
Abstract
Over the past 15 years, targeted therapy has revolutionized the systemic treatment of cancer. In parallel, there has been a growing debate on the choice of end points in clinical trials in oncology. This debate basically hinges on the choice between overall survival (OS) and progression-free survival (PFS). PFS is advantageous because it is measured earlier than OS, requires a smaller sample size than OS to achieve the desired power, and is not influenced by cross-over. On the other hand, PFS is prone to measurement error and bias, and may not capture the entire treatment effect on the outcomes of most interest to patients with an incurable disease: a prolonged survival and improved quality of life. Therefore, how can we choose between two imperfect end points? The answer to this question would certainly be made easier if PFS could be demonstrated to be a valid surrogate for OS. The validation of a surrogate end point is best made using individual-patient data (IPD) from randomized trials, which allows for standardized assessments of the patient-level and the trial-level correlations between surrogate and final end points. Proper IPD meta-analytical evaluations for targeted agents have still been rare, and to our knowledge only three studies on this topic are currently available in the metastatic setting: one in breast cancer, one in colorectal cancer and one in lung cancer. Although these three studies suffer from limitations inherent to the availability of IPD and the design of the original clinical trials, they have not been able to validate PFS as surrogate for OS, because only modest correlations were found between these two end points, both at the patient and at the trial level. Even if properly conducted surrogate-endpoint evaluations have thus far been unsuccessful, these evaluations are a step in the right direction and can be expected to be applied on a much larger scale in the era of data sharing of clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Overall survival and progression-free survival have advantages and disadvantages as end points for randomized trials in oncology; progression-free survival, which is more efficient as an end point, may replace overall survival if it is validated as a surrogate for the latter. |
Validation of a surrogate end point is more reliable if made using individual patient data in a meta-analysis of randomized clinical trials, but this process is time consuming and not always successful for reasons that are still under debate. |
Targeted agents have improved the outcomes for patients with various types of cancer, but there have been only a few attempts to validate progression-free survival as a surrogate for overall survival in the setting of targeted therapies. |
1 Introduction
Targeted therapy of cancer entails the use of agents that are capable of modulating the function of well-defined molecules with a critical role in tumor pathogenesis. A targeted approach is more likely to be effective when drugs are used alongside predictive biomarkers that allow for selection of patients more likely to benefit from therapy, a strategy nowadays referred to as personalized (or “precision” or “stratified”) therapy [1–7]. Targeted therapy has revolutionized the systemic treatment of cancer, and most of the drugs approved over the past 15 years are targeted agents. In parallel to the emergence of targeted therapy, there has been a growing debate on the choice of end points in clinical trials in oncology, because different end points display advantages and disadvantages in their ability to capture the improvement in prognosis brought on by novel therapies. This concern basically hinges on the choice between overall survival (OS) and progression-free survival (PFS) as primary end points in phase 3 trials in advanced cancer. Although the debate on the comparative merits of PFS and OS was already at play in the chemotherapy era [8, 9], it has been fueled in recent years by situations in which highly effective targeted agents (as judged by improvements in tumor response rates and PFS) were not found to improve OS in clinical trials [10, 11]. In these and other instances, gains in PFS have been considered as a sufficient basis for regulatory approval, but questions have remained about the ability of these agents to also improve OS. The disconnect between PFS and OS results within trials may be due to various factors, including cross-over or use of other treatments after disease progression, but it raises the question of whether PFS can be considered a surrogate for OS. Given the important role currently played by targeted therapies in the anticancer armamentarium, and the fact that many targeted therapies have been approved on the basis of gains in PFS (and tested in trials that were powered for PFS but not OS), the issue of surrogacy may gain increasing prominence in the era of targeted agents. In the current paper, we explore the potential role of PFS as a surrogate for OS in trials of targeted therapy for advanced cancer, the results obtained so far, and future lines of research on this topic. Of note, we do not discuss immunotherapy, a setting for which the relationship between PFS and OS is less clear at this point [12].
