well as at the blood-brain barrier although the composition and chemical properties of a protein meal could also have an impact on gastric emptying. A useful strategy to alleviate this is to advise patients to take their levodopa medication on an empty stomach, ideally at least 30–60 min before meals. Although protein restriction has also been proposed, this approach carries substantial potential side effects (dyskinesia, weight loss, malnutrition) and should therefore not be encouraged [78]. The use of gastro-prokinetic agents such as domperidone or camicinal may be worth exploring in patients with repeated delayed ‘On’ symptoms. These agents can enhance gastric emptying, encourage more rapid absorption of l-dopa in the small bowel and therefore result in reductions in ‘off’ time [79].
Levodopa is routinely combined with a dopa-decarboxylase inhibitor (carbidopa or benserazide), which improves its absorption and reduces levodopa peripheral metabolism [80]. Controlled-release formulations of levodopa–carbidopa have been manufactured with the aim to produce more consistent plasma levels than the IR formulations; however, these agents also seem to be marred by erratic gut absorption and therefore variable plasma concentrations [81,82,83]. A recent formulation of extended-release (ER) levodopa–carbidopa (IPX066/Rytary®/Numient®) is a capsule formulation in a 1:4 ratio of carbidopa:levodopa in four distinct dosages [84]. Levodopa in this agent is rapidly absorbed with a resultant concentration peak (Cmax) within 1 h [85]. A sustained-release of levodopa subsequently follows with stable concentrations for approximately 4–5 h; thus, theoretically reducing the pulsatile stimulation of dopamine receptors and this mechanism of contribution to motor complications [86]. IPX066 has been shown to reduce ‘off’ time as a percentage of waking hours in comparison to standard IR formulations as well as levodopa–carbidopa–entacapone preparations in patients with motor complications that were not improved by previously available prolonged-release levodopa–carbidopa formulations [87, 88]. Common adverse effects of IPX066 are similar to other levodopa formulations, as is their incidence. An similar agent IPX203 has also shown similar promise and is currently being evaluated in a Phase 3 study (NCT03670953).
DM-1992 is another novel long-acting Levodopa formulation that is under clinical investigation. This agent consists of an IR levodopa layer and a novel expanding core of ER Levodopa with stomach retention of up to 9 h, and therefore a more stable pharmacokinetic profile [89]. The crossing over of patients from IR Levodopa to DM-1992 in a small cohort seemed to yield a reduction of an hour in ‘off’ time although worsening of gait and the occurrence of dizziness was more common with DM-1992 [89, 90] and this has deterred further progress to Phase 3 efficacy trials.
The Accordion Pill is another slow-release agent currently being investigated [91]. This medication comprises multiple layers of carbidopa combined with both IR and controlled release (CR) levodopa with a stomach retention of up to 14 h [92]. Plasma levodopa levels were found to fluctuate less in healthy controls when compared to IR levodopa while expected benefits to ‘on’ time and reductions in total daily l-dopa dosages have been noted [93]. The ACCORDANCE Phase 3 trial [94] compared IR levodopa with the Accordion Pill. Despite known improvements in the variability of plasma levodopa levels with the pill, no superiority in daily ‘off’ time or ‘on’ time without troublesome dyskinesia was noted (ClinicalTrials.gov ID: NCT02605434). The role of this agent in future practice is currently uncertain considering this outcome.
3.2 Adjunctive Therapies
3.2.1 Catechol-O-Methyltransferase Inhibitors
A further strategy to reduce MF is to increase levodopa peak plasma concentrations and action duration. Adjunctive medications are a means to achieving this by interfering with dopamine and levodopa metabolism without needing to increase the total levodopa daily dose. Levodopa is metabolised peripherally by catechol-O-methyl transferase (COMT). Drugs inhibiting COMT increase the elimination half-life of levodopa thus increasing its bioavailability and stabilising plasma levels [95]. This provides more sustained brain dopaminergic stimulation thus decreasing ‘off’ time, and in some instances decreasing the required daily levodopa dose.
Entacapone is a selective, reversible inhibitor of COMT and primarily acts to block levodopa metabolism in the periphery and improve levodopa absorption [95]. Repeated administration of 200 mg taken at the same time as each dose of levodopa/carbidopa increases plasma levodopa concentration by approximately 30% [94]. Three randomised trials evaluating entacapone as an adjunctive treatment to levodopa in patients with wearing-‘off’ motor fluctuations, demonstrated superiority in ‘off’ times (on average an increase in daily ‘ON’ time by 0.8 h) and UPDRS motor scores [96,97,98]. However, this approach can result in a higher rate of dyskinesia (STRIDE-PD study), which may necessitate adjustment to individual patient levodopa doses [34, 95]. Formulations of l-dopa combined with entacapone (Stalevo®, Stanek®, Sastravi®) are also available and reduce the pill burden for patients and thus may ease administration, although their clinical outcomes are thought to be largely similar to levodopa/entacapone taken separately.
Tolcapone is an alternative COMT inhibitor with greater potency by virtue of its combined central and peripheral action on COMT [99] that improves ‘on’ time by approximately 1.8 h over a 24-h period [100]. This agent was however linked to three cases of hepatotoxicity [101, 102] and therefore requires regular monitoring of liver function tests to allow its safe use. This need for regular blood tests has greatly reduced its usage in clinical practice. Another recently available alternative is opicapone [103] which is a selective, peripherally acting, once-daily COMT inhibitor. At a dose of 50 mg daily, levodopa bioavailability is significantly higher than that achieved by entacapone[ [104]. Phase III studies demonstrated superiority over placebo and non-inferiority when compared to entacapone [105, 106]. Scrutiny of ‘off’ time reductions however suggests a modest benefit in favour of opicapone over entacapone (116.8 min vs 96.3 min) [106]. The medication is generally well tolerated though dyskinesias, orthostatic hypotension and hallucinations can occur though some of this can potentially be overcome by concurrent lowering of levodopa doses [105, 106].
3.2.2 Monoamine Oxidase-B Inhibitors
Monoamine oxidase-B inhibitors (MAO-Bi) selectively and irreversibly block MAO-B, the main enzyme responsible for degrading dopamine in the synaptic cleft, and therefore increase dopamine concentrations in the brain. Their effects on dopaminergic stimulation is relatively modest which is largely explicable by the additional presence of presynaptic dopamine transporters which rapidly remove dopamine from the synaptic cleft. Selegiline (Deprenyl®) was the first available MAO-Bi, followed by rasagiline (Azilect®) and these remain the most commonly prescribed options. The PRESTO trial randomised 472 patients experiencing motor fluctuations to receive either rasagiline 0.5 mg or 1 mg/day with a reduction in the mean total daily ‘off’ or an approximate gain of 0.85 h/day in ‘on’ time without troublesome dyskinesias when added to l-dopa [107, 108]. The safety and efficacy of rasagiline was further explored in the LARGO trial and found to be comparable with entacapone [107, 109]. Evidence for the use of selegiline relies on lower quality studies demonstrating a modest reduction in daily ‘off’ time [110]. The drug is extensively metabolised in the liver with a resultant 10% bioavailability, variable pharmacokinetics and high metabolite concentrations. Selegiline orally disintegrating tablets aim to mitigate this by allowing direct absorption into the systemic circulation via the oral mucosa and therefore bypassing the gastrointestinal system and the first-pass metabolism. Selegiline metabolism can produce amphetamine like compounds which has led to concerns in patients with pre-existing cardiac conditions [111].
MAO-Bi can rarely induce hallucinations, confusion, and hypertensive crises. Furthermore, simultaneous administration with tricyclic antidepressants, selective serotonin and serotonin–norepinephrine reuptake inhibitors potentially increases the risk of serotonin syndrome although this does not tend to occur at the low therapeutic doses used (higher doses result in loss of selectivity and resultant inhibition of MAO-B and MAO-A, with a resultant increased risk of serotonin syndrome) [112]. The concomitant use of rasagiline/selegiline and selective serotonin reuptake inhibitors (SSRIs) is not recommended/contraindicated according to product monographs/labels. Nonetheless, to date, the only report of serotonin syndrome in this context is from the Parkinson Study Group survey suggesting an incidence of 0.24%, which is consistent with the authors own experience of safely using these drugs in combination [113].
Safinamide is a recently available highly selective, reversible MAO-B inhibitor with additional effects of blocking voltage-dependent sodium and calcium channels as well as reducing glutamate release and transmission [114]. This agent is given orally (50–100 mg/day) and provides increases in daily on time without troublesome dyskinesias to a magnitude similar to rasagiline [115,116,117]. A recent meta-analysis suggested that safinamide may be less effective than both rasagiline and selegiline though had comparable efficacy to entacapone [118]. Available follow-up studies suggest however sustained responses and tolerability to Safinamide over 2 years and the possibility of lower occurrence of dyskinesia, while headache and hypertension can feature in a small proportion of cases albeit less frequently with the 50 mg dose [115, 116].
3.2.3 Dopamine Agonists
Dopamine agonists (DA) mimic the action of dopamine by stimulating striatal post-synaptic receptors [119]. Dopamine receptors are widely distributed in the central nervous system and periphery. These receptors can be divided into the D1 (D1 and D5 receptors) and D2 (D2–D4) families. The nigro-striatal pathway is largely mediated by D1–D3 receptors [119]. Agonists are classified as either ergot derived (e.g. bromocriptine, pergolide, cabergoline, lisuride) or non-ergot derived (e.g. apomorphine, ropinirole, pramipexole, and rotigotine) and each agent has a slightly variable affinity for different receptors [120, 121]. DAs can be valuable as adjunctive therapy to levodopa for patients with motor complications by offering a further avenue to improve ‘off’ times without increasing levodopa [122]. Pramipexole and ropinirole are examples of this class of medication that have been demonstrated to significantly reduce ‘off’ time when compared with placebo. While initial interest revolved around IR formulations of these agents, the more novel extended-release (ER) formulations offer the benefit of reductions in ‘off’ time by promoting more stable plasma levels while reducing the number of medication intakes. Ropinirole ER is one such example and has been demonstrated to be more efficacious in maintaining a reduction in ‘off’ time of up to 20 % when compared with ropinirole IR formulations [123, 124]. Pramipexole ER has also been demonstrated to deliver up to an additional hour of ‘on’ time per day when compared with placebo though appears to be approximately equivalent to the IR formulation while tolerability of these agents are largely similar [125, 126]. The choice may therefore depend on individual tolerability and preference. Rotigotine, is a topical, non-ergot dopamine agonist with broad affinity for dopamine (D1–D5) receptors [127]. Rotigotine is non-inferior to the other agonist options though use of this agent is limited in some individuals by the potential to develop application-site reactions. Rotigotine also offers a 24-h transdermal delivery (2–16 mg/24 h) which results in the additional value of improving early-morning motor dysfunction and nocturnal sleep disturbances [128, 129]. Rotigotine may also be effective in addressing swallowing dysfunction and improving gastric emptying potentially via a reduced inhibition of the myenteric plexus and central dopaminergic augmentation of D1 receptors on gastro-intestinal motility though further studies are necessary before this medication can be recommended for this indication [130, 131].
