
Abstract
Kinase activating missense mutations in leucine-rich repeat kinase 2 (LRRK2) are pathogenically linked to neurodegenerative Parkinson’s disease (PD). Over the past decade, substantial effort has been devoted to the development of potent and selective small molecule inhibitors of LRRK2, as well as their preclinical testing across different Parkinson’s disease models. This review outlines the genetic and biochemical evidence that pathogenic missense mutations increase LRRK2 kinase activity, which in turn provides the rationale for the development of small molecule inhibitors as potential PD therapeutics. An overview of progress in the development of LRRK2 inhibitors is provided, which in particular indicates that highly selective and potent compounds capable of clinical utility have been developed. We outline evidence from rodent- and human-induced pluripotent stem cell models that support a pathogenic role for LRRK2 kinase activity, and review the substantial experiments aimed at evaluating the safety of LRRK2 inhibitors. We address challenges still to overcome in the translational therapeutic pipeline, including biomarker development and clinical trial strategies, and finally outline the potential utility of LRRK2 inhibitors for other genetic forms of PD and ultimately sporadic PD. Collective evidence supports the ongoing clinical translation of LRRK2 inhibitors as a therapeutic intervention for PD is greatly needed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Increased LRRK2 kinase activity is implicated in both familial and sporadic forms of Parkinson’s disease. |
Aberrant LRRK2 kinase activity can be targeted with highly potent and selective small molecule inhibitors. |
Preclinical and early clinical testing to date support continued efforts to advance LRRK2 inhibitors to the clinic. |
1 Introduction
Parkinson’s disease (PD) is a common neurodegenerative movement disorder and a global health burden. The clinical course of PD is long, progressively disabling, and currently cannot be halted, reversed or prevented. The typical PD motor symptoms of bradykinesia, resting tremor and rigidity occur when approximately 50% of dopaminergic neurons, located in the substantia nigra, have degenerated [1, 2]. Current estimates suggest that PD actually commences at least a decade before the onset of motor symptoms [3]. PD is also associated with a host of non-motor symptoms including depression, anxiety, dementia, sleep dysfunction, gastrointestinal dysfunction and hyposmia [4]. The non-motor symptoms of PD contribute substantially to the burden of disease and are potentially related to the accumulation of pathological forms of the α-synuclein protein [5]. This accumulation of insoluble α-synuclein into neural cytoplasmic aggregates, called Lewy bodies, is a defining hallmark of PD [6]. However, exactly how the accumulation of pathogenic α-synuclein occurs still remains to be defined. Evidence suggests that pathogenic forms of α-synuclein are transmitted throughout multiple brain regions in a distinctive pattern based on Braak staging [7], although it should be noted that exceptions to this concept have been identified [8, 9]. Irrespective of mechanism, the current front-line treatment for PD is symptomatic relief from motor symptoms using dopamine replacement medication, or in some instances, deep-brain stimulation. Such treatments though, are not without side effects or complications and can become less effective as PD progresses. Moreover, current treatments do not address the non-motor symptoms, which remain a great unmet need for PD patients. For these reasons, disease-modifying treatments targeting pathogenic pathways in PD have been researched and developed over decades, and while much has been learnt, the underlying causes and pathological mechanisms of PD still remain unclear. Importantly however, a number of relatively recent pivotal discoveries have demonstrated an underlying role for genetics in PD. Missense mutations have been identified in a number of genes that are pathogenic for familial PD, that is PD that has been inherited in a recessive or dominant manner, whilst whole genome sequencing has identified polymorphisms that increase susceptibility to sporadic PD (for recent reviews on the genetics of PD see [10,11,12,13]). There is now much interest in understanding PD-linked genes and the proteins they encode, as they may not only provide insight into disease pathogenesis, but may also comprise potential novel therapeutic targets. One such PD-linked gene that has received substantial interest as a potential therapeutic target is leucine rich repeat kinase 2 (LRRK2).
First linked to autosomal dominantly inherited PD in 2004 [14, 15], LRRK2 has emerged as a key player in PD research. LRRK2 mutations account for nearly half of all genetic variants identified in database analyses of familial and sporadic PD cases [16, 17]. In addition, LRRK2-associated PD clinically closely resembles typical sporadic PD, in terms of onset age, symptoms and disease progression [18,19,20]. Genetic and clinical evidence therefore suggests that LRRK2 may play a general role in the disease mechanisms of both genetic and sporadic forms of PD. Most importantly, increased LRRK2 kinase activity has been demonstrated to mediate PD-associated pathogenic phenotypes, including inflammation, autophagy-lysosomal impairment, dysfunctional vesicle trafficking, defective neurite outgrowth and neuronal differentiation [21,22,23]. Thus, particular attention and intensive efforts have been paid to inhibiting LRRK2 kinase activity with small-molecule compounds. This review will outline the therapeutic rationale behind targeting LRRK2 and provide an update on current developments and challenges of this potentially exciting novel treatment for PD.
2 LRRK2 Kinase Hyperactivity in Parkinson’s Disease
2.1 Genetic and Biochemical Evidence
2.1.1 Overview of the LRRK2 Protein and Genetics
The LRRK2 gene is made up of 51 exons that encode a large protein of 2527 amino acids. The LRRK2 protein incorporates both active kinase and GTPase functions and this enzymatic core is surrounded by N-terminal armadillo repeats (ARM), ankyrin repeats (ANK), leucine-rich repeats (LRR) and C-terminal WD40 repeats (Fig. 1). More than 100 different LRRK2 sequence variants have been discovered in familial and sporadic PD patients, with six of these directly associated with PD pathogenesis (G2019S, R1441C, R1441G, R1441H, Y1699C and I2020T) [24, 25]. Interestingly, all six pathogenic mutations lie in the catalytic core of LRRK2 (Fig. 1), with the ROC domain containing R1441C/G/H mutations, the COR domain containing Y1699C, and the kinase domain harbouring G2019S and I2020T [26]. The LRRK2 G2019S mutation is by far the most frequent genetic determinant of PD, although its frequency varies widely across different ethnic populations [24, 25]. The highest reported frequency for the LRRK2 G2019S mutation, of up to 37%, has been reported in familial PD patients (and 3% in healthy controls) from North African Arab descent [27]. Mutations at the LRRK2 R1441 residue are the second most common pathogenic mutations, with the most frequent being R1441C, which was first discovered in Western Nebraska families [15]. The R1441G mutation was first reported in individuals from a Basque ethnic background [14] with the highest prevalence of 46% in familial Basque patients [28], whilst the R1441H mutation has been identified in a small number of PD families from the USA, western Europe and Australia [24]. The Y1669C mutation was identified in two independent large families of UK ancestry [14] and German–Canadian ancestry [15, 29], while the I2020T mutation is responsible for familial PD cases of German ancestry [15, 30] and a kindred from Sagamihara in Japan [31, 32]. The penetrance of LRRK2 is incomplete and dependent on age and ethnic background. Estimates of penetrance for the G2019S mutation range from 20 to 75% [25].

The LRRK2 protein. The domain structure of the LRRK2 protein is shown along with the pathogenic missense mutations that increase LRRK2 activity. Biomarker readouts of LRRK2 activity include phosphorylation sites on the LRRK2 protein, as well as downstream Rab GTPase substrates
In addition to the six known pathogenic mutations, several other LRRK2 variants have also been associated with increased PD risk (E334K, A419V, R1325Q, T1410M, R1628P, M1646T, N2081D and G2385R), or indeed protection (A221V, A1151T and N551K-R1398H-K1423K haplotype) [25]. The frequency of these risk polymorphisms again varies across populations, but they are much more common than pathogenic LRRK2 mutations and have a vastly reduced penetrance, potentially modulating PD risk by a few percent. The extent to which risk variants modulate LRRK2 kinase activity is still unclear; however, an understanding of both pathogenic risk and protective variants of LRRK2 can help to build genomic susceptibility profiles and stratify patient groups who might benefit most from targeted LRRK2 therapies.