2 Advantages and Disadvantages of OS and PFS
Historically, OS has been the gold-standard primary end point in phase 3 trials in advanced cancer. OS is the most objective end point for assessing the efficacy of anticancer treatment, and—alongside quality of life—the most relevant measure of patient benefit [13]. However, the use of OS as primary end point is increasingly challenging because of the large sample sizes and extended follow-up that are required in trials designed with the primary aim of assessing improvements in OS [14]. Moreover, the assessment of OS is increasingly confounded by post-progression or post-trial therapies, a problem now widely acknowledged in oncology [10, 11, 15]. As a result of these drawbacks, there has been increasing interest in developing and validating surrogate end points for OS in order to expedite drug development, approval and reimbursement [16, 17]. In advanced solid tumors, the end point more often used to replace OS is PFS, defined as the time elapsed between randomization and the occurrence of disease progression—as measured, e.g., by Response Evaluation Criteria in Solid Tumors [18]—or death from any cause. PFS, which is accepted by regulatory authorities in the US and Europe [19, 20], is preferable to time to progression (TTP), an end point for which patients who die with no prior documentation of disease progression are censored in the analysis. PFS is advantageous because it is measured earlier, often leads to more statistical power than OS at equivalent durations of follow-up, and is not influenced by cross-over [14]. On the other hand, PFS is far from perfect as an end point, as it is prone to measurement error and bias. Moreover, PFS may not capture the entire treatment effect on the outcomes of most interest to patients with an incurable disease: a prolonged survival and improved quality of life. Therefore, we are left with two imperfect end points, each having drawbacks and advantages. How are we to choose? The answer to this question is still a matter of debate, but it would certainly be made easier if PFS could be demonstrated to be a surrogate for OS; if that was the case, the advantages of PFS—an increased efficiency and the lack of influence from cross-over—could be combined with the ability to predict a more objective and clinically relevant end point, OS.
3 Validation of Surrogate End Points
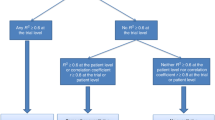
Surrogate end points are biomarkers (i.e., indicators of biologic or pathogenic processes or of response to treatment) intended to substitute for a final outcome that directly measure how patients feel, function or survive in clinical trials [21, 22]. Establishing a surrogate end point entails its evaluation from biological, clinical and statistical standpoints [23]. From the statistical point of view, the central issue is the validation of the end point as a surrogate. The formal process of validation has caused considerable controversy in the literature, but it is acknowledged that “the strength of the evidence for surrogacy depends upon (i) the biological plausibility of the relationship, (ii) the demonstration in epidemiological studies of the prognostic value of the surrogate for the clinical outcome, and (iii) evidence from clinical trials that treatment effects on the surrogate correspond to effects on the clinical outcome” [24]. The first condition implies that the surrogate needs to be in the causal pathway between treatment and the final outcome. This was first embodied in a set of reference criteria formulated by Prentice [25], which have been shown to be difficult to evaluate in a trial without making unverifiable assumptions [23]. More recently, the requirement that the surrogate be in the causal pathway between treatment and the final outcome has been dealt with using the causal-inference approach [26]. The second and third conditions mentioned above pertain to the statistical validation of a surrogate end point and may be rephrased as follows: for a surrogate to replace a final end point, there must be a high correlation between the surrogate end point and the final end point at the patient level (i.e., patients with improvements in the surrogate also tend to have improvements on the final end point)—this can also be interpreted as the prognostic role of the surrogate; and there must be a high correlation between the treatment effect on the surrogate end point and the treatment effect on the final end point (i.e., at the trial level the treatment effect on the surrogate must reliably predict the treatment effect on the final end point) [16]. Counterintuitively, these two correlations are independent, which implies that a claim of surrogacy requires stronger conditions than a mere correlation between the surrogate and the final end point [27]. The trial-level association is usually summarized by R 2, which measures the correlation between treatment effects on the surrogate and the true end points with values ranging from 0.00 to 1.00 (higher values indicate stronger correlation). Many biomarkers in oncology are prognostic, which often leads to high individual-level correlations, typically measured by a Spearman’s rank correlation (ρ, also ranging from 0.00 to 1.00). However, it is much more difficult to achieve a high trial-level correlation (R 2). Although there is no formal consensus on the minimum trial-level R 2 that is needed to validate a surrogate end point, values closer to 1.00 are desirable. If enough trials are available, the regression approach can make due allowance for estimation error in both the treatment effects estimated on the surrogate and the true end point (errors-in-variable regression) [23].