Major concerns with DAs relate to their adverse effects. These typically include mild peripheral oedema, excessive daytime sleepiness, constipation, hallucinations, nausea, and postural hypotension. Neuropsychiatric issues, are of particular concern and include Impulse Control Disorders (e.g. hypersexuality, pathological gambling, and compulsive shopping), punding, and dopamine dysregulation syndrome [132]. Patients should be routinely warned about the possibility of ICD prior to prescription of all DAs and their presence should be enquired about from patient/carer at all subsequent follow up appointments. Although these concerns can effectively be managed with a reduction and subsequent withdrawal of the agent, the concern of a dopamine agonist withdrawal syndrome (DAWS) can emerge if doses are reduced or withdrawn too precipitously. This condition includes withdrawal symptoms such as anxiety, panic, agoraphobia, fatigue, dysphoria, nausea, vomiting, diaphoresis, as well as suicidal ideation [133]. These symptoms do not seem to be ameliorated by replacement with levodopa and the re-introduction of the agonist may sometimes be in the only solution [134]. A further serious adverse event is of fibrotic reactions (heart valves, pleuropulmonary, retroperitoneal) which seem to relate to ergot DA agents specifically [135]. This has led to the discontinuation of these agents in many countries. In general, DAs are best avoided in the most elderly patients as they tend to be more prone to these adverse effects. Specifically, there is some suggestion that the rotigotine patch and ropinirole ER may result in lower rates of ICDs when compared to patients taking levodopa together with other agonists [136, 137] regardless of patients’ ages [138] though this isolated report is unlikely to change overall clinician aversion towards the use of these agents in elderly patients.
3.2.4 Other Agents
Amantadine is a non-competitive NMDA receptor inhibitor which selectively blocks activated open receptor channels with both direct and indirect downstream effects on glutamatergic and dopaminergic signalling [139]. Although primarily used to treat dyskinesia, Amantadine also has the potential to reduce ‘off’ time [140], and gait freezing [141]. A daily dose up to 200–400 mg is typically used. Higher doses are more likely to induce side effects such as hallucinations, dry eye, dry mouth, constipation, and cognitive dysfunction. Much of the data currently available are from open label studies and well-designed randomized long-term trials addressing the efficacy and safety of this drug are still needed [142, 143].
Amantadine ER is a novel extended-release once-daily formulation that is currently available in selected countries. At the recommended dosage, plasma amantadine concentrations are 1.4- to 2.0-fold higher than IR formulations while peak plasma concentrations are reached more gradually while remaining sustained throughout the day (prolonged Tmax) [144]. This agent has a once-daily bed-time dosing scheme while the switch from IR to ER consists of administering 137 mg for 1 week and increasing to the target dose of 274 mg thereafter. This has been shown to induce a significant primary reduction in the Unified Dyskinesia Rating Scale score, and a secondary increase in ‘on’ time without troublesome dyskinesias when compared to placebo [145, 146]. Amantadine ER can achieve an 18% reduction in dyskinesias with a resultant 2.8 h of increase in ‘on’ time without troublesome dyskinesia though the side effect profile seems to be similar to the IR formulation [147].
Istradefylline is a selective adenosine A2A receptor antagonist utilised for adjunctive treatment of motor fluctuations [148]. It was developed in Japan and the majority of the experience with this drug is in Japanese patients. This agent modulates striatopallidal GABAergic output neurons and reduces daily ‘off’ time by approximately 0.7 h though trial data of its benefits have been inconsistent [149, 150]. Post marketing surveillance however suggests improvements in ‘off’ time in approximately 40% of people [151]. Commonly reported adverse effects include dyskinesia and hallucinations [151, 152]. Currently this agent is not widely available although there are plans to launch it in Europe and the USA.
Zonisamide (25–50 mg/day) increases dopamine levels by activation of tyrosine hydroxylase in the striatum as well as via some moderate inhibition of MAO-B. The further inhibition of T-type calcium channels and glutamate release potentially also exerts inhibition on the indirect pathway in the basal ganglia. A placebo-controlled randomised trial over 1 year in patients with wearing-off symptoms suggested a modest benefit in reducing ‘off’ time, without increasing dyskinesia [153]. Zonisamide is now approved for the treatment of PD in Japan.
3.2.5 Rapid-Acting Agents
Despite best efforts to optimize oral dopaminergic therapy, chronic PD patients can suffer up to 2–3 h of ‘off’ time per day on average [26]. Rapid-acting medications can potentially bridge symptom control by addressing unpredictable ‘off’ periods, and dose failures, while prolonging the latency of l-dopa effectiveness. It should be stressed however, that while options currently available are efficacious, their reliability can be variable (oral preparations) or limiting from a logistical perspective (sub-cutaneous apomorphine). Also, clinicians should be cautious regarding the use of these options on a regular basis considering the theoretical concerns of potent pulsatile dopaminergic stimulation. Patients using these agents on a regular basis should therefore be considered for the more reliable and continuous device assisted treatments available.
Dispersible benserazide-l-Dopa, (Madopar®) was developed to enable PD patients with difficulties swallowing pills to receive medication in a liquid form. The drug has similar efficacy, and dosage equivalence to conventional levodopa formulations without side effect differences. The agent has a significantly shorter Tmax, arguably owing to its accelerated absorption from a lack of disintegration by gastric fluid [154]. This rapid acting agent has a much faster and more constant onset of action than the standard preparation (25 vs 46 min) [155] and can therefore be considered as first line for the treatment of EMO and unpredictable ‘off’ symptoms. This proven efficacy is often less reliable in clinical practice because of its shorter duration of action, necessitating the development of other potent, rapidly acting agents.
Apomorphine, an aporphine alkaloid derived from acidification of morphine, is a potent DA with a broad spectrum of effects on both D1- and D2-like receptors [156]. This is in contrast to the oral DAs that mainly bind to D2 and D3 receptors. In addition, apomorphine also has antagonistic properties towards serotonergic and adrenergic receptors [157]. It has limited oral bioavailability therefore parenteral administration has been favoured. Apomorphine has a similar volume of distribution, plasma clearance and half-life following subcutaneous infusion therefore intravenous infusion can be avoided [158,159,160]. A number of factors influence its subcutaneous absorption. These include the injection site (abdominal injections have better results), volume and depth of injection (greater volume reduces the Tmax), skin state (temperature, vascularization, body fat) and the presence of nodules (a common feature following chronic apomorphine use) which hinder absorption [161]. After a subcutaneous injection, peak blood concentration is typically reached within 10 min, with a maximum cerebrospinal fluid concentration achieved after 30 min [160] with a brain-to-blood concentration ratio of approximately 8:1 [162]. Also, the rapid metabolism and clearance of the drug results in a T½ of around 33 min [160]. Although intra-individual variability in its pharmacodynamic effect is low, inter-individual variability is high resulting in a need for individual titration to identify the optimal dose during the initiation process. On balance, a clinical response tends to be appreciated within 10 mins of an injection and can last for close to 1 h [163].
Currently available formulations are a subcutaneous multi-dose pen or a subcutaneous pump (continuous subcutaneous apomorphine infusion-CSAI-discussed in the section on device assisted therapies). The subcutaneous apomorphine pen is arguably the most potent rapid treatment approach for ‘off’ symptoms. Injections can be administered following either an inpatient dose titration process or an outpatient approach. Efficacy of the apomorphine pen has been demonstrated in several randomised evaluations [164,165,166]. The injections have been demonstrated to be more reliable than dispersible l-dopa in reducing time to ‘on’ [167]. Improvements have been noted in the treatment of early-morning akinesia [168] as well as daytime ‘off’ periods. Furthermore, observational and randomised controlled studies seem to suggest consequent gains in ‘on’ time of up to 2 h per day [164,165,166, 169,170,171,172,173]. While improvements in urinary dysfunction and pain [174] have been noted, convincing evidence for improvements in NMF are lacking. The apomorphine pen is not infrequently accompanied by side effects such as nausea, yawning, somnolence, dizziness, orthostatic hypotension and dyskinesia. While this can be an effective long term therapy in some people, up to one-third of patients discontinue apomorphine within a year of initiation [164,165,166, 172].
In addition to drug induced side effects, some patients struggle with apomorphine due to needle phobia, or difficulty administering a pen injection during an acute ‘off’ phase. Alternative less invasive delivery systems are in active development. An apomorphine powder formulation via an inhaler device (VR040) has been subjected to randomised trials. This device is well-tolerated, with reasonable efficacy in the management of MF [175]. Further studies have suggested rapid absorption (2–7 min) and reversal of the ‘off’ state (10 min) without significant pulmonary safety concerns [176, 177]. Sublingual apomorphine is a further option with comparable symptomatic effects to its subcutaneous counterpart. A novel bilayer film of sublingual apomorphine (APL-130277) helps reduce the time taken for its sublingual dissolution and absorption and it has been demonstrated in Phase 3 studies to allow full achievement of on states within 15–30 min of administration [25, 178]. Side effects noted are broadly similar to subcutaneous apomorphine although up to a third of patients may develop a combination of lip or oropharyngeal swelling and erythema. A Phase 3 crossover trial evaluating APL-130277 against subcutaneous apomorphine in patients with MF is currently underway (Clinical-Trials.gov identifier: NCT03391882) and will be crucial in informing its use in clinical practice.
A levodopa inhaled powder known as CVT-301 is a novel dry powder formulation in capsules which are inserted into and administered via a breath-actuated device [179]. This agent is rapidly absorbed through the pulmonary epithelium, avoiding first-pass metabolism and therefore achieving peak plasma concentrations within 15 min of inhalation [180]. Clinical effects are noted within 5–15 min with benefits lasting up to the fixed 90 min evaluation mark of the study [181]. This agent has been further demonstrated to achieve plasma levodopa concentrations more rapidly than oral levodopa while maintaining more stable concentrations over 60 min. On average, patients using two inhalations per day appeared to gain about 0.8 h/day in ‘ON’ time when compared to placebo [179,180,181]. The most common side effect is the predictable occurrence of a cough which does not seem to abate on lower doses while other respiratory side effects (upper respiratory tract infections, discoloured sputum, and throat irritation) occur less frequently and without any detrimental impact on lung function [182]. Hypotension and atrial fibrillation are potentially serious adverse events that have been less frequently described. Moving forwards, head-to-head comparisons between subcutaneous apomorphine, sublingual and inhaled formulations of apomorphine and l-dopa would be of value prior to determining the relative usefulness of these agents in the hierarchy of managing ‘off’ episodes, however it is unlikely that the commercial manufacturers would risk supporting randomised comparisons of any of these agents other than against placebo.
4 Device-Assisted Therapies
Despite best efforts with a mixture of approaches described thus far, a proportion of patients will continue to experience debilitating ‘off’ phenomena for significant proportions of their day. Device-assisted treatments (often termed advanced therapies) are crucial in managing these cases considering the potency of their effects, albeit with their greater invasive nature. While their modes of action vary, these treatments collectively capitalise on their superiority in delivering continuous dopaminergic (or electrical) stimulation. The exact timing for introducing these treatments into care remains controversial though expert consensus can be useful in this regard. In a Delphi-panel consensus [183], recommendations for device assisted therapies included for; patients with troublesome motor fluctuations (≥ 1 h of troublesome dyskinesia/day, ≥ 2 h “off” symptoms/day) on ≥ 5 oral levodopa doses/day with functional impairment and difficulty with activities of daily living [183]. Patients with good levodopa response, good cognition, and age < 70 years were deemed good candidates for the three main device-assisted therapies though other specific patient related considerations should also be factored into decision making processes when considering the most appropriate individualised option [183].
4.1 Infusions
Infusion therapies inevitably achieve more stable plasma drug concentrations in comparison to their more traditional oral counterparts. The Levodopa–carbidopa intestinal gel (LCIG) infusion is one such approach that provides a continuous jejunal infusion of levodopa/carbidopa [184]. LCIG is delivered by an external pump which administers small doses of levodopa/carbidopa approximately once every minute to the small intestine [185]. This bypasses the unpredictable nature of gastric emptying and therefore overcomes irregular absorption [186]. LCIG provides stable plasma concentrations throughout the day which is likely to be the explanation for its superiority to orally administered levodopa [185, 186]. For the majority of patients, the external pump is used to deliver the agent continuously over 16 h during the waking day via a percutaneous gastrojejunostomy tube (PEG-J). In a small proportion of cases where uncertainty of the degree of l-dopa responsiveness exists, a prior trial of the agent with a naso-jejunal phase may be necessary [187]. Some patients may benefit from 24 h administration of the infusion to improve nocturnal symptoms [188] and it has even been proposed to improve freezing of gait [189] although this phenomenon often eventually becomes refractory to l-dopa therapy of any form. While a small proportion continue on oral agents, monotherapy is achievable in the vast majority, perhaps supplemented by CR oral l-dopa to cover the night-time period.