2.1.2 Biochemical Evidence that Pathogenic LRRK2 Mutations Increase LRRK2 Kinase Activity
Most LRRK2 research has focused on the pathogenic missense mutations and in particular if/how these mutations affect the proteins enzymatic functions. Initial in vitro assays using autophosphorylation or optimised peptide substrates consistently demonstrated that the LRRK2 G2019S mutation, which is located in the activation loop of the kinase domain, had the greatest effect on LRRK2 kinase activity resulting in a twofold increase [33, 34]. The development of antibodies to LRRK2 autophosphorylation sites, in particular Ser1292, allowed for subsequent assessment of LRRK2 activity in cell and animal models. This revealed that all known pathogenic LRRK2 mutations increase the enzyme’s kinase activity [35]. This finding was confirmed following the identification and validation of a subset of Rab GTPase proteins as bona fide in vivo LRRK2 substrates [36, 37]. The Rab GTPase family is the largest family of small GTPase proteins and collectively are critical regulators of vesicle trafficking [38]. LRRK2 phosphorylates up to 14 members of this ~ 70 member GTPase family, with phosphorylation of Rab8 and Rab10 at Thr72 and Thr73, respectively, residues located in the effector-binding switch II domain, being particularly robust [37, 39]. Using these in vivo readouts of activity, it is evident that the pathogenic mutations in the ROC-COR domain (R1441C, R1441G, R1441H and Y1699C) have a greater effect on kinase activity than G2019S, consistently enhancing it by up to four-fold [35, 39, 40]. Why the LRRK2 ROC-COR mutations have a greater effect on LRRK2 kinase activity in vivo still remains to be fully elucidated. Recent evidence suggests that mutations in the ROC-COR domain cause LRRK2 to be more efficiently recruited to membrane-bound Rab proteins, which in turn exert an allosteric level of regulation on LRRK2 activity [41, 42]. A lack of direct association between in vitro and in vivo LRRK2 activity readouts is also illustrated by the G2385 risk variant, which has decreased LRRK2 activity when assayed in vitro [43], but not in vivo using Rab10 as a substrate [42]. Thus, despite the mechanistic complexities surrounding how, that all known pathogenic mutations increase LRRK2 kinase activity strongly implicates kinase activity as a main effector of LRRK2-mediated pathobiology.
2.2 Pathobiological Evidence Implicating LRRK2 Kinase Activity
Mounting evidence has indicated that LRRK2 kinase activity is critical for PD phenotypes induced by mutant LRRK2, and a reduction in kinase activity using small molecule compounds could have a neuroprotective impact in both animal and cell models. Here we will highlight some results from rodent and patient-specific induced pluripotent stem cell (iPSC) models. For additional reviews on LRRK2 pathobiology please refer to [44,45,46].
2.2.1 Brief Overview of Evidence from Rodent Models
Preclinical studies to date have largely employed transgenic rodent models to evaluate LRRK2 inhibitor efficacy in different PD models. Mice overexpressing the kinase activating LRRK2 G2019S showed progressive degeneration of dopaminergic neurons, which could be attenuated with early LRRK2 kinase inhibitors [47, 48]. Moreover, α-synuclein-induced dopaminergic neurodegeneration could also be effectively attenuated by inhibiting LRRK2 following adeno viral mediated expression of α-synuclein in LRRK2 G2019S transgenic rats [49]. In α-synuclein overexpressing Thy1 transgenic mice, LRRK2 kinase inhibition significantly reduced trans-axonal α-synuclein aggregates and α-synuclein propagation [50], and LRRK2 inhibitors could block potentiated pathology in G2019S overexpressing mice that had been inoculated with α-synuclein fibrils [51]. Inhibition of LRRK2 kinase activity also reversed neurite shortening in primary neuronal cultures from transgenic mice overexpressing LRRK2 G2019S or R1441G [52], and could enhance dopamine release and synaptic vesicle mobilisation/recycling in R1441G mice [53]. LRRK2 kinase inhibition has also been shown to attenuate proinflammatory microglial signalling in rats [54]. Collectively these results point to a key role for LRRK2 kinase activity in mediating neurodegenerative phenotypes, however it would be prudent to continue to evaluate this concept as both the rodent models and LRRK2 inhibitors continue to improve. Indeed, recent studies have described the characterisation of more physiological point mutation Knockin mice [55,56,57,58]. Phenotypes of these animals with regard to dopaminergic dysfunction and α-synuclein pathology are subtle compared to transgenic overexpression models and it will be of interest to determine the effectiveness of LRRK2 inhibitors under these conditions.
2.2.2 Brief Overview of Evidence from Induced Pluripotent Stem Cell Models
In the absence of readily available human neuron samples, IPSC models have emerged as important tools for evaluating LRRK2 inhibitors in primary human cell lines [59]. Somatic cells obtained from patients can be reprogrammed to disease-relevant iPSCs and then differentiated to neural stem cells and neurons. Liu and colleagues applied this technology to generate iPSC-derived neural stem cells from PD patients harbouring the LRRK2 G2019S mutation [60]. The mutant cells showed deficiencies in nuclear envelope organisation and potentiated senescence compared to control cells, and these phenotypes were rescued with LRRK2 inhibition [60]. Enhanced mitochondrial DNA damage is also evident in iPSC-derived lymphoblastoid cells from LRRK2 G2019S patients and notably, this damage can be restored with LRRK2 inhibitor treatment [61]. Reduced oxygen consumption and increased cytotoxicity after exposure to valinomycin were observed in a population of iPSC-derived neural cells with LRRK2 G2019S or R1441C mutations, and again these mitochondrial phenotypes could be reduced by pharmacological LRRK2 inhibition [62]. Sensory neurons with the LRRK2 G2019S mutation also showed altered calcium dynamics, which was again rescued with LRRK2 kinase inhibitors [63]. Thus, as for rodent studies, evidence can be found for a key role of LRRK2 kinase activity in mediating pathological events, and again, as cell models, inhibitors and understanding of LRRK2 biology improve, it will be important to strengthen this evidence.
3 Translation of LRRK2 Therapeutics to the Clinic
3.1 Overview of Drug Development
3.1.1 Early Non-selective and First Generation of Selective Compounds
Over the past decade, many compounds capable of inhibiting LRRK2 kinase activity have been identified through drug discovery studies, and up to now there have been four generations of advancement [64] (Table 1). For additional reviews on LRRK2 inhibitor development readers are referred to [21, 65,66,67]. Generation “0” are the earliest compounds, including H-1152, GW5074, staurosporine and sunitinib, and are largely non-selective pan-kinase inhibitors with relatively poor potency and poor blood–brain barrier (BBB) permeability [47, 68,69,70]. Their limited clinical and research application facilitated further development of more favourable compounds. Subsequent kinase profiling screens resulted in the first-generation selective compounds with improved potency including LRRK2-IN-1 [71], CZC-54252, CZC-25146 [72] and TTT-3002 [73]. These compounds could not robustly cross the BBB, but comprised useful in vitro tool compounds for accelerating understanding of LRRK2 biology and pharmacology.
3.1.2 Highly Potent and Selective Compounds
Second-generation inhibitors had improvements in all areas, including higher potency, higher selectivity, better brain penetrance and fewer off-target effects. HG-10-102-1 was the first selective LRRK2 inhibitor reported to block LRRK2 activity in mouse brain in a dose-dependent manner [74], with the pyrrolopyrimidine JH-II-127 another brain penetrant selective inhibitor [75]. A seemingly reasonable candidate, GSK2578215A, failed to inhibit LRRK2 in mouse brain via intraperitoneal injection, although being highly potent and BBB penetrant [76]. GNE-7915, GNE-0877 and GNE-9605 were developed later and showed enhanced potency even following oral administration [77, 78]. Moreover, GNE-0877 and GNE-9605 blocked LRRK2 in a robust, dose-dependent manner in the brain of LRRK2 G2019S mice [77]. PF-06447475, another characteristic second-generation LRRK2 inhibitor, revealed a decrease in LRRK2 activity after 90 min even with a low dose (3 mg/kg) in rodent brain and kidney [79]. Meanwhile, PF-06447475 had minimal effects on off-target enzymes and demonstrated exceptional brain permeability since it shows similar concentrations of unbound compound in the brain and plasma. This inhibitor also displays good tolerance with a relatively high dose (65 mg/kg) in a 2-week toxicological assessment [79].
3.1.3 Highly Potent and Selective Compounds with Blood–Brain Barrier Penetration
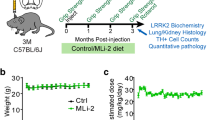
A more recently developed and structurally novel inhibitor, MLi-2, belongs to the third-generation inhibitors [64]. MLi-2 exhibited exceptional potency in vitro (half maximal inhibitory concentration (IC50) = 0.76–3.4 nM depending on LRRK2 biochemical assay employed) and a dose-dependent reduction in LRRK2 kinase activity in mice peripherally (IC50 = 0.8 nM) and in brain (IC50 = 1.1 nM) [80]. This inhibitor was shown to have exceptionally high selectivity for LRRK2 when screened against over 300 kinases and a number of receptors and ion channels [80, 81]. Moreover, it remained well tolerated over a 15-week period when administered daily at 30 mg/kg [80]. No adverse effects of MLi-2 were noticed on body weight, food intake, or behavioural performances at plasma exposures > 100 × the in vivo IC50 for brain LRRK2 kinase inhibition in the MitoPark mouse model of PD [80]. Another third-generation inhibitor, PF-06685360 (also known as PFE-360), also showed higher kinase selectivity, improved oral bioavailability and good brain permeability with IC50 of 3 nM [82]. A pharmacokinetic experiment indicated that an oral dose of 7.5 mg/kg was necessary for this inhibitor to achieve full peripheral LRRK2 inhibition at both 1 h and 12 h time points after dosing in rats [82].