The validation of a surrogate end point is best made using individual-patient data (IPD) from randomized trials, which should preferably be selected after a systematic review of the literature [16]. It is only through the use of IPD that is it possible to use exactly the same PFS definition in each trial and to use the same definition of analyses populations. Importantly, surrogacy depends on the clinical context, which in oncology may include at least the treatment, the tumor type, and the line of therapy. The IPD meta-analytical approach has been used to evaluate PFS as a surrogate for OS using cytotoxic agents in advanced colorectal cancer, advanced breast cancer, locally advanced or advanced lung cancer, locally advanced head and neck and nasopharyngeal cancer, and advanced gastric cancer [8, 9, 28–34]. Among the hematologic malignancies, this approach has also been used to evaluate leukemia-free survival as a surrogate for OS in acute myelogenous leukemia [35]. PFS has been shown to be an appropriate surrogate for evaluation of cytotoxic effects in most of the solid tumors assessed to date, but in metastatic breast cancer, PFS was not shown to be a valid surrogate for OS in a meta-analysis of 3953 patients from 11 trials that compared an anthracycline (alone or in combination) with a taxane (alone or in combination with an anthracycline) [8]. However, relationships between surrogate and final end points for one drug do not necessarily apply to a drug with a different mode of action for treating the same disease [24], a further reason why surrogacy evaluation for targeted therapies is warranted. On the other hand, there are no specific statistical issues that differentially affect surrogacy validation between targeted therapies and agents from other classes.
4 PFS as Surrogate for OS in Advanced Solid Tumors Treated with Targeted Therapies
PFS has often been used as the primary end point in phase 3 clinical trials in oncology, as well as the basis of approval by the Food and Drug Administration [36]. Unfortunately, no recent reviews by that Agency are currently available, but a brief survey of the literature discloses various recent instances in which the accelerated or regular approval of targeted agents has been based on PFS [37–40]. Although studies on the patient-level role of PFS or TTP as potential surrogates for OS among patients with specific tumors treated with targeted agents have been published [41, 42], proper IPD meta-analytical evaluations have still been rare in this setting, as we identified only three studies on this topic in the literature: one in breast cancer, one in colorectal cancer, and one in non-small cell lung cancer (see Table 1).
In the meta-analysis to evaluate surrogacy for anti-HER2 targeted agents in advanced breast cancer [43], IPD from 1839 patients enrolled in eight randomized trials were centrally analyzed. All but one trial were sponsored by the pharmaceutical industry, but they agreed to provide IPD to the central academic data center for the purpose of the project. Seven of the eight trials evaluated the addition of either trastuzumab or lapatinib to a backbone of chemotherapy or hormone therapy, and one trial compared two trastuzumab regimens; six of the eight trials were conducted in the first-line setting. In that meta-analysis, PFS was shown to be moderately correlated with OS at the individual level (Spearman correlation ρ = 0.67; 95% confidence interval [CI] 0.66–0.67). Treatment effects (log hazard ratios) on PFS also correlated moderately with treatment effects on OS in a linear regression model weighted by trial size (R 2 = 0.51; 95% CI 0.22–0.81). This means that in the weighted regression model, only about half of the variation in treatment effect on OS is explained by treatment effects on PFS. The linear regression model weighted by trial size was used because of difficulties in making an error-in-variable regression model converge that accounted for estimation error in both the treatment effects on PFS and OS, and to adjust at least approximately for measurement errors. Of note, the estimated individual level correlation (0.67) in this study of anti-HER2 targeted agents was almost identical to the estimated individual level correlation in the meta-analysis of anthracyclines versus taxanes cited above (0.69) [8]. Taking these two studies together, it is fair to conclude that surrogacy of PFS for OS in metastatic breast cancer is still not validated for either cytostatic or cytotoxic agents.