In a randomised, double blind trial, LCIG was found to increase ‘on’ time without troublesome dyskinesia by approximately 2 h per day with significant quality of life gains [190]. Although some non-motor symptom improvements have been demonstrated [191], the response of non-motor symptoms to LCIG are less predictable and specific studies demonstrating efficacy for NMF are lacking, therefore this treatment should only be routinely used in patients with significant MF. Despite the substantial advantages in MF seen with this treatment, the use of LCIG may be limited by device- and procedure-related complications such as blocked tubes, tube dislocations, stoma complications, superficial infections, peritonitis, and pneumoperitoneum [190, 192]. These issues can result in a proportion of patients discontinuing treatment, though systemic and procedural modifications have been demonstrated to minimise these occurrences [193,194,195]. Polyneuropathy has been noted in a small proportion of patients using LCIG, therefore prior surveillance for vitamin B12 deficiency (including measurement of homocysteine and methyl malonic acid) and parenteral B12 replacement may be useful in this regard [196, 197]. In view of the substantial undertakings and costs involved, some current guidelines suggest that the LCIG should only be utilised in cases of advanced PD where patients have either failed or do not qualify for alternative device-assisted modalities [198].
Continuous subcutaneous apomorphine infusion (CSAI) is a proven alternative to LCIG that is less invasive and considerably less expensive and thus should be discussed as an alternative treatment option with all patients considering LCIG. This infusion is administered via a portable pump system delivering a continuous dose, with the possibility of intermittent rescue boluses as needed. Infusions typically last 12–16 h (waking time), but a 24-h regimen can also be programmed to treat nocturnal hypokinesia [199, 200].
Suitable patients include those who have ‘off’ periods no longer controlled with optimised oral therapy, those who need apomorphine pen rescue doses too frequently or those who require an alternative to surgical or enteral therapy [90]. Patients starting CSAI should be administered domperidone 10 mg (or trimethobenzamide in countries where domperidone is not available) three times daily from 1 day before initiation to 3–7 days in total to prevent nausea. Prior to initiation of domperidone, an electrocardiogram demonstrating a normal QT interval should be performed (in view of an increased risk of ventricular tachyarrhythmia and sudden cardiac death with domperidone) particularly in patients with PD and pre-existing cardiac disease [201]. Apomorphine infusions are typically started at a dose of 0.5 or 1 mg/h while anecdotally slow up-titrations (0.5 or 1 mg/h daily increments) targeting an infusion rate ranging from 4 to 7 mg/h tend to achieve better outcomes. A concomitant reduction of oral antiparkinsonian drugs is usual. Titration can also be achieved in the outpatient setting (after the initial exclusion of potential adverse reactions) with an inpatient test dose, although a slower increase process tends to be more typical [200].
It is crucial to note that apomorphine is the only agent shown to have similar efficacy to levodopa when treating PD motor symptoms, although as would be expected, patients experience a shorter duration of effect [167, 202]. Multiple open-label series have confirmed the efficacy of CSAI in reversing severe, sudden ‘off’ states in advanced PD despite optimised oral therapy [169, 172, 203]. A recent randomised, double-blind trial of a 16-h daily CSAI infusion has further demonstrated similar improvements in ‘on’ time to LCIG (i.e. approximately 2-h reduction in ‘off’ time) while patients were also able to reduce their daily levodopa equivalent medication by more than 300 mg [204]. Some emerging evidence also suggests efficacy in the management of non-motor symptoms, in particular sleep, mood, gastrointestinal, perceptual problems and urinary domains [174, 205], although approaches using CSAI purely to treat NMF cannot be recommended currently without further conclusive evidence of benefit in this regard.
Skin nodules, nausea, and somnolence can commonly occur, although less common adverse effects such as severe hypotension, hallucinations, confusion, and infusion-site cellulitis can potentially be more concerning. Haemolytic anaemia and leucopenia have rarely been described [206,207,208,209]. The long-term tolerability of CSAI is variable with up to two-thirds of patients ceasing therapy after an average of 17.9 months due to complications such as hallucinations, and less common reports of impulse control disorder, and dopamine dysregulation [210,211,212]. An argument has been mounted by some suggesting an either neutral or remission effect of CSAI on psychotic symptoms and hallucinations due to the drugs shared piperidine moiety structure with antipsychotic agents, although this is not universally seen and the available evidence is insufficient to prompt clinicians to adopt this approach [210, 213, 214]. Although ICDs are a major concern with dopamine agonists, the lower D3:D2 ratio of apomorphine compared to oral agonists along with the lower incidence of ICDs noted in a number of studies suggests that patients should not be precluded from being considered for this treatment, even if they have experienced ICD symptoms previously [211, 215, 216], although more cautious titration approaches should be employed.
Difficulties related to the injection system (i.e. the technical aspects of the pump), the complexity of the initial titration schedule, the difficulty of tailing off levodopa and other oral dopamine agonists, the shorter duration of benefit in some people, and the risk of skin nodules and ulceration may account for its limited acceptance and explain why it is not used more widely. However, some of these cutaneous risks can be reduced through improved skin hygiene, changing the site of injection, using newer styles of needles, and using localised massage and ultrasound therapy [208].
A subcutaneous carbidopa-levodopa infusion (ND0612) through a small pump-patch is currently under investigation with preliminary pharmacokinetic studies suggesting stable plasma levodopa concentrations are achievable over 24 h. A number of Phase 2 studies have established that this agent provides more stable plasma levels of levodopa compared to oral medications, while one study suggests that ND0612 may result in higher total plasma levodopa, i.e. higher bioavailability [217, 218] and less inter- and intra-subject plasma level variability compared to LCIG [219,220,221]. Similar to LCIG and CSAI, these same studies also suggest that up to 2 h of reduction in ‘off’ time compared to optimal oral therapy is achievable with ND0612. A further finding showed an up to 80% reduction in oral l-dopa intake while only a small proportion of subjects were able to achieve monotherapy with the infusion. Significant improvements in sleep quality and early-morning motor symptoms in patients running infusions for 24 h is encouraging, although similar cutaneous concerns seem to occur in patients as with apomorphine [222]. A Phase 3 multicentre, randomised, double-blind placebo-controlled trial of ND0612 (INDIGO) is currently ongoing (ClinicalTrials.gov ID: NCT2726386) [223].
4.2 Neurosurgical Approaches
Deep brain stimulation (DBS) is a proven treatment for reduction in the severity and duration of MF in PD. It requires the surgical implantation of intracranial electrodes and a pacemaker device, which provides continuous stimulation to deep structures of the brain [224]. The exact mechanism of action in PD treatment is unclear, although modulation of pathological neuronal firing patterns within the cortico-basal ganglia networks is potentially implicated in the motor symptom improvements noted [225, 226]. The merits of this approach in addressing l-dopa-associated motor fluctuations is considerable. Compared to best medical therapy, DBS improves ‘on’ time without troublesome dyskinesia by approximately 4–5 h, with up to two-thirds of patients achieving meaningful improvements in motor fluctuations by 6 months [227, 228]. These positive effects appear to last longer than 10 years in several long-term studies, although the net effects on quality of life in comparison to pre-operative status seem to wane after 5 years, presumably because of the inexorable progression of the disease [229, 230]. There has been recent interest in this treatment being considered earlier in the course of disease [231] and for DBS to be utilised as the first choice of device-assisted therapies when this is clinically appropriate. The optimal timing for DBS requires individual discussions with a considered balanced between the potential risks and benefits, as well as consideration of a patient’s expectations and lifestyle.
The subthalamic nucleus (STN) and globus pallidus interna (GPi) are the most commonly targeted structures for DBS in PD patients, although the preferences are largely dependent on the clinical scenario being addressed [232, 233]. Although STN DBS improves the severity of the motor symptoms of PD and has the potential to reduce dopaminergic medication requirements, it is also associated with an increased risk of cognitive and psychiatric complications [234,235,236]. Conversely, GPi stimulation is less likely to negatively impact mood and cognitive processing while resulting in greater reductions in dyskinesia [227,228,229]. However, GPi DBS rarely allows for a major reduction in dopaminergic therapies [227,228,229]. Potential complications relate to the surgical procedure and hardware such as wound infections or erosions, lead migration/malposition, lead or extension fractures and component malfunctions [237, 238]. Other rare risks include the occurrence of post-implantation seizures, oedema and symptomatic intracerebral haemorrhage [239].
While reduction in medication requirement is usual after STN DBS, the majority of patients do not stop all their dopaminergic replacement and ‘on’ and ‘off’ motor states and decline in motor function can begin to recur with time, likely due to disease progression and development of stimulation and l-dopa-resistant symptoms [240]. The therapeutic effects noted can also be limited by inaccurate placement of electrodes as can side effects such as speech disturbance, gait impairment, paraesthesia, and diplopia [241]. While some of these aspects can be mitigated by adjustments to stimulation [242], the additional utilisation of longer-acting formulations or continuous infusions can at times be of value. An open-label study comparing DBS to LCIG and CSAI suggested superior control of dyskinesia and less procedure- or device-related complications [243] with DBS, although the lack of randomisation makes interpretation of this study less generalisable. Novel mechanisms to improve stimulation delivery are currently being developed and could potentially make the clinical benefits noted more durable into the future [244, 245].
5 Specific NMF Considerations
The management of non-motor symptoms is crucial in improving patients’ quality of life. While each symptom provides unique management challenges, it is beyond the scope of this review to explore these finer aspects, although a recent Movement Disorder Society sanctioned evidence-based critique is a worthwhile source in this regard [246]. These guidelines do not address the treatment of NMF, which continues to lack a specific evidence base. Fluctuations in NMF severity in accordance with dopamine replacement therapy is highly suggestive that the NMF have a dopaminergic origin. Although current approaches towards NMF largely rely on principles established for motor fluctuations, specific pathophysiological considerations imply that some additional strategies are potentially warranted. Potential overarching principles could encompass targeting respective NMS with the combination of continuous dopaminergic stimulation and supplementary treatments targeting non-dopaminergic mechanisms when necessary [247, 248]. While non-dopaminergic symptom-oriented approaches should be trialled (e.g. antidepressant drugs for depressive mood fluctuations), many NMFs do not particularly respond to such treatment strategies. A secondary imperative is to establish if the different dopaminergic thresholds for improvement (i.e. the required level of medication adaptation) are similar for both MF and NMF [249]. This can be achieved by implementing the principles outlined thus far for management of MF but with clear attention to the fluctuation of NMS. Examples of this include medication reductions with the aim of improving neuropsychiatric symptoms and orthostatic hypotension or the utility of add-on therapy to improve sleep. Specific examples of useful add-on therapy include the potential benefit of safinamide [117] and tolcapone [250,251,252] in improving depression and sleep as well as dopamine agonists in addressing depression, sleep, apathy and pain [129, 166]. It is important to consider the potential risk of dopa dysregulation in patients with NMF, in whom escalating levels of dopaminergic medication can be seen despite little objective evidence of any motor disability.