Taken together, LRRK2 kinase inhibitors have advanced from relatively non-selective inhibitors through to highly selective and potent compounds, and now, to those also capable of penetrating the BBB with few off-target effects. The development of newer LRRK2 kinase inhibitors with better efficacy profiles has provided impetus for further translation, including into human clinical trials.
3.2 Preclinical Safety of LRRK2 Kinase Inhibitors
One important aspect for translation of LRRK2 inhibitors to the clinic is a comprehensive understanding of any potential safety liabilities of such compounds. This is complicated as the exact biological function of LRRK2 has remained elusive. Moreover, LRRK2 is not a brain-specific protein, being also highly expressed in kidney, lung and immune cells in particular [21].
3.2.1 Kidney Lysosomal Phenotypes
To begin to address safety concerns, LRRK2 knockout (KO) rodents were employed to assess the physiological effects of complete LRRK2 loss of function. Combined studies of LRRK2 KO rodents suggest that LRRK2 has little effect on lifespan or general health, but aberrant lysosomal phenotypes can emerge in the kidney [83,84,85,86]. Enlarged vacuoles in renal tubules and impaired lysosomal-autophagy function with a subsequent accumulation of lipids in the kidney have been observed in LRRK2 KO rodents, but their renal function was still unremarkable [84,85,86]. Importantly, kidney phenotypes have not been observed in heterozygous LRRK2 KO mice [86], and no renal toxicity has been observed in rodents or nonhuman primates after treatment with second-generation LRRK2 kinase inhibitors for up to 29 days [87]. In contrast, rats treated with PF-06685360 administered orally at 7.5 mg/kg for up to 12 weeks, exhibited noticeable kidney darkening as well as progressive accumulation of hyaline droplets in the renal proximal tubules, resembling the pathology of the LRRK2 KO rats [82]. These changes were detectable following 2 weeks of treatment and were partially reversible within a 30-day treatment-free period [82]. In addition, no evidence of renal tubular injury or abnormalities in the urinalysis and serum kidney markers were observed after 12 weeks of treatment suggesting that PF-06685360 may not have kidney toxicity or adverse consequences for kidney function [82]. Moreover, MitoPark mice receiving MLi-2 at 30 mg/kg per day for 15 weeks did not exhibit kidney darkening or accumulation of pigment in renal cortical tubules [80], and in nonhuman primates, LRRK2 inhibitor treatment did not induce kidney deficits [87]. One caveat in using LRRK2 KO models to mimic LRRK2 kinase inhibitor-induced phenotypes is that kinase activity is only one of the main functions of LRRK2 and phenotypes observed in KO animals might be attributed to the loss of its non-kinase functions such as GTPase, dimerisation or scaffolding. Collective evidence thus suggests that aberrant kidney phenotypes are unlikely to be a significant liability for LRRK2 kinase inhibitors.
3.2.2 Lung Lysosomal Phenotypes
In contrast to kidney, preclinical toxicity studies showed that a number of earlier LRRK2 kinase inhibitors could induce morphological changes and abnormal cytoplasmic accumulation of lysosome-related lamellar bodies in type II pneumocytes in lungs of rodents and nonhuman primates [86, 87]. Although, not all inhibitors, like the second-generation GNE-7915, GNE-0877 or PF-06447475, revealed pathological changes in the lungs of rodents [49, 87]. MitoPark mice receiving MLi-2 at 30 mg/kg per day for 15 weeks displayed randomly scattered and very slight enlargement of type II pneumocytes in the lungs [80]. Non-human primates receiving either of three structurally diverse compounds MLi-2, PF-06685360 and GNE-7915, also developed mild accumulation of lamellar bodies in type II pneumocytes following 2 weeks of high-dose treatment [88]. Collectively, these results indicate this phenotype as an on-target effect of LRRK2 kinase inhibition. Importantly however, lower doses of PF-06685360 and MLi-2, that still substantially inhibited LRRK2 in brain, failed to induce lung pathology in the nonhuman primates [88]. Moreover, lung pathology induced by high-dose treatment was reversible upon drug withdrawal and no deficits in pulmonary function were detected, even at the highest doses employed [88]. Thus, preclinical toxicology studies do not preclude the ongoing clinical study of LRRK2 inhibitor therapeutics and suggest that a safety margin can be achieved in which brain LRRK2 is inhibited without effects on lung pathology.
3.2.3 Immune Cell Phenotypes
The highest expression of LRRK2 appears to be in inflammatory regulating immune cells, particularly monocytes and neutrophils [89,90,91]. Intriguingly, LRRK2 variants have also been implicated in an increased risk of developing Crohn’s disease [92, 93] and to an increased susceptibility to infection [94, 95], suggesting a biological function of LRRK2 in immunity [96]. Indeed, LRRK2 has been implicated in monocyte maturation and function [97,98,99], innate immune signalling pathways [93, 100, 101] and pathogen clearance [89, 102, 103]. An initial study showed reduced LRRK2 expression in peripheral mononuclear cells from a small cohort of subjects with the T2397M Crohn’s disease risk mutation [93]. That LRRK2 loss of function may be associated with immune phenotypes has further been demonstrated using LRRK2 KO mice, which are more susceptible to models of inflammatory bowel disease and Salmonella infection [93, 102]. However, LRRK2-mediated immune regulation is likely a more complicated story. Indeed, a recent study has demonstrated that LRRK2 KO mice are better able to clear Mycobacterium tuberculosis [103]. Moreover, treatment of murine macrophages with the second-generation GSK2578215A LRRK2 inhibitor promoted clearance of M. tuberculosis [103], but in another study, the same inhibitor impaired clearance of Salmonella typhimurium [102]. Meanwhile, the activating LRRK2 G2019S mutation has been associated with potentiated peripheral inflammation in asymptomatic carriers [104] and potentiated neuroinflammation in an overexpressing mouse model [105], with some studies, but certainly not all, reporting anti-inflammatory effects of LRRK2 inhibitors. That both activation and inhibition of LRRK2 are implicated in immune function suggests a potential, and likely complicated role for LRRK2 in immune homeostasis. The extent to which this is of concern for advancing LRRK2 inhibitors as PD therapeutics is still unclear and remains an interesting and active area of research.
In summary, preclinical findings to date do not necessarily preclude the advancement of LRRK2 kinase inhibitors to the next stages of drug development. However, a better understanding of LRRK2 biology would further aid in anticipating side effects, and it may be important to determine a therapeutic window of drug administration and/or consideration of cyclic therapies, particularly for long-term use as a treatment for PD. Indeed, determining safely tolerated levels of LRRK2 inhibitor is currently a goal of the first Phase 1 clinical trials currently being performed.
3.3 Early Phase 1 Clinical Trials
Although a majority of LRRK2 kinase inhibitors remain in late stages of preclinical evaluation, favourable candidates have now entered phase 1 clinical trials to determine the safety and dosage profiles in healthy volunteers. Recently, the DNL201 LRRK2 inhibitor from Denali Therapeutics reported positive results from its safety, pharmacokinetic, and pharmacodynamic data [106]. In this randomised, double-blind, placebo-controlled first in human phase 1a study, 122 healthy subjects received either single or multiple ascending oral doses of DNL201 or placebo. The treatment was safe and well tolerated without serious adverse events at a drug level that resulted in a greater than 90% of inhibition of peripheral LRRK2 activity. This inhibitor also reached high concentrations in cerebrospinal fluid (CSF), suggesting good brain penetration [106]. Detailed clinical data from this study are currently unavailable but outcomes disclosed to date suggest a promising outlook for further drug development and clinical testing. Indeed, an ongoing DNL201 phase 1b study (NCT03710707) is proposed to be performed in mild-to-moderate PD patients with and without a LRRK2 mutation and is designed to last for 28 days [107]. Meanwhile, Denali Therapeutics has announced that its second LRRK2 inhibitor, DNL-151, is now being tested in a phase 1 dose escalation study of healthy volunteers in the Netherlands [106]. Understandably there is substantial interest in the outcomes of these trials and whether LRRK2 inhibitors can progress to phase 2.