In the study on colorectal cancer [28], IPD from 7323 patients from 12 randomized trials were centrally analyzed through an independent academic collaboration of the Analysis and Research in Cancers of the Digestive System (ARCAD). The included trials evaluated the targeted agents bevacizumab, panitumumab, or cetuximab, which were typically combined with standard chemotherapy backbones in the first-line setting. The individual-level correlation between PFS and OS was given by ρ = 0.55 (95% CI 0.54–0.56), and the trial-level correlation of treatment effects on PFS and OS by R 2 = 0.45 (95% CI 0.16–0.75), which are both insufficient to make a strong claim of surrogacy. Of note, this meta-analysis used the regression technique that allows for estimation errors of the treatment effects on both the surrogate and the true end point, an approach that is theoretically appropriate.
The third example evaluated the surrogacy of PFS for OS in an IPD meta-analytical setting in advanced non-small cell lung cancer. This time, the independent analysis was performed under the auspices of the Food and Drug Administration, using IPD submissions for drug approval between 2003 and 2013. The 15 trials evaluated the effect on survival end points of the targeted agents afatinib, bevacizumab, cetuximab, crizotinib, gefitinib, and vandetanib, as well as of various chemotherapy agents in the first- or second-line setting among 12,567 patients. While an individual-level correlation was not provided, the trial-level correlation, calculated using a weighted linear regression of hazard ratios estimated by Cox regression models, was estimated as 0.08 (95% CI 0.00–0.31), suggesting an almost negligible trial-level surrogacy. Interestingly, the trial-level correlation was estimated as 0.35 (95% CI 0.00–0.72) when three trials on targeted therapy with molecularly-enriched populations were excluded.
These three studies suffer from drawbacks inherent to the limited availability of the IPD and the design of the original clinical trials. The breast cancer study included trials in the first and second line of therapy. As noted above, surrogacy is often context-dependent, and it remains uncertain if different correlations could exist for trastuzumab and lapatinib in first and second lines of therapy. Moreover, the association between PFS and OS may have been attenuated by treatment cross-over or effective second-line treatments; unfortunately, the paucity of data on such treatments in the data collection of clinical trials in oncology precluded further analyses investigating their potential role in attenuating the correlation at the trial level. The colorectal cancer study was restricted to the first line, but, as mentioned above, in this disease PFS was initially found to be a good surrogate for OS both at the patient level and at the trial level with fluoropyrimidine-based therapy, used in the 1980s and 1990s [9]. The weaker correlation found for targeted agents in colorectal cancer suggests that the trial-level correlation was attenuated when second-line treatments became available, which was the case when more contemporary treatment regimens were analyzed [28]. Finally, the non-small cell lung cancer example included trials that evaluated together various targeted agents on different biological pathways and chemotherapies in different lines of treatment. Moreover, no separate results were provided for trials on targeted therapy.
5 Conclusion
The most appropriate approach to evaluate surrogate end points in randomized clinical trials is through the use of the IPD meta-analysis technique, which allows to evaluate a candidate surrogate end point both at the individual and the trial level. It has recently been suggested that surrogate end points successfully evaluated using the meta-analytical IPD approach will also be appealing from a causal-inference perspective [44].
As a result of the various possible settings for surrogacy analyses—cancer type, therapy class, treatment line, etc.—, the process of validation is time-consuming and largely dependent on the willingness of original investigators to share clinical-trial data. In the advanced solid-tumor setting, only three IPD studies have evaluated PFS as a surrogate end point for OS in trials of targeted treatments [28, 40, 43]. When individual-level surrogacy results were available [28, 43], PFS was found to be only moderately correlated with OS, and in all cases the treatments effects on PFS were insufficient to make claims of surrogacy for OS at the trial level. Even if properly conducted surrogate-endpoint evaluations have thus far been unsuccessful, so that truly validated surrogates are currently rare in medical oncology [17, 45], these evaluations are a step in the right direction [46] and can be expected to be applied on a much larger scale in the era of data sharing of clinical trials [47, 48]. On the other hand, we believe that lack of formal validation should not be considered as a reason to abandon the use of end points, such as PFS, which have proven useful in drug development. Thus, we believe PFS will continue to be used in future trials of targeted therapy, in spite of its lack of formal validation as a reliable surrogate for OS, until such time as more reliable end points can replace or be used alongside OS.
References
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–92.
Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–42.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80.
O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004.
Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–57.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–94.