Finally, although the evidence base for device-assisted therapies improving NMS is growing [205, 228, 253], specific improvements in NMF are largely limited to a small cohort study of STN-DBS suggesting a reduction in NMF of approximately 60% with greatest promise seen with pain fluctuations [254]. The dearth of specific evidence available for the management of NMF is a result of a poor understanding of pathophysiology, and a lack of quality detection tools [255]. Controlled trials targeting NMF as primary endpoints similar to those currently available motor fluctuations should be encouraged moving forward considering their known impact on patients’ quality of life.
6 Conclusion
Motor fluctuations develop almost universally in PD patients with the progression of disease. While levodopa has not been conclusively determined to be causative of this phenomenon, patients generally only manifest with these episodes in its presence. Modifications of formulations and the mode of administration of this medication, in conjunction with adjunctive options, can initially be of value although many patients will ultimately require device-assisted therapies to manage these symptoms, provided they are otherwise fit enough to withstand their invasive nature/side effects. Future studies should focus on improving our understanding of the disease biology contributing to these occurrences as preventative approaches will be crucial moving forward.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
Mayeux R, et al. The frequency of idiopathic Parkinson’s disease by age, ethnic group, and sex in northern Manhattan, 1988–1993. Am J Epidemiol. 1996;143(5):528 (Am. J. Epidemiol: 1995).
Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of parkinson’s disease. Arch Neurol. 1993;50(2):140–8.
Poewe W. Non-motor symptoms in Parkinson’s disease. Eur J Neurol. 2008. https://doi.org/10.1111/j.1468-1331.2008.02056.x.
Poewe W, et al. Parkinson disease. Nat Rev Dis Prim. 2017. https://doi.org/10.1038/nrdp.2017.13.
Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism—chronic treatment with L-dopa. N Engl J Med. 1969. https://doi.org/10.1056/NEJM196902132800701.
Cotzias GC, Van Woert MH, Schiffer LM. “Aromatic amino acids and modification of parkinsonism. N Engl J Med. 1967. https://doi.org/10.1056/NEJM196702162760703.
Fahn S. The history of dopamine and levodopa in the treatment of Parkinson’s disease. Mov Disord. 2008. https://doi.org/10.1002/mds.22028.
Obeso JA, Rodriguez-Oroz MC, Chana P, Lera G, Rodriguez M, Olanow CW. The evolution and origin of motor complications in Parkinson’s disease. Neurology. 2000;55(11 Suppl 4):S13–20.
Jankovic J. Motor fluctuations and dyskinesias in Parkinson’s disease: clinical manifestations. Mov Disord. 2005. https://doi.org/10.1002/mds.20458.
Martinez-Martin P, et al. Prevalence of nonmotor symptoms in Parkinson’s disease in an international setting; study using nonmotor symptoms questionnaire in 545 patients. Mov Disord. 2007. https://doi.org/10.1002/mds.21586.
Shulman LM, Taback RL, Bean J, Weiner WJ. Comorbity of the nonmotor symptoms of Parkinson’s disease. Mov Disord. 2001. https://doi.org/10.1002/mds.1099.
Marsden CD, Parkes JD. Success and problems of long-term levodopa therapy in Parkinson’s disease. Lancet. 1977. https://doi.org/10.1016/S0140-6736(77)91146-1.
Merello M, Lees AJ. Beginning-of-dose motor deterioration following the acute administration of levodopa and apomorphine in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1992. https://doi.org/10.1136/jnnp.55.11.1024.
Melamed E, Bitton V, Zelig O. Delayed onset of responses to single doses of L-Dopa in Parkinsonian fluctuators on long-term L-Dopa therapy. Clin Neuropharmacol. 1986. https://doi.org/10.1097/00002826-198604000-00009.
Witjas T, et al. Nonmotor fluctuations in Parkinson’s disease: frequent and disabling. Neurology. 2002. https://doi.org/10.1212/WNL.59.3.408.
Stacy M. The wearing-off phenomenon and the use of questionnaires to facilitate its recognition in Parkinson’s disease. J Neural Transm. 2010. https://doi.org/10.1007/s00702-010-0424-5.
Chaudhuri KR, et al. The nondeclaration of nonmotor symptoms of Parkinson’s disease to health care professionals: an international study using the nonmotor symptoms questionnaire. Mov Disord. 2010. https://doi.org/10.1002/mds.22868.
Martínez-Fernández R, Schmitt E, Martinez-Martin P, Krack P. The hidden sister of motor fluctuations in Parkinson’s disease: a review on nonmotor fluctuations. Mov Disord. 2016. https://doi.org/10.1002/mds.26731.
Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001. https://doi.org/10.1002/mds.1090.
Katzenschlager R, Head J, Schrag A, Ben-Shlomo Y, Evans A, Lees AJ. Fourteen-year final report of the randomized PDRG-UK trial comparing three initial treatments in PD. Neurology. 2008. https://doi.org/10.1212/01.wnl.0000310812.43352.66.
Hauser RA, et al. Ten-year follow-up of Parkinson’s disease patients randomized to initial therapy with ropinirole or levodopa. Mov Disord. 2007. https://doi.org/10.1002/mds.21743.
Hely MA, Reid WGJ, Adena MA, Halliday GM, Morris JGL. The Sydney Multicenter Study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord. 2008. https://doi.org/10.1002/mds.21956.
Rizos A, et al. Characterizing motor and non-motor aspects of early-morning off periods in Parkinson’s disease: an international multicenter study. Parkinsonism Relat Disord. 2014. https://doi.org/10.1016/j.parkreldis.2014.09.013.
Merims D, Djaldetti R, Melamed E. Waiting for ON: a major problem in patients with Parkinson disease and ON/OFF motor fluctuations. Clin Neuropharmacol. 2003. https://doi.org/10.1097/00002826-200307000-00009.
Hauser RA, et al. Sublingual apomorphine (APL-130277) for the acute conversion of OFF to ON in Parkinson’s disease. Mov Disord. 2016. https://doi.org/10.1002/mds.26697.
Van Lunen B, Anthony J, Spears J. Implications of motor fluctuations in Parkinson’s disease patients on chronic therapy (IMPACT): results from an observational registry. Mov Disord. 2005;20:S146.
Storch A, et al. Nonmotor fluctuations in Parkinson disease: severity and correlation with motor complications. Neurology. 2013. https://doi.org/10.1212/WNL.0b013e318285c0ed.
Brown RG, et al. Depression and anxiety related subtypes in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2011. https://doi.org/10.1136/jnnp.2010.213652.
Marras C, Chaudhuri KR. Nonmotor features of Parkinson’s disease subtypes. Mov Disord. 2016. https://doi.org/10.1002/mds.26510.
Chaudhuri KR, Poewe W, Brooks D. Motor and nonmotor complications of levodopa: phenomenology, risk factors, and imaging features. Mov Disord. 2018. https://doi.org/10.1002/mds.27386.
Hillen ME, Sage JI. Nonmotor fluctuations in patients with Parkinson’s disease. Neurology. 1996. https://doi.org/10.1212/WNL.47.5.1180.
Picillo M, et al. Gender and non motor fluctuations in Parkinson’s disease: a prospective study. Parkinsonism Relat Disord. 2016. https://doi.org/10.1016/j.parkreldis.2016.04.001.
Fahn S, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004. https://doi.org/10.1056/NEJMoa033447.
Stocchi F, Rascol O, Kieburtz K, Poewe W, Jankovic J, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol. 2010;68(1):18–27.
Olanow CW, et al. Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson’s disease. Mov Disord. 2013. https://doi.org/10.1002/mds.25364.
Schrag A, Quinn N. Dyskinesias and motor fluctuations in Parkinson’s disease: a community-based study. Brain. 2000. https://doi.org/10.1093/brain/123.11.2297.
Foltynie T, et al. BDNF val66met influences time to onset of levodopa induced dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2009. https://doi.org/10.1136/jnnp.2008.154294.
De Lau LML, Verbaan D, Marinus J, Heutink P, Van Hilten JJ. Catechol-O-methyltransferase Val158Met and the risk of dyskinesias in Parkinson’s disease. Mov Disord. 2012. https://doi.org/10.1002/mds.23805.
Lee JY, Cho J, Lee EK, Park SS, Jeon BS. Differential genetic susceptibility in diphasic and peak-dose dyskinesias in Parkinson’s disease. Mov Disord. 2011. https://doi.org/10.1002/mds.23400.
Schumacher-Schuh AF, et al. Association of common genetic variants of HOMER1 gene with levodopa adverse effects in Parkinson’s disease patients. Pharmacogenom J. 2014. https://doi.org/10.1038/tpj.2013.37.
Politis M, Wu K, Molloy S, Bain PG, Chaudhuri KR, Piccini P. Parkinson’s disease symptoms: the patient’s perspective. Mov Disord. 2010. https://doi.org/10.1002/mds.23135.
Chapuis S, Ouchchane L, Metz O, Gerbaud L, Durif F. Impact of the motor complications of Parkinson’s disease on the quality of life. Mov Disord. 2005. https://doi.org/10.1002/mds.20279.
Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2010. https://doi.org/10.1136/jnnp.2009.173286.
Storch A, et al. Quantitative assessment of non-motor fluctuations in Parkinson’s disease using the Non-Motor Symptoms Scale (NMSS). J Neural Transm. 2015. https://doi.org/10.1007/s00702-015-1437-x.
Nutt JG, Woodward WR, Carter JH, Gancher ST. Effect of long-term therapy on the pharmacodynamics of levodopa: relation to on-off phenomenon. Arch Neurol. 1992. https://doi.org/10.1001/archneur.1992.00530350037016.
Nutt JG, Holford NHG. The response to levodopa in Parkinson’s disease: imposing pharmacological law and order. Ann Neurol. 1996. https://doi.org/10.1002/ana.410390504.
Nutt JG, Carter JH, Lea ES, Sexton GJ. Evolution of the response to levodopa during the first 4 years of therapy. Ann Neurol. 2002. https://doi.org/10.1002/ana.10189.
Zappia M, et al. Loss of long-duration response to levodopa over time in PD: implications for wearing-off. Neurology. 1999. https://doi.org/10.1212/wnl.52.4.763.
Stocchi F. The hypothesis of the genesis of motor complications and continuous dopaminergic stimulation in the treatment of Parkinson’s disease. Parkinsonism Relat Disord. 2009. https://doi.org/10.1016/S1353-8020(09)70005-7.
Nutt JG, Woodward WR, Hammerstad JP, Carter JH, Anderson JL. The On–Off phenomenon in Parkinson’s disease: relation to levodopa absorption and transport. N Engl J Med. 1984. https://doi.org/10.1056/NEJM198402233100802.
Edwards LL, Quigley EMM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease: frequency and pathophysiology. Neurology. 1992. https://doi.org/10.1212/wnl.42.4.726.
Baruzzi A, et al. Infuence of meal ingestion time on pharmacokinetics of orally administered levodopa in parkinsonian patients. Clin Neuropharmacol. 1987. https://doi.org/10.1097/00002826-198712000-00004.
Leon AS, Spiegel HE. The effect of antacid administration on the absorption and metabolism of levodopa. J Clin Pharmacol New Drugs. 1972. https://doi.org/10.1002/j.1552-4604.1972.tb00053.x.
Valenzuela JE. Dopamine as a possible neurotransmitter in gastric relaxation. Gastroenterology. 1976. https://doi.org/10.1016/S0016-5085(76)80051-0.
Frackowiak RS, Leenders KL, Poewe WH, Palmer AJ, Brenton DP. Inhibition of L-18Ffluorodopa uptake into human brain by amino acids demonstrated by positron emission tomography. Ann Neurol. 1986;20(2):258–62.