3.4 Some Remaining Challenges for Clinical Translation
3.4.1 LRRK2 Activity Readouts and Biomarkers
In spite of the great progress in clinical development of LRRK2 kinase inhibitors, ongoing translation is still facing hurdles. One of which is a lack of suitable biomarkers that could be monitored to assess the target engagement and subsequent efficacy of LRRK2 inhibitors. To date, the most widely used pharmacodynamic biomarker of LRRK2 target engagement is to measure LRRK2 phosphorylation levels on serine residues Ser910 and Ser935 [108]. These two residues, which regulate LRRK2 binding to 14-3-3 family adaptor proteins [109], are constitutively phosphorylated on LRRK2 and even though they are not autophosphorylation sites, constitutive phosphorylation of wild-type LRRK2 requires kinase activity [108]. Consequently, all tested LRRK2 inhibitors cause loss of phosphorylation at these sites in around 1–2 h, and in a dose-dependent manner in all cell and animal models tested. Two additional residues located nearby, Ser955 and Ser973 appear to be regulated in a similar manner [110], and all four of these phosphorylation sites have been demonstrated to be LRRK2 inhibitor sensitive in peripheral blood mononuclear cells from control and sporadic PD subjects treated ex vivo with LRRK2 inhibitors [111]. However, exactly how LRRK2 kinase activity regulates LRRK2 phosphorylation at these residues is largely unknown. It has been suggested as an indirect regulation via either phosphatases [112], or upstream kinases other than LRRK2 itself [100, 113, 114]. Thus, LRRK2 dephosphorylation at Ser910, Ser935, Ser955 and Ser973 remains an indirect measure of LRRK2 kinase activity and requires careful validation owing to the intricate regulation. Additionally, pathogenic LRRK2 mutations in the ROC-COR domain result in a loss of constitutive phosphorylation of the Ser910, Ser935, Ser955 and Ser973 residues, making this an unsuitable readout of inhibitor engagement in these patients [108, 109]. Another method for assessing LRRK2 kinase activity is to measure levels of LRRK2 phosphorylation at Ser1292 [35]. In contrast to Ser910, Ser935, Ser955 and Ser973, Ser1292 is a physiological autophosphorylation site with its dephosphorylation thus a direct consequence of LRRK2’s own kinase activity. Although Ser1292 phosphorylation could be detected in urinary exosomes from sporadic PD patients [115], the very low stoichiometry of phosphorylation on this residue and consequential difficulties with its robust detection has limited its practical utilisation in other biospecimens. Most recently, phosphorylation of the direct LRRK2 substrate Rab10 at Thr73 has been evaluated as a biomarker readout of LRRK2 activity. Rab10 Thr73 phosphorylation decreases in a dose-dependent manner with LRRK2 inhibitors in cell and animal models, including in peripheral immune cells from both healthy controls [36] and PD patients [91, 116]. Indeed, the use of Rab10 Thr73 phosphorylation in combination with LRRK2 Ser935 provides a seemingly robust readout of peripheral LRRK2 inhibitor target engagement. However, the extent to which Rab10 Thr73 phosphorylation is a biomarker of increased LRRK2 activity is less clear. As outlined above, LRRK2-mediated Rab phosphorylation contains a number of potential rate-limiting steps including membrane recruitment, allosteric interactions and guanidine nucleotide binding [41, 42]. Moreover, Rab10 phosphorylation was not significantly increased in peripheral immune cells from a small cohort of LRRK2 G2019S [91], and in peripheral immune cells from sporadic PD patients there was no correlation between LRRK2 levels and Rab10 phosphorylation [116]. It is also important to note that there are currently no validated central readouts of LRRK2 activity or inhibitor target engagement. In this regard, the development of CSF biomarkers is still ongoing, as are efforts to generate imaging agents [117, 118]. In addition to direct measures of target engagement, there is also much interest in developing specific assays that can determine the extent to which inhibitors are impacting on LRRK2-mediated pathobiology. Again, such biomarkers remain to be fully validated and LRRK2 biology to be better understood, but mitochondrial function, lysosomal function and inflammation are continually implicated in LRRK2 pathobiology and may provide key readouts of LRRK2 inhibitor efficacy suitable for a clinical trial time frame.
3.4.2 Who to Recruit for Trials and When
There are some important considerations for patient recruitment in clinical trials, specifically regarding suitable patient populations and the appropriate therapeutic window for intervention. That is, will LRRK2 inhibitors have disease-modifying effects at later stages of PD, will they be most efficacious upon diagnosis of PD, or indeed should they be given before the onset of clinical symptoms? The incomplete understanding of LRRK2 inhibitor mechanisms of action makes it difficult to determine the extent to which inhibiting LRRK2 will be efficacious in advanced disease with substantial Lewy pathology, neuroinflammation and neurodegeneration. Additionally, careful assessment of LRRK2 levels in post-mortem brain tissue of PD patients with and without LRRK2 mutations suggests a reduction in LRRK2 protein at end-stage PD, decreasing the size of the LRRK2 pool that might be actively targeted with inhibitor therapy [119, 120]. Declining LRRK2 protein levels in brain were associated with increasing disease duration, while interestingly, an upregulation of LRRK2 protein in brain tissue from patients with restricted Lewy body disease was observed [119, 120]. Restricted Lewy body cases are a potential prodromal PD cohort [121] and the prospect of early treatment to prevent neurodegeneration is clearly exciting. However, the treatment of mutation carriers before the onset of clinical symptoms is complicated by the relatively low and incomplete disease penetrance of LRRK2 mutations [22, 122, 123]. Thus, to facilitate asymptomatic treatment, predictors for pheno-conversion to clinical PD first need to be identified. Currently proposed clinical prodromal markers involve subtle non-motor symptoms [124], arm swing asymmetry [125], axial rotation smoothness [125] and dopaminergic nigrostriatal denervation [126], while inflammatory profiling or uric acid levels may also identify LRRK2 G2019S mutation carriers at higher risk of developing PD [104, 127]. However, longitudinal follow up of mutation cohorts is still required to validate the sensitivity and specificity of any identified markers. Finally, there are also considerations regarding the LRRK2 mutation carriers themselves. The pathogenic mutations in the ROC-COR domain increase LRRK2 activity the most, and potentially subjects with these mutations will show the most benefit with LRRK2 inhibitors over a clinical trial timeframe. However, these mutations are relatively rare and finding a sufficient cohort for a properly powered trial may be difficult. Indeed, even with G2019S subjects included, recruitment for a clinical trial is likely to require a coordinated international effort. Consequently, there is also interest in identifying other PD patient populations that may benefit from LRRK2 inhibitor therapy.
4 LRRK2 Inhibitors as Potential Therapeutics Beyond LRRK2-Mutation Carriers
4.1 LRRK2 Inhibitors in Other Genetic Forms of PD
LRRK2 has been implicated to share common biological pathways with other PD-associated genes, leading to suggestions that LRRK2 therapeutic strategies may have potential for PD patients beyond those carrying LRRK2 mutations.
4.1.1 LRRK2 and VPS35
Missense mutations in vacuolar protein sorting associated protein 35 (VPS35) were linked to PD in 2011 [128, 129]. VPS35 is a member of the retromer complex, which controls endosomal protein trafficking, and is particularly important for membrane protein recycling between endosomes and the trans Golgi network [130]. Like pathogenic LRRK2 mutations, VPS35 mutations are autosomal dominant and carriers share close clinical phenotypes, in terms of age at onset and clinical symptoms, to sporadic PD [131]. The most common pathogenic VPS35 mutation is D620N, which is proposed to induce a loss of function [132, 133]. Drosophila studies have functionally linked LRRK2 and VPS35 indicating convergence on the same intracellular vesicle trafficking pathway in the endosomal-lysosomal/Golgi sorting system to mediate neurodegeneration [134, 135]. Altered VPS35 levels and evidence of retromer dysfunction have also been observed in post-mortem brain tissue from LRRK2 mutation carriers [120]. Importantly, biochemical studies show that LRRK2-mediated Rab10 phosphorylation is enhanced in the presence of D620N mutant VPS35, and suppressed in the absence of VPS35 [136]. That mutant VPS35 may directly modulate LRRK2 activity suggests that PD patients with VPS35 mutations may also potentially benefit from LRRK2 inhibitor treatment.