Burzykowski T, Buyse M, Piccart-Gebhart MJ, Sledge G, Carmichael J, Luck HJ, et al. Evaluation of tumor response, disease control, progression-free survival, and time to progression as potential surrogate end points in metastatic breast cancer. J Clin Oncol. 2008;26(12):1987–92.
Buyse M, Burzykowski T, Carroll K, Michiels S, Sargent DJ, Miller LL, et al. Progression-free survival is a surrogate for survival in advanced colorectal cancer. J Clin Oncol. 2007;25(33):5218–24.
Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol. 2011;29(21):2866–74.
Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–77.
Pilotto S, Carbognin L, Karachaliou N, Garassino M, Cuppone F, Petraglia S, et al. Moving towards a customized approach for drug development: lessons from clinical trials with immune checkpoint inhibitors in lung cancer. Transl Lung Cancer Res. 2015;4(6):704–12.
Johnson JR, Temple R. Food and drug administration requirements for approval of new anticancer drugs. Cancer Treat Rep. 1985;69(10):1155–9.
Saad ED, Buyse M. Statistical controversies in clinical research: end points other than overall survival are vital for regulatory approval of anticancer agents. Ann Oncol. 2016;27(3):373–8.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–46.
Buyse M, Molenberghs G, Burzykowski T, Renard D, Geys H. The validation of surrogate endpoints in meta-analyses of randomized experiments. Biostatistics. 2000;1(1):49–67.
Ciani O, Davis S, Tappenden P, Garside R, Stein K, Cantrell A, et al. Validation of surrogate endpoints in advanced solid tumors: systematic review of statistical methods, results, and implications for policy makers. Int J Technol Assess Health Care. 2014;30(3):312–24.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47.
US Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research. Center for Biologics Evaluation and Research. Guidance for industry: clinical trial endpoints for the approval of cancer drugs and biologics. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071590.pdf. Accessed 16 Aug 2016.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Appendix 1 to the Guideline on the Evaluation of Anticancer Medicinal Products in Man (CHMP/ewp/205/95 rev.3). Methodological considerations for using progression-free survival (PFS) as primary endpoint in confirmatory trials for registration. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/12/WC500017736.pdf. Accessed 16 Aug 2016.
Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89–95.
De Gruttola VG, Clax P, DeMets DL, Downing GJ, Ellenberg SS, Friedman L, et al. Considerations in the evaluation of surrogate endpoints in clinical trials. summary of a National Institutes of Health workshop. Control Clin Trials. 2001;22(5):485–502.
Buyse M, Molenberghs G, Paoletti X, Oba K, Alonso A, Van der Elst W, et al. Statistical evaluation of surrogate endpoints with examples from cancer clinical trials. Biom J Biometrische Zeitschrift. 2016;58(1):104–32.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use. ICH Harmonised Tripartite Guideline. Statistical principles for clinical trials. Federal Register 63, No. 179, 49583. Available at: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf. Accessed 18 Aug 2016.
Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8(4):431–40.
Vanderweele TJ. Surrogate measures and consistent surrogates. Biometrics. 2013;69(3):561–9.
Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996;125(7):605–13.
Shi Q, de Gramont A, Grothey A, Zalcberg J, Chibaudel B, Schmoll HJ, et al. Individual patient data analysis of progression-free survival versus overall survival as a first-line end point for metastatic colorectal cancer in modern randomized trials: findings from the analysis and research in cancers of the digestive system database. J Clin Oncol. 2015;33(1):22–8.
Laporte S, Squifflet P, Baroux N, Fossella F, Georgoulias V, Pujol JL, et al. Prediction of survival benefits from progression-free survival benefits in advanced non-small-cell lung cancer: evidence from a meta-analysis of 2334 patients from 5 randomised trials. BMJ Open. 2013;3(3).
Foster NR, Renfro LA, Schild SE, Redman MW, Wang XF, Dahlberg SE, et al. Multitrial evaluation of progression-free survival as a surrogate end point for overall survival in first-line extensive-stage small-cell lung cancer. J Thorac Oncol. 2015;10(7):1099–106.