Çamcı G, Oğuz S. Association between Parkinson’s disease and Helicobacter pylori. J Clin Neurol (Korea). 2016. https://doi.org/10.3988/jcn.2016.12.2.147.
van Kessel SP, El Aidy S. Contributions of gut bacteria and diet to drug pharmacokinetics in the treatment of Parkinson’s disease. Front Neurol. 2019. https://doi.org/10.3389/fneur.2019.01087.
Fabbnni G, Mouradian MM, Junecos JL, Schlegel J, Mohr E, Chase TN. Motor fluctuations in Parkinson’s disease: central pathophysiological mechanisms, Part I. Ann Neurol. 1988. https://doi.org/10.1002/ana.410240303.
Mouradian MM, Junecos JL, Fabbrini G, Schlegel J, Bartko JJ, Chase TN. Motor fluctuations in Parkinson’s disease: central pathophysiological mechanisms, part II. Ann Neurol. 1988. https://doi.org/10.1002/ana.410240304.
Bravi D, Mouradian MM, Roberts JW, Davis TL, Sohn YH, Chase TN. Wearing-off fluctuations in Parkinson’s disease: contribution of postsynaptic mechanisms. Ann Neurol. 1994. https://doi.org/10.1002/ana.410360108.
Barbato L, et al. The long-duration action of levodopa may be due to a postsynaptic effect. Clin Neuropharmacol. 1997. https://doi.org/10.1097/00002826-199710000-00003.
Stocchi F, Vacca L, Berardelli A, De Pandis F, Ruggieri S. Long-duration effect and the postsynaptic compartment: study using a dopamine agonist a short half-life. Mov Disord. 2001. https://doi.org/10.1002/mds.1070.
Bergstrom BP, Garris PA. ‘Passive stabilization’ of striatal extracellular dopamine across the lesion spectrum encompassing the presymptomatic phase of Parkinson’s disease: a voltammetric study in the 6-OHDA-lesioned rat. J Neurochem. 2003. https://doi.org/10.1046/j.1471-4159.2003.02104.x.
Abercrombie ED, Bonatz AE, Zigmond MJ. Effects of l-DOPA on extracellular dopamine in striatum of normal and 6-hydroxydopamine-treated rats. Brain Res. 1990. https://doi.org/10.1016/0006-8993(90)91318-B.
Chase TN, Baronti F, Fabbrini G, Heuser IJ, Juncos JL, Mouradian MM. Rationale for continuous dopaminomimetic therapy of Parkinson’s disease. Neurology. 1989;39(11 Suppl 2):7–10.
De La Fuente-Fernndez R, et al. Biochemical variations in the synaptic level of dopamine precede motor fluctuations in Parkinson’s disease: PET evidence of increased dopamine turnover. Ann Neurol. 2001. https://doi.org/10.1002/ana.65.
Obeso JA, et al. The origin of motor fluctuations in Parkinson’s disease: importance of dopaminergic innervation and basal ganglia circuits. Neurology. 2004. https://doi.org/10.1212/wnl.62.1_suppl_1.s17.
Chaudhuri KR, Rizos A, Sethi KD. Motor and nonmotor complications in Parkinson’s disease: an argument for continuous drug delivery? J Neural Transm. 2013. https://doi.org/10.1007/s00702-013-0981-5.
Brown P, Marsden CD. What do the basal ganglia do? Lancet. 1998. https://doi.org/10.1016/S0140-6736(97)11225-9.
Remy P, Doder M, Lees A, Turjanski N, Brooks D. Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain. 2005. https://doi.org/10.1093/brain/awh445.
Halliday GM, Leverenz JB, Schneider JS, Adler CH. The neurobiological basis of cognitive impairment in Parkinson’s disease. Mov Disord. 2014. https://doi.org/10.1002/mds.25857.
Lim SY, Fox SH, Lang AE. Overview of the extranigral aspects of Parkinson disease. Arch Neurol. 2009. https://doi.org/10.1001/archneurol.2008.561.
Verschuur CVM, et al. Randomized delayed-start trial of levodopa in Parkinson’s disease. N Engl J Med. 2019. https://doi.org/10.1056/NEJMoa1809983.
Hsu A, Yao HM, Gupta S, Modi NB. Comparison of the pharmacokinetics of an oral extended-release capsule formulation of carbidopa-levodopa (IPX066) with immediate-release carbidopa-levodopa (Sinemet®), sustained-release carbidopa-levodopa (Sinemet® CR), and carbidopa–levodopa–entacapone (Stalevo®). J Clin Pharmacol. 2015. https://doi.org/10.1002/jcph.514.
Goetze O, Wieczorek J, Mueller T, Przuntek H, Schmidt WE, Woitalla D. Impaired gastric emptying of a solid test meal in patients with Parkinson’s disease using 13C-sodium octanoate breath test. Neurosci Lett. 2005. https://doi.org/10.1016/j.neulet.2004.11.007.
Destée A, Rérat K, Bourdeix I. Is there a difference between levodopa/dopa-decarboxylase inhibitor and entacapone and levodopa/dopa-decarboxylase inhibitor dose fractionation strategies in Parkinson’s disease patients experiencing symptom re-emergence due to wearing-off? The honeymoon study. Eur Neurol. 2009. https://doi.org/10.1159/000177938.
Carter JH, Nutt JG, Woodward WR, Hatcher LF, Trotman TL. Amount and distribution of dietary protein affects clinical response to levodopa in Parkinson’s disease. Neurology. 1989. https://doi.org/10.1212/wnl.39.4.552.
Cereda E, Barichella M, Pedrolli C, Pezzoli G. Low-protein and protein-redistribution diets for Parkinson’s disease patients with motor fluctuations: a systematic review. Mov Disord. 2010. https://doi.org/10.1002/mds.23226.
Burn DJ, et al. Decreased ‘OFF’ time and improved Parkinson’s disease symptoms with the gastroprokinetic camicinal as an adjunct to L-DOPA based treatment; a pilot study. Mov Disord. 2015. https://doi.org/10.1002/mds.26295.
Cotzias GC, Papavasiliou PS, Gellene R. Experimental treatment of parkinsonism with L-Dopa. Neurology. 1968;18(3):276–7.
LeWitt PA, et al. Pharmacokinetic–pharmacodynamic crossover comparison of two levodopa extension strategies. Mov Disord. 2009. https://doi.org/10.1002/mds.22587.
Dupont E, et al. Sustained-release Madopar HBS® compared with standard Madopar® in the long-term treatment of de novo parkinsonian patients. Acta Neurol Scand. 2009. https://doi.org/10.1111/j.1600-0404.1996.tb00163.x.
Block G, Liss C, Reines S, Irr J, Nibbelink D. Comparison of immediate-release and controlled release carbidopa/levodopa in Parkinson’s disease. A multicenter 5-year study. The CR First Study Group. Eur Neurol. 1997;37(1):23–7.
Mittur A, Gupta S, Modi NB. Pharmacokinetics of Rytary®, an extended-release capsule formulation of carbidopa–levodopa. Clin Pharmacokinet. 2017. https://doi.org/10.1007/s40262-017-0511-y.
Yao HM, Hsu A, Gupta S, Modi NB. Clinical pharmacokinetics of IPX066: evaluation of dose proportionality and effect of food in healthy volunteers. Clin Neuropharmacol. 2016. https://doi.org/10.1097/WNF.0000000000000126.
Morgan JC, Dhall R, Rubens R, Khanna S, Gupta S. Dosing patterns during conversion to IPX066, extended-release carbidopa–levodopa (ER CD-LD), in Parkinson’s disease with motor fluctuations. Parkinsons Dis. 2018. https://doi.org/10.1155/2018/9763057.
Hauser RA, et al. Extended-release carbidopa–levodopa (IPX066) compared with immediate-release carbidopa-levodopa in patients with Parkinson’s disease and motor fluctuations: a phase 3 randomised, double-blind trial. Lancet Neurol. 2013. https://doi.org/10.1016/S1474-4422(13)70025-5.
Pahwa R, et al. Randomized trial of IPX066, carbidopa/levodopa extended release, inearly Parkinson’s disease. Parkinsonism Relat Disord. 2014. https://doi.org/10.1016/j.parkreldis.2013.08.017.
Metman LV, Stover N, Chen C, Cowles VE, Sweeney M. Gastroretentive carbidopa/levodopa, DM-1992, for the treatment of advanced Parkinson’s disease. Mov Disord. 2015. https://doi.org/10.1002/mds.26219.
LeWitt PA. New levodopa therapeutic strategies. Parkinsonism Relat Disord. 2016. https://doi.org/10.1016/j.parkreldis.2015.09.021.
LeWitt PA, et al. Accordion pill carbidopa/levodopa (AP-CD/LD) for treatment of advanced PD. Mov Disord. 2014. https://doi.org/10.1002/mds.25914.
Navon N, Gendreau R, Meckler J. Gastric retention of the accordion Pill™: results from MRI studies with Parkinson’s disease patients and healthy volunteers [abstract]. Mov Disord. 2018;33(suppl 2).
LeWitt PA, Giladi N, Navon N. Pharmacokinetics and efficacy of a novel formulation of carbidopa-levodopa (Accordion Pill®) in Parkinson’s disease. Parkinsonism Relat Disord. 2019. https://doi.org/10.1016/j.parkreldis.2019.05.032.
LeWitt P, Gendreau R, Meckler J, Navon N. Design of a phase 3 efficacy and safety trial of accordion Pill™ carbidopa/levodopa for Parkinson’s disease (PD) patients experiencing motor fluctuations [abstract]. Mov Disord. 2018;33(suppl 2).
Kuoppamäki M, Leinonen M, Poewe W. Efficacy and safety of entacapone in levodopa/carbidopa versus levodopa/benserazide treated Parkinson’s disease patients with wearing-off. J Neural Transm. 2015. https://doi.org/10.1007/s00702-015-1449-6.
Rinne UK, Larsen JP, Siden Å, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Neurology. 1998. https://doi.org/10.1212/WNL.51.5.1309.
Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M. Efficacy and safety of entacapone in Parkinson’s disease patients with suboptimal levodopa response: a 6-month randomized placebo controlled double blind study in Germany and Austria (Celomen study). Acta Neurol Scand. 2002. https://doi.org/10.1034/j.1600-0404.2002.1o174.x.
Brooks DJ, Sagar H. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind, six month study. J Neurol Neurosurg Psychiatry. 2003. https://doi.org/10.1136/jnnp.74.8.1071.
Ceravolo R, Piccini P, Bailey DL, Jorga KM, Bryson H, Brooks DJ. 18F-dopa PET evidence that tolcapone acts as a central COMT inhibitor in Parkinson’s disease. Synapse. 2002. https://doi.org/10.1002/syn.10034.
Adler CH, et al. Randomized, placebo-controlled study of tolcapone in patients with fluctuating Parkinson disease treated with levodopa–carbidopa. Arch Neurol. 1998. https://doi.org/10.1001/archneur.55.8.1089.
Assal F, Spahr L, Hadengue A, Rubbici-Brandt L, Burkhard PR. Tolcapone and fulminant hepatitis. Lancet. 1998. https://doi.org/10.1016/S0140-6736(05)61511-5.
Ferreira JJ, et al. Summary of the recommendations of the EFNS/MDS-ES review on therapeutic management of Parkinson’s disease. Eur J Neurol. 2013. https://doi.org/10.1111/j.1468-1331.2012.03866.x.
Ferreira JJ, et al. Effect of opicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity and motor fluctuations in patients with Parkinson’s disease. Eur J Neurol. 2015. https://doi.org/10.1111/ene.12666.
Rocha JF, et al. Effect of opicapone and entacapone upon levodopa pharmacokinetics during three daily levodopa administrations. Eur J Clin Pharmacol. 2014. https://doi.org/10.1007/s00228-014-1701-2.