4.1.2 LRRK2 and PINK1/Parkin
Loss of function missense mutations in both PTEN-induced kinase 1 (PINK1) and Parkin are the most common cause of recessively inherited early-onset forms of PD [12, 137], with both proteins important for the regulation of mitophagy and mitochondrial quality control [138]. LRRK2 has been linked to mitochondrial impairments in PD patients with the G2019S mutation [139], and also to the regulation of PINK1/Parkin-dependent mitophagy in patient derived fibroblast and induced pluripotent stem cell models [140, 141]. Importantly, the LRRK2-IN-1 inhibitor was able to attenuate impaired PINK1/Parkin-mediated mitophagy in fibroblasts from LRRK2 G2019S mutation patients [140]. The expression of LRRK2 has been reported to be modulated by levels of PINK1, being increased in fibroblasts from PINK1 mutation carriers and decreased upon PINK1 overexpression [142]. LRRK2 inhibitors can also reduce valinomycin-induced mitochondrial dysfunction in cells derived from patients carrying PINK1, Parkin or LRRK2 mutations [62, 143]. Intriguingly, Rab GTPases are also downstream substrates of PINK1 [144] suggesting further convergence with the LRRK2 pathway. Thus, LRRK2 kinase inhibitors may also have potential for rescuing mitochondrial defects in PINK1/Parkin-associated PD patients.
4.1.3 LRRK2 and GBA
Another potentially interesting subcategory of PD patients are those with missense mutations in lysosomal glucocerebrosidase (GBA). Homozygous GBA mutations cause the lysosomal storage disorder Gaucher’s disease, which also results in a higher incidence of PD [145,146,147]. Indeed, heterozygous missense mutations in GBA are now considered one of the more common established risk factors for PD, with a penetrance similar to LRRK2 mutations [148, 149]. Apart from a higher risk of cognitive decline [150], GBA mutation carriers again show a similar clinical disease course to both sporadic and LRRK2-associated PD. Although GBA and LRRK2 are yet to be directly functionally linked, it is apparent that both proteins show substantial biological overlap. Both enzymes are highly expressed in monocytes [89, 90, 151,152,153,154], both have been implicated in (i) immune function and inflammation [92, 93, 96, 153, 155, 156], (ii) lysosomal function [157,158,159,160,161], (iii) α-synuclein pathology [51, 161, 162], and missense mutations alter the activities of both enzymes increasing the risk of developing PD. Whether there is a direct interaction between these two proteins is an active area of investigation. If LRRK2 kinase inhibitors also benefit PD patients with GBA mutations, then the pool of patients that may be therapeutically targeted will be substantially increased.
4.2 LRRK2 Inhibitors in Sporadic PD
As outlined in Sect. 2, genome-wide association studies have identified variations in non-coding regions in and around the LRRK2 locus that comprise increased risk factors for sporadic PD, with the prevalence ranging from around 2 to 40% in certain populations [163, 164]. Given that LRRK2-associated PD patients also present with clinical and neuropathological profiles largely indistinguishable from late-onset idiopathic PD [19], an exciting prospect is whether more common sporadic PD could also potentially be treated with LRRK2 kinase inhibitors. Technical challenges have made assessing LRRK2 activity difficult, but recent advances do allude to a potential role in sporadic PD. In particular, Di Maio et al. developed a proximity ligation assay to improve detection of LRRK2 Ser1292 and show that its phosphorylation was increased in the remaining dopamine neurons from sporadic PD post-mortem brain tissue [165]. Cross validation of these results and extension to other brain regions is still required, moreover it remains unknown if brain LRRK2 activity is increased earlier in PD pathogenesis or just at end-stage disease. The Ser1292 proximity ligation assay also revealed that LRRK2 activity in rodent brain was increased by oligomeric α-synuclein [165]. Additionally, studies in sporadic PD-relevant cell and rodent α-synuclein models suggest a neuroprotective effect of LRRK2 inhibitors [49, 50, 143, 166], although this has not always been observed [167, 168]. LRRK2 protein levels were also found to be increased in sporadic PD patient immune cells [116, 169], although the specificity for PD and the way in which increased immune cell LRRK2 relates to PD pathogenesis, still remains to be determined.
Also of interest to sporadic PD is the emerging link between LRRK2 and Rab29 (also known as Rab7L1) risk variants. Genome-wide association studies first identified the PARK16 region as modulating the risk of sporadic PD [163, 170, 171]. The PARK16 locus contains a number of candidate genes, and although not conclusive, functional evidence implicates Rab29 as a candidate risk gene. In particular, elegant functional studies in cell and drosophila models demonstrated that modulating Rab29 expression could rescue PD phenotypes induced by LRRK2 G2019S [135]. Moreover, Rab29 was found to directly interact with LRRK2 [135]. The LRRK2–Rab29 interaction occurred at the Golgi apparatus, an observation that has been independently confirmed by a number of research groups [41, 172,173,174]. Pathogenic LRRK2 variants are more efficiently recruited to Golgi bound Rab29 and impact upon Golgi morphology, retromer trafficking, autophagy and centrosomal function, with a number of phenotypes dependent on LRRK2 kinase activity [41, 135, 172, 173]. Further strengthening this interaction is that Rab29 protein levels can increase LRRK2 activity [42], and that Rab29 is in turn phosphorylated by LRRK2 [41]. Importantly, genetic evidence suggests that LRRK2 and Rab29 variants function together to modulate PD, further implicating LRRK2 dysfunction in sporadic PD, or at least in a subset of patients [135]. It is certainly early days with regard to the potential use of LRRK2 inhibitors in sporadic PD; however, assessing LRRK2 activity and function in sporadic PD patients will be an important area of investigation as clinical trials of LRRK2 inhibitors progress.
5 Summary
Accumulating evidence suggests that increased LRRK2 kinase activity may play an important role in PD pathogenesis. Consequently, there is substantial interest in blocking LRRK2 kinase activity as a potential treatment for PD. Through a concerted international effort involving close relationships between academia and industry, over the past decade LRRK2 drug development has progressed remarkably, to a point where potent and selective inhibitors have yielded positive initial results in preclinical studies and early-stage clinical trials. Preclinical studies suggest a neuroprotective role of these drugs, and potential safety concerns in peripheral tissues such as lung, appear manageable although a complete understanding of LRRK2 pathobiology and mechanisms of action of LRRK2 inhibitors remain elusive. In addition, biomarker strategies to measure efficacy of LRRK2 inhibitors need to improve to ensure successful implementation of drugs in clinical trials. Moreover, it will also be important to define patient populations, safe doses and therapeutic windows for optimal design of clinical trials for treating PD patients with, and possibly beyond, LRRK2 mutations. In particular, further work examining LRRK2 activity and function in sporadic PD is warranted to determine if this major PD population, or at least a subset thereof, may also benefit from LRRK2 inhibitor-based therapies.
References
Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909.
Ross GW, Petrovitch H, Abbott RD, Nelson J, Markesbery W, Davis D, et al. Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann Neurol. 2004;56(4):532–9.
Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896–912.
Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 2006;5(3):235–45.
Halliday G, Lees A, Stern M. Milestones in Parkinson’s disease—clinical and pathologic features. Mov Disord. 2011;26(6):1015–21.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–40.
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211.
Jellinger KA. A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol. 2008;116(1):1–16.
Parkkinen L, Pirttila T, Alafuzoff I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008;115(4):399–407.
Gasser T, Hardy J, Mizuno Y. Milestones in PD genetics. Mov Disord. 2011;26(6):1042–8.
Houlden H, Singleton AB. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012;124(3):325–38.
Lill CM. Genetics of Parkinson’s disease. Mol Cell Probes. 2016;30(6):386–96.
Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9(8):445–54.
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7.
Domingo A, Klein C. Genetics of Parkinson disease. Handb Clin Neurol. 2018;147:211–27.
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat. 2010;31(7):763–80.
Paisan-Ruiz C, Lewis PA, Singleton AB. LRRK2: cause, risk, and mechanism. J Parkinsons Dis. 2013;3(2):85–103.
Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–90.
Haugarvoll K, Rademakers R, Kachergus JM, Nuytemans K, Ross OA, Gibson JM, et al. Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology. 2008;70(16 Pt 2):1456–60.
Atashrazm F, Dzamko N. LRRK2 inhibitors and their potential in the treatment of Parkinson’s disease: current perspectives. Clin Pharmacol. 2016;8:177–89.
Cookson MR. LRRK2 pathways leading to neurodegeneration. Curr Neurol Neurosci Rep. 2015;15(7):42.
Harvey K, Outeiro TF. The role of LRRK2 in cell signalling. Biochem Soc Trans. 2019;47(1):23–44.
Monfrini E, Di Fonzo A. Leucine-rich repeat kinase (LRRK2) genetics and Parkinson’s disease. Adv Neurobiol. 2017;14:3–30.
Hernandez DG, Reed X, Singleton AB. Genetics in Parkinson disease: mendelian versus non-mendelian inheritance. J Neurochem. 2016;139(Suppl 1):59–74.
Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci. 2010;11(12):791–7.
Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med. 2006;354(4):422–3.
Gorostidi A, Ruiz-Martinez J, de Munain LA, Alzualde A, Marti Masso JF. LRRK2 G2019S and R1441G mutations associated with Parkinson’s disease are common in the Basque Country, but relative prevalence is determined by ethnicity. Neurogenetics. 2009;10(2):157–9.
Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology. 2004;62(9):1619–22.
Berg D, Schweitzer KJ, Leitner P, Zimprich A, Lichtner P, Belcredi P, et al. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease. Brain. 2005;128(Pt 12):3000–11.
Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, Yagishita S. Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord. 2009;15(4):300–6.
Ujiie S, Hatano T, Kubo S, Imai S, Sato S, Uchihara T, et al. LRRK2 I2020T mutation is associated with tau pathology. Parkinsonism Relat Disord. 2012;18(7):819–23.
Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, et al. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J. 2007;405(2):307–17.
West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, et al. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005;102(46):16842–7.
Sheng Z, Zhang S, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med. 2012;4(164):164ra1.
Thirstrup K, Dachsel JC, Oppermann FS, Williamson DS, Smith GP, Fog K, et al. Selective LRRK2 kinase inhibition reduces phosphorylation of endogenous Rab10 and Rab12 in human peripheral mononuclear blood cells. Sci Rep. 2017;7(1):10300.
Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016. https://doi.org/10.7554/elife.12813.
Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–25.
Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife. 2017. https://doi.org/10.7554/elife.31012.
Ito G, Katsemonova K, Tonelli F, Lis P, Baptista MA, Shpiro N, et al. Phos-tag analysis of Rab10 phosphorylation by LRRK2: a powerful assay for assessing kinase function and inhibitors. Biochem J. 2016;473(17):2671–85.
Liu Z, Bryant N, Kumaran R, Beilina A, Abeliovich A, Cookson MR, et al. LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum Mol Genet. 2018;27(2):385–95.
Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, et al. Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J. 2018;37(1):1–18.
Rudenko IN, Kaganovich A, Hauser DN, Beylina A, Chia R, Ding J, et al. The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem J. 2012;446(1):99–111.
Daniel G, Moore DJ. Modeling LRRK2 pathobiology in Parkinson’s disease: from yeast to rodents. Curr Top Behav Neurosci. 2015;22:331–68.
Martin I, Kim JW, Dawson VL, Dawson TM. LRRK2 pathobiology in Parkinson’s disease. J Neurochem. 2014;131(5):554–65.
Cookson MR. Mechanisms of mutant LRRK2 neurodegeneration. Adv Neurobiol. 2017;14:227–39.
Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med. 2010;16(9):998–1000.
Xiong Y, Neifert S, Karuppagounder SS, Liu Q, Stankowski JN, Lee BD, et al. Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc Natl Acad Sci USA. 2018;115(7):1635–40.
Daher JP, Abdelmotilib HA, Hu X, Volpicelli-Daley LA, Moehle MS, Fraser KB, et al. Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates alpha-synuclein gene-induced neurodegeneration. J Biol Chem. 2015;290(32):19433–44.
Bae EJ, Kim DK, Kim C, Mante M, Adame A, Rockenstein E, et al. LRRK2 kinase regulates alpha-synuclein propagation via RAB35 phosphorylation. Nat Commun. 2018;9(1):3465.
Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JP, Milnerwood AJ, et al. G2019S-LRRK2 expression augments alpha-synuclein sequestration into inclusions in neurons. J Neurosci. 2016;36(28):7415–27.
Lavalley NJ, Slone SR, Ding H, West AB, Yacoubian TA. 14-3-3 Proteins regulate mutant LRRK2 kinase activity and neurite shortening. Hum Mol Genet. 2016;25(1):109–22.
Qin Q, Zhi LT, Li XT, Yue ZY, Li GZ, Zhang H. Effects of LRRK2 inhibitors on nigrostriatal dopaminergic neurotransmission. CNS Neurosci Ther. 2017;23(2):162–73.
Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, et al. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. 2012;32(5):1602–11.
Longo F, Mercatelli D, Novello S, Arcuri L, Brugnoli A, Vincenzi F, et al. Age-dependent dopamine transporter dysfunction and Serine129 phospho-alpha-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol Commun. 2017;5(1):22.
Giesert F, Glasl L, Zimprich A, Ernst L, Piccoli G, Stautner C, et al. The pathogenic LRRK2 R1441C mutation induces specific deficits modeling the prodromal phase of Parkinson’s disease in the mouse. Neurobiol Dis. 2017;105:179–93.
Novello S, Arcuri L, Dovero S, Dutheil N, Shimshek DR, Bezard E, Morari M. G2019S LRRK2 mutation facilitates alpha-synuclein neuropathology in aged mice. Neurobiol Dis. 2018;120:21–33.
Volta M, Beccano-Kelly DA, Paschall SA, Cataldi S, MacIsaac SE, Kuhlmann N, et al. Initial elevations in glutamate and dopamine neurotransmission decline with age, as does exploratory behavior, in LRRK2 G2019S knock-in mice. Elife. 2017;6:e28377.
Weykopf B, Haupt S, Jungverdoben J, Flitsch LJ, Hebisch M, Liu GH, et al. Induced pluripotent stem cell-based modeling of mutant LRRK2-associated Parkinson’s disease. Eur J Neurosci. 2019;49(4):561–89.
Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature. 2012;491(7425):603–7.
Howlett EH, Jensen N, Belmonte F, Zafar F, Hu X, Kluss J, et al. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum Mol Genet. 2017;26(22):4340–51.
Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012;4(141):141ra90.
Schwab AJ, Ebert AD. Neurite aggregation and calcium dysfunction in iPSC-derived sensory neurons with parkinson’s disease-related LRRK2 G2019S mutation. Stem Cell Rep. 2015;5(6):1039–52.
West AB. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp Neurol. 2017;298(Pt B):236–45.
Christensen KV, Smith GP, Williamson DS. Development of LRRK2 inhibitors for the treatment of parkinson’s disease. Prog Med Chem. 2017;56:37–80.
Hatcher JM, Choi HG, Alessi DR, Gray NS. Small-molecule inhibitors of LRRK2. Adv Neurobiol. 2017;14:241–64.
Galatsis P. Leucine-rich repeat kinase 2 inhibitors: a patent review (2014–2016). Expert Opin Ther Pat. 2017;27(6):667–76.
Nichols RJ, Dzamko N, Hutti JE, Cantley LC, Deak M, Moran J, et al. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson’s disease. Biochem J. 2009;424(1):47–60.
Covy JP, Giasson BI. Identification of compounds that inhibit the kinase activity of leucine-rich repeat kinase 2. Biochem Biophys Res Commun. 2009;378(3):473–7.
Anand VS, Reichling LJ, Lipinski K, Stochaj W, Duan W, Kelleher K, et al. Investigation of leucine-rich repeat kinase 2: enzymological properties and novel assays. FEBS J. 2009;276(2):466–78.
Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat Chem Biol. 2011;7(4):203–5.
Ramsden N, Perrin J, Ren Z, Lee BD, Zinn N, Dawson VL, et al. Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson’s disease-related toxicity in human neurons. ACS Chem Biol. 2011;6(10):1021–8.
Yao C, Johnson WM, Gao Y, Wang W, Zhang J, Deak M, et al. Kinase inhibitors arrest neurodegeneration in cell and C. elegans models of LRRK2 toxicity. Hum Mol Genet. 2013;22(2):328–44.
Choi HG, Zhang J, Deng X, Hatcher JM, Patricelli MP, Zhao Z, et al. Brain penetrant LRRK2 inhibitor. ACS Med Chem Lett. 2012;3(8):658–62.
Hatcher JM, Zhang J, Choi HG, Ito G, Alessi DR, Gray NS. Discovery of a pyrrolopyrimidine (JH-II-127), a highly potent, selective, and brain penetrant LRRK2 inhibitor. ACS Med Chem Lett. 2015;6(5):584–9.
Reith AD, Bamborough P, Jandu K, Andreotti D, Mensah L, Dossang P, et al. GSK2578215A; a potent and highly selective 2-arylmethyloxy-5-substitutent-N-arylbenzamide LRRK2 kinase inhibitor. Bioorg Med Chem Lett. 2012;22(17):5625–9.
Estrada AA, Chan BK, Baker-Glenn C, Beresford A, Burdick DJ, Chambers M, et al. Discovery of highly potent, selective, and brain-penetrant aminopyrazole leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J Med Chem. 2014;57(3):921–36.
Estrada AA, Liu X, Baker-Glenn C, Beresford A, Burdick DJ, Chambers M, et al. Discovery of highly potent, selective, and brain-penetrable leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J Med Chem. 2012;55(22):9416–33.