Lees AJ, et al. Opicapone as adjunct to levodopa therapy in patients with Parkinson disease and motor fluctuations a randomized clinical trial. JAMA Neurol. 2017. https://doi.org/10.1001/jamaneurol.2016.4703.
Ferreira JJ, Lees A, Rocha JF, Poewe W, Rascol O, Soares-da-Silva P. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2016. https://doi.org/10.1016/S1474-4422(15)00336-1.
Elmer LW. Rasagiline adjunct therapy in patients with Parkinson’s disease: Posthoc analyses of the PRESTO and LARGO trials. Parkinsonism Relat Disord. 2013. https://doi.org/10.1016/j.parkreldis.2013.06.001.
Schwid SR. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol. 2005. https://doi.org/10.1001/archneur.62.2.241.
Rascol O, et al. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet. 2005. https://doi.org/10.1016/S0140-6736(05)71083-7.
Fox SH, et al. International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2018. https://doi.org/10.1002/mds.27372.
Heinonen EH, Myllylä V. Safety of selegiline (Deprenyl) in the treatment of Parkinson’s disease. Drug Saf. 1998. https://doi.org/10.2165/00002018-199819010-00002.
Richard IH, Kurlan R, Tanner C, Factor S, Hubble J, Suchowersky O, Waters C. Serotonin syndrome and the combined use of deprenyl and an antidepressant in Parkinson's disease. Neurology. 1997;48(4):1070–7.
Panisset M, Chen JJ, Rhyee SH, Conner J, Mathena J. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014. https://doi.org/10.1002/phar.1500.
Caccia C, et al. Safinamide: From molecular targets to a new anti-Parkinson drug. Neurology. 2006. https://doi.org/10.1212/wnl.67.7_suppl_2.s18.
Borgohain R, et al. Randomized trial of safinamide add-on to levodopa in Parkinson’s disease with motor fluctuations. Mov Disord. 2014. https://doi.org/10.1002/mds.25751.
Schapira AHV, et al. Assessment of safety and efficacy of safinamide as a levodopa adjunct in patients with Parkinson disease and motor fluctuations a randomized clinical trial. JAMA Neurol. 2017. https://doi.org/10.1001/jamaneurol.2016.4467.
Borgohain R, et al. Two-year, randomized, controlled study of safinamide as add-on to levodopa in mid to late Parkinson’s disease. Mov Disord. 2014. https://doi.org/10.1002/mds.25961.
Binde CD, Tvete IF, Gåsemyr J, Natvig B, Klemp M. A multiple treatment comparison meta-analysis of monoamine oxidase type B inhibitors for Parkinson’s disease. Br J Clin Pharmacol. 2018. https://doi.org/10.1111/bcp.13651.
Kvernmo T, Härtter S, Burger E. A review of the receptor-binding and pharmacokinetic properties of dopamine agonists. Clin Ther. 2006. https://doi.org/10.1016/j.clinthera.2006.08.004.
Jenner P. Pharmacology of dopamine agonists in the treatment of Parkinson’s disease. Neurology. 2002. https://doi.org/10.1212/wnl.58.suppl_1.s1.
Piercey MF. Pharmacology of pramipexole, a dopamine D3-preferring agonist useful in treating Parkinson’s disease. Clin Neuropharmacol. 1998;21(3):141–51.
Hoehn MMM, Elton RL. Low dosages of bromocriptine added to levodopa in Parkinson’s disease. Neurology. 1985. https://doi.org/10.1212/wnl.35.2.199.
Stocchi F, et al. Ropinirole 24-hour prolonged release and ropinirole immediate release in early Parkinson’s disease: a randomized, double-blind, non-inferiority crossover study. Curr Med Res Opin. 2008. https://doi.org/10.1185/03007990802387130.
Pahwa R, et al. Ropinirole 24-hour prolonged release: randomized, controlled study in advanced Parkinson disease. Neurology. 2007. https://doi.org/10.1212/01.wnl.0000258660.74391.c1.
Poewe W, et al. Extended-release pramipexole in early Parkinson disease A 33-week randomized controlled trial. Neurology. 2011. https://doi.org/10.1212/WNL.0b013e31822affb0.
Schapira AHV, et al. Extended-release pramipexole in advanced Parkinson disease: a randomized controlled trial. Neurology. 2011. https://doi.org/10.1212/WNL.0b013e31822affdb.
Elshoff JP, Cawello W, Andreas JO, Mathy FX, Braun M. An update on pharmacological, pharmacokinetic properties and drug-drug interactions of rotigotine transdermal system in Parkinson’s disease and restless legs syndrome. Drugs. 2015. https://doi.org/10.1007/s40265-015-0377-y.
Trenkwalder C, et al. Rotigotine effects on early morning motor function and sleep in Parkinson’s disease: a double-blind, randomized, placebo-controlled study (RECOVER). Mov Disord. 2011. https://doi.org/10.1002/mds.23441.
Kassubek J, et al. Rotigotine transdermal system and evaluation of pain in patients with Parkinson’s disease: a post hoc analysis of the RECOVER study. BMC Neurol. 2014. https://doi.org/10.1186/1471-2377-14-42.
Hirano M, Isono C, Sakamoto H, Ueno S, Kusunoki S, Nakamura Y. Rotigotine transdermal patch improves swallowing in dysphagic patients with Parkinson’s disease. Dysphagia. 2015. https://doi.org/10.1007/s00455-015-9622-5.
Tateno H, et al. Transdermal dopamine agonist ameliorates gastric emptying in Parkinson’s disease. J Am Geriatr Soc. 2015. https://doi.org/10.1111/jgs.13800.
Lee JY, et al. Association between the dose of dopaminergic medication and the behavioral disturbances in Parkinson disease. Parkinsonism Relat Disord. 2010. https://doi.org/10.1016/j.parkreldis.2009.12.002.
Rabinak CA, Nirenberg MJ. Dopamine agonist withdrawal syndrome in parkinson disease. Arch Neurol. 2010. https://doi.org/10.1001/archneurol.2009.294.
Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging. 2013. https://doi.org/10.1007/s40266-013-0090-z.
Antonini A, Poewe W. Fibrotic heart-valve reactions to dopamine-agonist treatment in Parkinson’s disease. Lancet Neurol. 2007. https://doi.org/10.1016/S1474-4422(07)70218-1.
Rizos A, et al. A European multicentre survey of impulse control behaviours in Parkinson’s disease patients treated with short- and long-acting dopamine agonists. Eur J Neurol. 2016. https://doi.org/10.1111/ene.13034.
Garcia-Ruiz PJ, et al. Impulse control disorder in patients with Parkinson’s disease under dopamine agonist therapy: a multicentre study. J Neurol Neurosurg Psychiatry. 2014. https://doi.org/10.1136/jnnp-2013-306787.
Shulman LM, Minagar A, Rabinstein A, Weiner WJ. The use of dopamine agonists in very elderly patients with Parkinson’s disease. Mov Disord. 2000. https://doi.org/10.1002/1531-8257(200007)15:4%3c664:AID-MDS1010%3e3.0.CO;2-D.
Saint-Cyr JA, Trépanier LL. Beneficial effects of amantodine on L-dopa-induced dyskinesias in Parkinson’s disease. Mov Disord. 2000. https://doi.org/10.1002/1531-8257(200009)15:5%3c873:AID-MDS1017%3e3.0.CO;2-I.
Metman LV, Del Dotto P, Van Den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology. 1998. https://doi.org/10.1212/wnl.50.5.1323.
Malkani R, Zadikoff C, Melen O, Videnovic A, Borushko E, Simuni T. Amantadine for freezing of gait in patients with parkinson disease. Clin Neuropharmacol. 2012. https://doi.org/10.1097/WNF.0b013e31826e3406.
Thomas A, Iacono D, Luciano AL, Armellino K, Di Iorio A, Onofrj M. Duration of amantadine benefit on dyskinesia of severe Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2004;75(1):141–3.
Ory-Magne F, et al. Withdrawing amantadine in dyskinetic patients with Parkinson disease: the AMANDYSK trial. Neurology. 2014. https://doi.org/10.1212/WNL.0000000000000050.
Hauser RA, et al. Pharmacokinetics of ADS-5102 (Amantadine) extended release capsules administered once daily at bedtime for the treatment of dyskinesia. Clin Pharmacokinet. 2019. https://doi.org/10.1007/s40262-018-0663-4.
Pahwa R, et al. ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson disease (EASE LID study): a randomized clinical trial. JAMA Neurol. 2017. https://doi.org/10.1001/jamaneurol.2017.0943.
Oertel W, et al. Randomized, placebo-controlled trial of ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (EASE LID 3). Mov Disord. 2017. https://doi.org/10.1002/mds.27131.
Pahwa R, et al. Amantadine extended release for levodopa-induced dyskinesia in Parkinson’s disease (EASED Study). Mov Disord. 2015. https://doi.org/10.1002/mds.26159.
Kase H. Industry forum: progress in pursuit of therapeutic A2A antagonists—the adenosine A2A receptor selective antagonist KW6002: research and development toward a novel nondopaminergic therapy for Parkinson’s disease. Neurology. 2003. https://doi.org/10.1212/01.wnl.0000095219.22086.31.
Hauser RA, et al. Study of istradefylline in patients with Parkinson’s disease on levodopa with motor fluctuations. Mov Disord. 2008. https://doi.org/10.1002/mds.22095.
Mizuno Y, Kondo T. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov Disord. 2013. https://doi.org/10.1002/mds.25418.
Takahashi M, Fujita M, Asai N, Saki M, Mori A. Safety and effectiveness of istradefylline in patients with Parkinson’s disease: interim analysis of a post-marketing surveillance study in Japan. Expert Opin Pharmacother. 2018. https://doi.org/10.1080/14656566.2018.1518433.
Kondo T, Mizuno Y. A long-term study of istradefylline safety and efficacy in patients with parkinson disease. Clin Neuropharmacol. 2015. https://doi.org/10.1097/WNF.0000000000000073.
Murata M, et al. Zonisamide improves wearing-off in Parkinson’s disease: a randomized, double-blind study. Mov Disord. 2015. https://doi.org/10.1002/mds.26286.
Fornadi F, Milani F, Werner M. Madopar dispersible in the treatment of advanced Parkinson’s disease. Clin Neuropharmacol. 1995. https://doi.org/10.1097/00002826-199417003-00003.
Jansson Y, Eriksson B, Johnels B. Dispersible levodopa has a fast and more reproducible onset of action than the conventional preparation in Parkinson’s disease. A study with optoelectronic movement analysis. Parkinsonism Relat Disord. 1998. https://doi.org/10.1016/s1353-8020(98)00036-4.
Auffret M, Drapier S, Vérin M. Pharmacological insights into the use of apomorphine in Parkinson’s disease: clinical relevance. Clin Drug Investig. 2018. https://doi.org/10.1007/s40261-018-0619-3.
Millan MJ, Maiofiss L, Cussac D, Audinot V, Boutin JA, Newman-Tancredi A. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J Pharmacol Exp Ther. 2002. https://doi.org/10.1124/jpet.102.039867.
Jenner P, Katzenschlager R. Apomorphine—pharmacological properties and clinical trials in Parkinson’s disease. Parkinsonism Relat Disord. 2016. https://doi.org/10.1016/j.parkreldis.2016.12.003.
Gancher ST, Nutt JG, Woodward WR. Absorption of apomorphine by various routes in parkinsonism. Mov Disord. 1991. https://doi.org/10.1002/mds.870060304.
Nicolle E, et al. Pharmacokinetics of apomorphine in parkinsonian patients. Fundam Clin Pharmacol. 1993. https://doi.org/10.1111/j.1472-8206.1993.tb00238.x.