Henderson JL, Kormos BL, Hayward MM, Coffman KJ, Jasti J, Kurumbail RG, et al. Discovery and preclinical profiling of 3-[4-(morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a highly potent, selective, brain penetrant, and in vivo active LRRK2 kinase inhibitor. J Med Chem. 2015;58(1):419–32.
Fell MJ, Mirescu C, Basu K, Cheewatrakoolpong B, DeMong DE, Ellis JM, et al. MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J Pharmacol Exp Ther. 2015;355(3):397–409.
Scott JD, DeMong DE, Greshock TJ, Basu K, Dai X, Harris J, et al. Discovery of a 3-(4-pyrimidinyl) Indazole (MLi-2), an orally available and selective leucine-rich repeat kinase 2 (LRRK2) inhibitor that reduces brain kinase activity. J Med Chem. 2017;60(7):2983–92.
Andersen MA, Wegener KM, Larsen S, Badolo L, Smith GP, Jeggo R, et al. PFE-360-induced LRRK2 inhibition induces reversible, non-adverse renal changes in rats. Toxicology. 2018;395:15–22.
Baptista MA, Dave KD, Frasier MA, Sherer TB, Greeley M, Beck MJ, et al. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS One. 2013;8(11):e80705.
Tong Y, Giaime E, Yamaguchi H, Ichimura T, Liu Y, Si H, et al. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol Neurodegener. 2012;7:2. https://doi.org/10.1186/1750-1326-7-2.
Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ 3rd, et al. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci USA. 2010;107(21):9879–84.
Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20(21):4209–23.
Fuji RN, Flagella M, Baca M, Baptista MA, Brodbeck J, Chan BK, et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci Transl Med. 2015;7(273):273ra15.
Baptista M, Merchant K, Barret T, Bryce D, Ellis M, Estrada A, Fell M, Fiske B, et al. LRRK2 kinase inhibitors induce a reversible effect in the lungs of non-human primates with no measurable pulmonary deficits. bioRxiv. 2018. https://doi.org/10.1101/390815.
Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185(9):5577–85.
Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, et al. Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm. 2011;118(5):795–808.
Fan Y, Howden AJM, Sarhan AR, Lis P, Ito G, Martinez TN, et al. Interrogating Parkinson’s disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils. Biochem J. 2018;475(1):23–44.
Hui KY, Fernandez-Hernandez H, Hu J, Schaffner A, Pankratz N, Hsu NY, et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med. 2018;10(423):eaa17795.
Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol. 2011;12(11):1063–70.
Marcinek P, Jha AN, Shinde V, Sundaramoorthy A, Rajkumar R, Suryadevara NC, et al. LRRK2 and RIPK2 variants in the NOD 2-mediated signaling pathway are associated with susceptibility to Mycobacterium leprae in Indian populations. PLoS One. 2013;8(8):e73103.
Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, et al. Genomewide association study of leprosy. N Engl J Med. 2009;361(27):2609–18.
Dzamko NL. LRRK2 and the immune system. Adv Neurobiol. 2017;14:123–43.
Bliederhaeuser C, Zondler L, Grozdanov V, Ruf WP, Brenner D, Melrose HL, et al. LRRK2 contributes to monocyte dysregulation in Parkinson’s disease. Acta Neuropathol Commun. 2016;4(1):123.
Speidel A, Felk S, Reinhardt P, Sterneckert J, Gillardon F. Leucine-rich repeat kinase 2 influences fate decision of human monocytes differentiated from induced pluripotent stem cells. PLoS One. 2016;11(11):e0165949.
Thevenet J, Pescini Gobert R, van Huijsduijnen HR, Wiessner C, Sagot YJ. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One. 2011;6(6):e21519.
Dzamko N, Inesta-Vaquera F, Zhang J, Xie C, Cai H, Arthur S, et al. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS One. 2012;7(6):e39132.
Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet. 2014;23(16):4201–14.
Liu W, Liu X, Li Y, Zhao J, Liu Z, Hu Z, et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J Exp Med. 2017;214(10):3051–66.
Hartlova A, Herbst S, Peltier J, Rodgers A, Bilkei-Gorzo O, Fearns A, et al. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J. 2018;37(12):e98694.
Dzamko N, Rowe DB, Halliday GM. Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Mov Disord. 2016;31(6):889–97.
Kozina E, Sadasivan S, Jiao Y, Dou Y, Ma Z, Tan H, et al. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain. 2018;141(6):1753–69.
Denali Therapeutics Announces Positive Clinical Results From LRRK2 Inhibitor Program For Parkinson’s Disease. http://investors.denalitherapeutics.com/news-releases/news-release-details/denali-therapeutics-announces-positive-clinical-results-lrrk2-ir-pages. Denali Therapeutics Inc. 2018. Accessed 26 Feb 2019.
Denali Therapeutics Announces First Patient Dosed in Phase 1b Study of DNL201 for Parkinson’s Disease. https://globenewswire.com/news-release/2018/12/10/1664447/0/en/Denali-Therapeutics-Announces-First-Patient-Dosed-in-Phase-1b-Study-of-DNL201-for-Parkinson-s-Disease.html. Denali Therapeutics Inc. 2018. Accessed 26 Feb 2019.
Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR, et al. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J. 2010;430(3):405–13.
Nichols RJ, Dzamko N, Morrice NA, Campbell DG, Deak M, Ordureau A, et al. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem J. 2010;430(3):393–404.
Doggett EA, Zhao J, Mork CN, Hu D, Nichols RJ. Phosphorylation of LRRK2 serines 955 and 973 is disrupted by Parkinson’s disease mutations and LRRK2 pharmacological inhibition. J Neurochem. 2012;120(1):37–45.
Perera G, Ranola M, Rowe DB, Halliday GM, Dzamko N. Inhibitor treatment of peripheral mononuclear cells from Parkinson’s disease patients further validates LRRK2 dephosphorylation as a pharmacodynamic biomarker. Sci Rep. 2016;6:31391.
Lobbestael E, Zhao J, Rudenko IN, Beylina A, Gao F, Wetter J, et al. Identification of protein phosphatase 1 as a regulator of the LRRK2 phosphorylation cycle. Biochem J. 2013;456(1):119–28.
Chia R, Haddock S, Beilina A, Rudenko IN, Mamais A, Kaganovich A, et al. Phosphorylation of LRRK2 by casein kinase 1alpha regulates trans-Golgi clustering via differential interaction with ARHGEF7. Nat Commun. 2014;5:5827.
Muda K, Bertinetti D, Gesellchen F, Hermann JS, von Zweydorf F, Geerlof A, et al. Parkinson-related LRRK2 mutation R1441C/G/H impairs PKA phosphorylation of LRRK2 and disrupts its interaction with 14-3-3. Proc Natl Acad Sci USA. 2014;111(1):E34–43.
Fraser KB, Rawlins AB, Clark RG, Alcalay RN, Standaert DG, Liu N, et al. Ser(P)-1292 LRRK2 in urinary exosomes is elevated in idiopathic Parkinson’s disease. Mov Disord. 2016;31(10):1543–50.
Atashrazm F, Hammond D, Perera G, Bolliger MF, Matar E, Halliday GM, et al. LRRK2-mediated Rab10 phosphorylation in immune cells from Parkinson’s disease patients. Mov Disord. 2019;34(3):406–15.
Malik N, Gifford AN, Sandell J, Tuchman D, Ding YS. Synthesis and in vitro and in vivo evaluation of [(3)H]LRRK2-IN-1 as a novel radioligand for LRRK2. Mol Imaging Biol. 2017;19(6):837–45.
Malik N, Tuchman D, Sandell J, Gifford A, Ding Y-S. Development of novel radioligands for imaging LRRK2 in Parkinson’s disease. J Nucl Med. 2018;59(supplement 1):1020.
Dzamko N, Gysbers AM, Bandopadhyay R, Bolliger MF, Uchino A, Zhao Y, et al. LRRK2 levels and phosphorylation in Parkinson’s disease brain and cases with restricted Lewy bodies. Mov Disord. 2017;32(3):423–32.
Zhao Y, Perera G, Takahashi-Fujigasaki J, Mash DC, Vonsattel JPG, Uchino A, et al. Reduced LRRK2 in association with retromer dysfunction in post-mortem brain tissue from LRRK2 mutation carriers. Brain. 2018;141(2):486–95.
DelleDonne A, Klos KJ, Fujishiro H, Ahmed Z, Parisi JE, Josephs KA, et al. Incidental Lewy body disease and preclinical Parkinson disease. Arch Neurol. 2008;65(8):1074–80.
Latourelle JC, Sun M, Lew MF, Suchowersky O, Klein C, Golbe LI, et al. The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s disease: the GenePD study. BMC Med. 2008;6:32.
Marder K, Wang Y, Alcalay RN, Mejia-Santana H, Tang MX, Lee A, et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology. 2015;85(1):89–95.
Mirelman A, Alcalay RN, Saunders-Pullman R, Yasinovsky K, Thaler A, Gurevich T, et al. Nonmotor symptoms in healthy Ashkenazi Jewish carriers of the G2019S mutation in the LRRK2 gene. Mov Disord. 2015;30(7):981–6.
Mirelman A, Bernad-Elazari H, Thaler A, Giladi-Yacobi E, Gurevich T, Gana-Weisz M, et al. Arm swing as a potential new prodromal marker of Parkinson’s disease. Mov Disord. 2016;31(10):1527–34.
Bergareche A, Rodriguez-Oroz MC, Estanga A, Gorostidi A, de Munain LA, Castillo-Trivino T, et al. DAT imaging and clinical biomarkers in relatives at genetic risk for LRRK2 R1441G Parkinson’s disease. Mov Disord. 2016;31(3):335–43.
Johansen KK, Wang L, Aasly JO, White LR, Matson WR, Henchcliffe C, et al. Metabolomic profiling in LRRK2-related Parkinson’s disease. PLoS One. 2009;4(10):e7551.
Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89(1):162–7.
Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89(1):168–75.
Seaman MN. The retromer complex—endosomal protein recycling and beyond. J Cell Sci. 2012;125(Pt 20):4693–702.
Trinh J, Zeldenrust FMJ, Huang J, Kasten M, Schaake S, Petkovic S, et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord. 2018;33(12):1857–70.
Malik BR, Godena VK, Whitworth AJ. VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with parkin. Hum Mol Genet. 2015;24(21):6106–17.
Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, et al. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun. 2014;5:3828.
Inoshita T, Arano T, Hosaka Y, Meng H, Umezaki Y, Kosugi S, et al. Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum Mol Genet. 2017;26(15):2933–48.
MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77(3):425–39.
Mir R, Tonelli F, Lis P, Macartney T, Polinski NK, Martinez TN, et al. The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem J. 2018;475(11):1861–83.
Kilarski LL, Pearson JP, Newsway V, Majounie E, Knipe MD, Misbahuddin A, et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease. Mov Disord. 2012;27(12):1522–9.
Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14.
Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75(22):2017–20.
Bonello F, Hassoun SM, Mouton-Liger F, Shin YS, Muscat A, Tesson C, et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson’s disease. Hum Mol Genet. 2019. https://doi.org/10.1093/hmg/ddz004.
Hsieh CH, Shaltouki A, Gonzalez AE, da Cruz BA, Burbulla LF, St Lawrence E, et al. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell. 2016;19(6):709–24.
Azkona G, de Maturana LR, Del Rio P, Sousa A, Vazquez N, Zubiarrain A, et al. LRRK2 expression is deregulated in fibroblasts and neurons from parkinson patients with mutations in PINK1. Mol Neurobiol. 2018;55(1):506–16.
Smith GA, Jansson J, Rocha EM, Osborn T, Hallett PJ, Isacson O. Fibroblast biomarkers of sporadic parkinson’s disease and LRRK2 kinase inhibition. Mol Neurobiol. 2016;53(8):5161–77.
Lai YC, Kondapalli C, Lehneck R, Procter JB, Dill BD, Woodroof HI, et al. Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. EMBO J. 2015;34(22):2840–61.
Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S, et al. The risk of Parkinson’s disease in type 1 Gaucher disease. J Inherit Metab Dis. 2010;33(2):167–73.
Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2004;351(19):1972–7.
Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, Barranger JA, et al. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N Engl J Med. 1987;316(10):570–5.
Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. 2012;78(6):417–20.
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–61.
Lerche S, Schulte C, Srulijes K, Pilotto A, Rattay TW, Hauser AK, et al. Cognitive impairment in glucocerebrosidase (GBA)-associated PD: not primarily associated with cerebrospinal fluid Abeta and Tau profiles. Mov Disord. 2017;32(12):1780–3.
Rieckmann JC, Geiger R, Hornburg D, Wolf T, Kveler K, Jarrossay D, et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol. 2017;18(5):583–93.
Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138(Pt 9):2648–58.
Atashrazm F, Hammond D, Perera G, Dobson-Stone C, Mueller N, Pickford R, et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci Rep. 2018;8(1):15446.
Berger J, Lecourt S, Vanneaux V, Rapatel C, Boisgard S, Caillaud C, et al. Glucocerebrosidase deficiency dramatically impairs human bone marrow haematopoiesis in an in vitro model of Gaucher disease. Br J Haematol. 2010;150(1):93–101.
Liu J, Halene S, Yang M, Iqbal J, Yang R, Mehal WZ, et al. Gaucher disease gene GBA functions in immune regulation. Proc Natl Acad Sci USA. 2012;109(25):10018–23.
Mizukami H, Mi Y, Wada R, Kono M, Yamashita T, Liu Y, et al. Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J Clin Investig. 2002;109(9):1215–21.
Henry AG, Aghamohammadzadeh S, Samaroo H, Chen Y, Mou K, Needle E, et al. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet. 2015;24(21):6013–28.
Roosen DA, Cookson MR. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol Neurodegener. 2016;11(1):73. https://doi.org/10.1186/s13024-016-0140-1.
Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci USA. 2018;115(39):E9115–24.
Gan-Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy. 2015;11(9):1443–57.
Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, et al. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain. 2014;137(Pt 3):834–48.
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16(4):394–406.
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303–7.
Sharma M, Ioannidis JP, Aasly JO, Annesi G, Brice A, Van Broeckhoven C, et al. Large-scale replication and heterogeneity in Parkinson disease genetic loci. Neurology. 2012;79(7):659–67.
Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci Transl Med. 2018;10(451):eaar5429.
Andersen MA, Christensen KV, Badolo L, Smith GP, Jeggo R, Jensen PH, et al. Parkinson’s disease-like burst firing activity in subthalamic nucleus induced by AAV-alpha-synuclein is normalized by LRRK2 modulation. Neurobiol Dis. 2018;116:13–27.
Daher JP, Pletnikova O, Biskup S, Musso A, Gelhaar S, Gatler D, Troncoso JC, Lee MK, Dawson TM, Dawson VL, Moore DJ. Neurodegenerative phenotypes in an A53T alpha-synuclein transgenic mouse model are independent of LRRK2. Hum Mol Genet. 2012;21(11):2420–31.
Henderson MX, Sengupta M, McGeary I, Zhang B, Olufemi MF, Brown H, Trojanowski JQ, Lee VMY. LRRK2 inhibition does not impart protection from alpha-synuclein pathology and neuron death in non-transgenic mice. Acta Neuropathol Commun. 2019;7(1):28.
Cook DA, Kannarkat GT, Cintron AF, Butkovich LM, Fraser KB, Chang J, et al. LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinsons Dis. 2017;3:11.
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–12.
Tucci A, Nalls MA, Houlden H, Revesz T, Singleton AB, Wood NW, et al. Genetic variability at the PARK16 locus. Eur J Hum Genet. 2010;18(12):1356–9.
Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci USA. 2014;111(7):2626–31.
Madero-Perez J, Fernandez B, Lara Ordonez AJ, Fdez E, Lobbestael E, Baekelandt V, et al. RAB7L1-Mediated relocalization of LRRK2 to the golgi complex causes centrosomal deficits via RAB8A. Front Mol Neurosci. 2018;11:417.
Fujimoto T, Kuwahara T, Eguchi T, Sakurai M, Komori T, Iwatsubo T. Parkinson’s disease-associated mutant LRRK2 phosphorylates Rab7L1 and modifies trans-Golgi morphology. Biochem Biophys Res Commun. 2018;495(2):1708–15.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was specifically received for the publication of this review
Conflict of interest
ND receives funding for Parkinson’s disease research, including on LRRK2, from the National Health and Medical Research Council (NHMRC) (#1103757), the Michael J Fox Foundation for Parkinson’s disease research (MJFF), the Shake It Up Australia Foundation and the University of Sydney. In the past 12 months ND has received travel support from Denali Therapeutics, Neuropore therapies and MJFF and received payments for Grant reviews from the NHMRC and IRC. ND is a co-inventor and has received royalties from a patent on the use of LRRK2 phosphorylation sites as pharmacodynamic biomarkers (WO2011131980A1). YZ has no conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Zhao, Y., Dzamko, N. Recent Developments in LRRK2-Targeted Therapy for Parkinson’s Disease. Drugs 79, 1037–1051 (2019). https://doi.org/10.1007/s40265-019-01139-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-019-01139-4