Gancher ST, Woodward WR, Boucher B, Nutt JG. Peripheral pharmacokinetics of apomorphine in humans. Ann Neurol. 1989. https://doi.org/10.1002/ana.410260209.
Corboy DL, Wagner ML, Sage JI. Apomorphine for motor fluctuations and freezing in Parkinson’s disease. Ann Pharmacother. 1995. https://doi.org/10.1177/106002809502900310.
LeWitt PA. Subcutaneously administered apomorphine: pharmacokinetics and metabolism. Neurology. 2004. https://doi.org/10.1212/wnl.62.6_suppl_4.s8.
Dewey RB, Hutton JT, LeWitt PA, Factor SA. A randomized, double-blind, placebo-controlled trial of subcutaneously injected apomorphine for parkinsonian off-state events. Arch Neurol. 2001. https://doi.org/10.1001/archneur.58.9.1385.
Gunzler SA, Koudelka C, Carlson NE, Pavel M, Nutt JG. Effect of low concentrations of apomorphine on parkinsonism in a randomized, placebo-controlled, crossover study. Arch Neurol. 2008. https://doi.org/10.1001/archneurol.2007.58.
Pahwa R, Koller WC, Trosch RM, Sherry JH. Subcutaneous apomorphine in patients with advanced Parkinson’s disease: a dose-escalation study with randomized, double-blind, placebo-controlled crossover evaluation of a single dose. J Neurol Sci. 2007. https://doi.org/10.1016/j.jns.2007.03.013.
Merello M, Pikielny R, Cammarota A, Leiguarda R. Comparison of subcutaneous apomorphine versus dispersible madopar latency and effect duration in Parkinson’s disease patients: a double-blind single-dose study. Clin Neuropharmacol. 1997. https://doi.org/10.1097/00002826-199704000-00008.
Isaacson S, Lew M, Ondo W, Hubble J, Clinch T, Pagan F. Apomorphine subcutaneous injection for the management of morning akinesia in Parkinson’s disease. Mov Disord Clin Pract. 2017. https://doi.org/10.1002/mdc3.12350.
Stibe CMH, Kempster PA, Lees AJ, Stern GM. Subcutaneous apomorphine in parkinsonian on–off oscillations. Lancet. 1988. https://doi.org/10.1016/S0140-6736(88)91193-2.
Stibe C, Lees A, Stern G. Subcutaneous infusion of apomorphine and lisuride in the treatment of parkinsonian on-off fluctuations. Lancet. 1987. https://doi.org/10.1016/S0140-6736(87)91660-6.
Frankel JP, Lees AJ, Kempster PA, Stern GM. Subcutaneous apomorphine in the treatment of Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1990. https://doi.org/10.1136/jnnp.53.2.96.
LeWitt PA, Ondo WG, Van Lunen B, Bottini PB. Open-label study assessment of safety and adverse effects of subcutaneous apomorphine injections in treating ‘off’ episodes in advanced Parkinson disease. Clin Neuropharmacol. 2009. https://doi.org/10.1097/WNF.0b013e31816d91f9.
Lees A. Dopamine agonists in Parkinson’s disease: a look at apomorphine. Fundam Clin Pharmacol. 1993. https://doi.org/10.1111/j.1472-8206.1993.tb00226.x.
Martinez-Martin P, et al. Chronic subcutaneous infusion therapy with apomorphine in advanced Parkinson’s disease compared to conventional therapy: a real life study of non motor effect. J Parkinsons Dis. 2011. https://doi.org/10.3233/JPD-2011-11037.
Grosset KA, Malek N, Morgan F, Grosset DG. Phase IIa randomized double-blind, placebo-controlled study of inhaled apomorphine as acute challenge for rescuing ‘off’ periods in patients with established Parkinson’s disease. Eur J Neurol. 2013. https://doi.org/10.1111/ene.12091.
Grosset KA, Malek N, Morgan F, Grosset DG. Inhaled dry powder apomorphine (VR040) for ’off ’ periods in Parkinson’s disease: an in-clinic double-blind dose ranging study. Acta Neurol Scand. 2013. https://doi.org/10.1111/ane.12107.
Grosset KA, Malek N, Morgan F, Grosset DG. Inhaled apomorphine in patients with ‘on-off’ fluctuations: a randomized, double-blind, placebo-controlled, clinic and home based, parallel-group study. J Parkinsons Dis. 2013. https://doi.org/10.3233/JPD-120142.
Olanow CW, et al. Apomorphine sublingual film for off episodes in Parkinson’s disease: a randomised, double-blind, placebo-controlled phase 3 study. Lancet Neurol. 2020. https://doi.org/10.1016/S1474-4422(19)30396-5.
Tambasco N, Romoli M, Calabresi P. Levodopa in Parkinson’s disease: current status and future developments. Curr Neuropharmacol. 2017. https://doi.org/10.2174/1570159x15666170510143821.
Taddei RN, Spinnato F, Jenner P. New symptomatic treatments for the management of motor and nonmotor symptoms of Parkinson’s disease. Int Rev Neurobiol. 2017;132:407–52.
LeWitt PA, et al. A randomized trial of inhaled levodopa (CVT-301) for motor fluctuations in Parkinson’s disease. Mov Disord. 2016. https://doi.org/10.1002/mds.26611.
Grosset D, et al. Long-term pulmonary safety of inhaled levodopa in Parkinson’s disease subjects with motor fluctuations: interim results of a phase 3 study. Parkinsonism Relat Disord. 2018. https://doi.org/10.1016/j.parkreldis.2017.11.287.
Antonini A, et al. Developing consensus among movement disorder specialists on clinical indicators for identification and management of advanced Parkinson’s disease: a multi-country Delphi-panel approach. Curr Med Res Opin. 2018. https://doi.org/10.1080/03007995.2018.1502165.
Bredberg E, et al. Intraduodenal infusion of a water-based levodopa dispersion for optimisation of the therapeutic effect in severe Parkinson’s disease. Eur J Clin Pharmacol. 1993. https://doi.org/10.1007/BF00315491.
Nyholm D, et al. Optimizing levodopa pharmacokinetics: intestinal infusion versus oral sustained-release tablets. Clin Neuropharmacol. 2003. https://doi.org/10.1097/00002826-200305000-00010.
Nyholm D, et al. Pharmacokinetics of levodopa, carbidopa, and 3-O-methyldopa following 16-hour jejunal infusion of levodopa-carbidopa intestinal gel in advanced Parkinson’s disease patients. AAPS J. 2013. https://doi.org/10.1208/s12248-012-9439-1.
Vijiaratnam N, et al. Levodopa–carbidopa intestinal gel: is the naso-jejunal phase a redundant convention? Intern Med J. 2018. https://doi.org/10.1111/imj.13754.
Zibetti M, et al. Sleep improvement with levodopa/carbidopa intestinal gel infusion in Parkinson disease. Acta Neurol Scand. 2013. https://doi.org/10.1111/ane.12075.
Chang FCF, et al. 24h Levodopa–carbidopa intestinal gel may reduce falls and ‘unresponsive’ freezing of gait in Parkinson’s disease. Parkinsonism Relat Disord. 2015. https://doi.org/10.1016/j.parkreldis.2014.12.019.
Olanow CW, et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 2014. https://doi.org/10.1016/S1474-4422(13)70293-X.
Standaert DG, et al. Effect of levodopa–carbidopa intestinal gel on non-motor symptoms in patients with advanced Parkinson’s disease. Mov Disord Clin Pract. 2017. https://doi.org/10.1002/mdc3.12526.
Fernandez HH, et al. Levodopa-carbidopa intestinal gel in advanced Parkinson’s disease: final 12-month, open-label results. Mov Disord. 2015. https://doi.org/10.1002/mds.26123.
Vijiaratnam N, Sue CM. Levodopa-carbidopa intestinal gel: ‘dismantling the road blocks of a journey’. Intern Med J. 2018. https://doi.org/10.1111/imj.13757.
Vijiaratnam N, Sue CM. How do i manage patients with the levodopa/carbidopa intestinal gel? Mov Disord Clin Pract. 2019. https://doi.org/10.1002/mdc3.12701.
Vijiaratnam N, Sue CM. Maximizing benefits of the levodopa/carbidopa intestinal gel: systematic considerations, challenging convention and individualizing approaches. Basal Ganglia. 2018. https://doi.org/10.1016/j.baga.2018.12.002.
Merola A, Romagnolo A, Zibetti M, Bernardini A, Cocito D, Lopiano L. Peripheral neuropathy associated with levodopa–carbidopa intestinal infusion: a long-term prospective assessment. Eur J Neurol. 2016. https://doi.org/10.1111/ene.12846.
Rispoli V, et al. Peripheral neuropathy in 30 duodopa patients with vitamins B supplementation. Acta Neurol Scand. 2017. https://doi.org/10.1111/ane.12783.
Rogers G, Davies D, Pink J, Cooper P. Parkinson’s disease: summary of updated NICE guidance. BMJ. 2017. https://doi.org/10.1136/bmj.j1951.
Bhidayasiri R, Sringean J, Anan C, Boonpang K, Thanawattano C, Chaudhuri KR. Quantitative demonstration of the efficacy of night-time apomorphine infusion to treat nocturnal hypokinesia in Parkinson’s disease using wearable sensors. Parkinsonism Relat Disord. 2016. https://doi.org/10.1016/j.parkreldis.2016.11.016.
Trenkwalder C, et al. Expert Consensus Group report on the use of apomorphine in the treatment of Parkinson’s disease—clinical practice recommendations. Parkinsonism Relat Disord. 2015. https://doi.org/10.1016/j.parkreldis.2015.06.012.
Renoux C, et al. Ventricular tachyarrhythmia and sudden cardiac death with domperidone use in Parkinson’s disease. Br J Clin Pharmacol. 2016. https://doi.org/10.1111/bcp.12964.
Kempster PA, Frankel JP, Stern GM, Lees AJ. Comparison of motor response to apomorphine and levodopa in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1990. https://doi.org/10.1136/jnnp.53.11.1004.
Gervason CL, Pollak PR, Limousin P, Perret JE. Reproducibility of motor effects induced by successive subcutaneous apomorphine injections in Parkinson’s disease. Clin Neuropharmacol. 1993. https://doi.org/10.1097/00002826-199304000-00003.
Katzenschlager R, et al. Apomorphine subcutaneous infusion in patients with Parkinson’s disease with persistent motor fluctuations (TOLEDO): a multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2018. https://doi.org/10.1016/S1474-4422(18)30239-4.
Martinez-Martin P, et al. EuroInf: a multicenter comparative observational study of apomorphine and levodopa infusion in Parkinson’s disease. Mov Disord. 2015. https://doi.org/10.1002/mds.26067.
Deleu D, Hanssens Y, Northway MG. Subcutaneous apomorphine: an evidence-based review of its use in Parkinson’s disease. Drugs Aging. 2004. https://doi.org/10.2165/00002512-200421110-00001.
Acland KM, Churchyard A, Fletcher CL, Turner K, Lees A, Dowd PM. Panniculitis in association with apomorphine infusion. Br J Dermatol. 1998. https://doi.org/10.1046/j.1365-2133.1998.02128.x.
Skorvanek M, Bhatia KP. The skin and Parkinson’s disease: review of clinical, diagnostic, and therapeutic issues. Mov Disord Clin Pract. 2017. https://doi.org/10.1002/mdc3.12425.
Pietz K, Hagell P, Odin P. Subcutaneous apomorphine in late stage Parkinson’s disease: a long term follow up. J Neurol Neurosurg Psychiatry. 1998. https://doi.org/10.1136/jnnp.65.5.709.
Rosa-Grilo M, Qamar MA, Evans A, Chaudhuri KR. The efficacy of apomorphine—a non-motor perspective. Parkinsonism Relat Disord. 2016. https://doi.org/10.1016/j.parkreldis.2016.11.020.
Barbosa P, Lees AJ, Magee C, Djamshidian A, Warner TT. A retrospective evaluation of the frequency of impulsive compulsive behaviors in Parkinson’s disease patients treated with continuous waking day apomorphine pumps. Mov Disord Clin Pract. 2017. https://doi.org/10.1002/mdc3.12416.
Todorova A, Samuel M, Brown RG, Chaudhuri KR. Infusion therapies and development of impulse control disorders in advanced Parkinson disease: clinical experience after 3 years’ follow-up. Clin Neuropharmacol. 2015. https://doi.org/10.1097/WNF.0000000000000091.
Borgemeester RWK, Lees AJ, van Laar T. Parkinson’s disease, visual hallucinations and apomorphine: a review of the available evidence. Parkinsonism Relat Disord. 2016. https://doi.org/10.1016/j.parkreldis.2016.04.023.
Ellis C, et al. Use of apomorphine in Parkinsonian patients with neuropsychiatric complications to oral treatment. Parkinsonism Relat Disord. 1997. https://doi.org/10.1016/S1353-8020(97)00009-6.
Moore TJ, Glenmullen J, Mattison DR. Reports of pathological gambling, hypersexuality, and compulsive shopping associated with dopamine receptor agonist drugs. JAMA Intern Med. 2014. https://doi.org/10.1001/jamainternmed.2014.5262.
Samuel M, et al. Impulse control disorders in Parkinson’s disease:management, controversies, and potential approaches HHS public access. Mov Disord. 2015. https://doi.org/10.1002/mds.26099.Impulse.
Giladi N, Caraco Y, Gurevich T, Djaldetti R, Adar L, Rachmilewitz Minei T, Oren S. ND0612 (levodopa/carbidopa for subcutaneous infusion) achieves stable levodopa plasma levels when administered in low and high doses in patients with PD [abstract]. Mov Disord. 2017;32(suppl:2).
Giladi N, et al. Pharmacokinetic profile of ND0612 (levodopa/carbidopa for subcutaneous infusion) in Parkinson’s disease (PD) patients with motor fluctuations: results of a phase IIa dose finding study. Eur J Neurol. 2015. https://doi.org/10.1111/ene.12805.
Adar L, Minei TR, Cohen Y. Pharmacokinetic profile of continuous levodopa/carbidopa delivery when administered subcutaneously (ND0612) versus duodenal infusion (levodopa/carbidopa intestinal gel). Mov Disord. 2017. https://doi.org/10.1002/mds.27087.
Adar L, Rachmilewitz Minei T, Cohen Y, Oren S. Pharmacokinetic profile of continuous levodopa/carbidopa delivery when administered subcutaneously (ND0612) versus duodenal infusion (levodopa/carbidopa intestinal gel) [abstract]. Mov Disord. 2017;32(suppl:2).
Oren S, Kieburtz K, Olanow CW, Cohen Y. A randomized controlled clinical study to evaluate the efficacy and safety of subcutaneous levodopa/carbidopa (ND0612H) in patients with advanced Parkinson’s disease. J Parkinsons Dis. 2016. https://doi.org/10.3233/JPD-169900.
Adar L, Minei TR. Identification of the optimal carbidopa concentration in subcutaneously administered ND0612. Mov Disord. 2017. https://doi.org/10.1002/mds.27087.
L. P. et al. Indigo: a multicenter, randomized, double-blind, placebocontrolled, study of continuous ND0612 infusion with adjunct oral levodopa in fluctuating Parkinson’s disease. Mov Disord. 2018. http://dx.doi.org/10.1002/mds.27434.
Okun MS. Deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2012. https://doi.org/10.1056/NEJMct1208070.
Herrington TM, Cheng JJ, Eskandar EN. Mechanisms of deep brain stimulation. J Neurophysiol. 2016. https://doi.org/10.1152/jn.00281.2015.
Eusebio A, et al. Deep brain stimulation can suppress pathological synchronisation in parkinsonian patients. J Neurol Neurosurg Psychiatry. 2011. https://doi.org/10.1136/jnnp.2010.217489.
Okun MS, et al. Subthalamic deep brain stimulation with a constant-current device in Parkinson’s disease: an open-label randomised controlled trial. Lancet Neurol. 2012. https://doi.org/10.1016/S1474-4422(11)70308-8.
Deuschl G, et al. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2006. https://doi.org/10.1056/NEJMoa060281.
Lachenmayer ML, et al. Stimulation of the globus pallidus internus in the treatment of Parkinson’s disease: long-term results of a monocentric cohort. Parkinsonism Relat Disord. 2019. https://doi.org/10.1016/j.parkreldis.2019.03.009.
Limousin P, Foltynie T. Long-term outcomes of deep brain stimulation in Parkinson disease. Nat Rev Neurol. 2019. https://doi.org/10.1038/s41582-019-0145-9.
Schuepbach WMM, et al. Neurostimulation for Parkinson’s disease with early motor complications. N Engl J Med. 2013. https://doi.org/10.1056/NEJMoa1205158.
Obeso JA, Olanow CW, Rodriguez-Oroz MC, Krack P, Kumar R, Lang AE. Deep-brain stimulation of the subthalamic nucleus or the pars interna of the globus pallidus in Parkinson’s disease. N Engl J Med. 2001. https://doi.org/10.1056/NEJMoa000827.
Bronstein JM, et al. Deep brain stimulation for Parkinson disease an expert consensus and review of key issues. Arch Neurol. 2011. https://doi.org/10.1001/archneurol.2010.260.
Anderson VC, Burchiel KJ, Hogarth P, Favre J, Hammerstad JP. Pallidal vs subthalamic nucleus deep brain stimulation in Parkinson disease. Arch Neurol. 2005. https://doi.org/10.1001/archneur.62.4.554.
Odekerken VJJ, et al. Subthalamic nucleus versus globus pallidus bilateral deep brain stimulation for advanced Parkinson’s disease (NSTAPS study): a randomised controlled trial. Lancet Neurol. 2013. https://doi.org/10.1016/S1474-4422(12)70264-8.
Ramirez-Zamora A, Ostrem JL. Globus pallidus interna or subthalamic nucleus deep brain stimulation for Parkinson disease a review. JAMA Neurol. 2018. https://doi.org/10.1001/jamaneurol.2017.4321.
Lyons KE, Wilkinson SB, Overman J, Pahwa R. Surgical and hardware complications of subthalamic stimulation: a series of 160 procedures. Neurology. 2004. https://doi.org/10.1212/01.WNL.0000134650.91974.1A.
Oh MY, et al. Long-term hardware-related complications of deep brain stimulation. Neurosurgery. 2002. https://doi.org/10.1097/00006123-200206000-00017.
Fenoy AJ, Simpson RK. Risks of common complications in deep brain stimulation surgery: Management and avoidance—clinical article. J Neurosurg. 2014. https://doi.org/10.3171/2013.10.JNS131225.
Constantinescu R, et al. Key clinical milestones 15 years and onwards after DBS-STN surgery—a retrospective analysis of patients that underwent surgery between 1993 and 2001. Clin Neurol Neurosurg. 2017. https://doi.org/10.1016/j.clineuro.2017.01.010.
Fasano A, et al. Motor and cognitive outcome in patients with Parkinson’s disease 8 years after subthalamic implants. Brain. 2010. https://doi.org/10.1093/brain/awq221.
Dayal V, Limousin P, Foltynie T. Subthalamic nucleus deep brain stimulation in Parkinson’s disease: the effect of varying stimulation parameters. J Parkinson’s Dis. 2017. https://doi.org/10.3233/JPD-171077.
Dafsari HS, et al. EuroInf 2: subthalamic stimulation, apomorphine, and levodopa infusion in Parkinson’s disease. Mov Disord. 2019. https://doi.org/10.1002/mds.27626.
Habets JGV, Heijmans M, Kuijf ML, Janssen MLF, Temel Y, Kubben PL. An update on adaptive deep brain stimulation in Parkinson’s disease. Mov Disord. 2018. https://doi.org/10.1002/mds.115.
Contarino MF, et al. Directional steering: a novel approach to deep brain stimulation. Neurology. 2014. https://doi.org/10.1212/WNL.0000000000000823.
Seppi K, et al. Update on treatments for nonmotor symptoms of Parkinson’s disease—an evidence-based medicine review. Mov Disord. 2019. https://doi.org/10.1002/mds.27602.
Schrag A, Sauerbier A, Chaudhuri KR. New clinical trials for nonmotor manifestations of Parkinson’s disease. Mov Disord. 2015. https://doi.org/10.1002/mds.26415.
Zesiewicz TA, et al. Practice parameter: Treatment of nonmotor symptoms of Parkinson disease: report of the quality standards subcommittee of the American academy of neurology. Neurology. 2010. https://doi.org/10.1212/WNL.0b013e3181d55f24.
Evans AH, Farrell MJ, Gibson SJ, Helme RD, Lim SY. Dyskinetic patients show rebound worsening of affect after an acute L-dopa challenge. Parkinsonism Relat Disord. 2012. https://doi.org/10.1016/j.parkreldis.2012.01.020.
Antonini A, et al. COMT inhibition with tolcapone in the treatment algorithm of patients with Parkinson’s disease (PD): Relevance for motor and non-motor features. Neuropsychiatr Dis Treat. 2008. https://doi.org/10.2147/ndt.s2404.
Ebersbach G, Hahn K, Lorrain M, Storch A. Tolcapone improves sleep in patients with advanced Parkinson’s disease (PD). Arch Gerontol Geriatr. 2010. https://doi.org/10.1016/j.archger.2010.03.008.
Muller T. Tolcapone addition improves Parkinson’s disease associated nonmotor symptoms. Ther Adv Neurol Disord. 2014. https://doi.org/10.1177/1756285613512392.
Wolz M, Hauschild J, Koy J, Fauser M, Klingelhöfer L, Schackert G, Reichmann H, Storch A. Immediate effects of deep brain stimulation of the subthalamic nucleus on nonmotor symptoms in Parkinson's disease. Parkinsonism Relat Disord. 2012;18(8):994–7.
Witjas T, et al. Effects of chronic subthalamic stimulation on nonmotor fluctuations in Parkinson’s disease. Mov Disord. 2007. https://doi.org/10.1002/mds.21602.
Odin P, et al. Collective physician perspectives on non-oral medication approaches for the management of clinically relevant unresolved issues in Parkinson’s disease: consensus from an international survey and discussion program. Parkinsonism Relat Disord. 2015. https://doi.org/10.1016/j.parkreldis.2015.07.020.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
NV’s research time and position is funded by the Janet Owens charitable foundation. No funding was received specifically for this review.
Conflict of interest
NV has received educational support from Abbvie, Stada, Ipsen and Merc, speaker’s honorarium from Abbvie & Stada and served on advisory boards for Abbvie & Britannia Pharmaceuticals. TF has received grant support form Cure Parkinson’s trust, Michael J Fox Foundation, John Black Charitable Foundation, Van Andel Institute, Defeat MSA, Innovate UK, National Institute for Health Research. He has received honoraria for speaking at meetings sponsored by Bial, Profile Pharma, Britannia and Boston Scientific. He has served on advisory boards for Living Cell Technologies, Voyager Therapeutics and Oxford Biomedica.
Rights and permissions
About this article

Cite this article
Vijiaratnam, N., Foltynie, T. Therapeutic Strategies to Treat or Prevent Off Episodes in Adults with Parkinson’s Disease. Drugs 80, 775–796 (2020). https://doi.org/10.1007/s40265-020-01310-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01310-2