Mauguen A, Pignon JP, Burdett S, Domerg C, Fisher D, Paulus R, et al. Surrogate endpoints for overall survival in chemotherapy and radiotherapy trials in operable and locally advanced lung cancer: a re-analysis of meta-analyses of individual patients’ data. Lancet Oncol. 2013;14(7):619–26.
Michiels S, Le Maitre A, Buyse M, Burzykowski T, Maillard E, Bogaerts J, et al. Surrogate endpoints for overall survival in locally advanced head and neck cancer: meta-analyses of individual patient data. Lancet Oncol. 2009;10(4):341–50.
Paoletti X, Oba K, Bang YJ, Bleiberg H, Boku N, Bouche O, et al. Progression-free survival as a surrogate for overall survival in advanced/recurrent gastric cancer trials: a meta-analysis. J Natl Cancer Inst. 2013;105(21):1667–70.
Rotolo F, Pignon JP, Bourhis J, et al. Surrogate endpoints for overall survival in loco-regionally advanced nasopharyngeal carcinoma: an individual patient data meta-analysis. J Natl Cancer Inst. 2016;109(4):pii:djw239.
Buyse M, Michiels S, Squifflet P, Lucchesi KJ, Hellstrand K, Brune ML, et al. Leukemia-free survival as a surrogate end point for overall survival in the evaluation of maintenance therapy for patients with acute myeloid leukemia in complete remission. Haematologica. 2011;96(8):1106–12.
Sridhara R, Johnson JR, Justice R, Keegan P, Chakravarty A, Pazdur R. Review of oncology and hematology drug product approvals at the US Food and Drug Administration between July 2005 and December 2007. J Natl Cancer Inst. 2010;102(4):230–43.
Nair A, Lemery SJ, Yang J, Marathe A, Zhao L, Zhao H, et al. FDA approval summary: lenvatinib for progressive, radio-iodine-refractory differentiated thyroid cancer. Clin Cancer Res. 2015;21(23):5205–8.
Kazandjian D, Blumenthal GM, Chen HY, He K, Patel M, Justice R, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist. 2014;19(10):e5–11.
Khozin S, Blumenthal GM, Jiang X, He K, Boyd K, Murgo A, et al. U.S. Food and Drug Administration approval summary: erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist. 2014;19(7):774–9.
Blumenthal GM, Cortazar P, Zhang JJ, Tang S, Sridhara R, Murgo A, et al. FDA approval summary: sunitinib for the treatment of progressive well-differentiated locally advanced or metastatic pancreatic neuroendocrine tumors. Oncologist. 2012;17(8):1108–13.
Halabi S, Rini B, Escudier B, Stadler WM, Small EJ. Progression-free survival as a surrogate endpoint of overall survival in patients with metastatic renal cell carcinoma. Cancer. 2014;120(1):52–60.
Zabor EC, Heller G, Schwartz LH, Chapman PB. Correlating surrogate endpoints with overall survival at the individual patient level in BRAFV600E-mutated metastatic melanoma patients treated with vemurafenib. Clin Cancer Res. 2016;22(6):1341–7.
Michiels S, Pugliano L, Marguet S, Grun D, Barinoff J, Cameron D, et al. Progression-free survival as surrogate end point for overall survival in clinical trials of HER2-targeted agents in HER2-positive metastatic breast cancer. Ann Oncol. 2016;27(6):1029–34.
Alonso A, Van der Elst W, Molenberghs G, Buyse M, Burzykowski T. On the relationship between the causal-inference and meta-analytic paradigms for the validation of surrogate endpoints. Biometrics. 2015;71(1):15–24.
Prasad V, Kim C, Burotto M, Vandross A. The strength of association between surrogate end points and survival in oncology: a systematic review of trial-level meta-analyses. JAMA Intern Med. 2015;175(8):1389–98.
Buyse M. Contributions of meta-analyses based on individual patient data to therapeutic progress in colorectal cancer. Int J Clin Oncol. 2009;14(2):95–101.
Christakis DA, Zimmerman FJ. Rethinking reanalysis. JAMA. 2013;310(23):2499–500.
Strom BL, Buyse M, Hughes J, Knoppers BM. Data sharing, year 1—access to data from industry-sponsored clinical trials. N Engl J Med. 2014;371(22):2052–4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Ligue Nationale Contre le Cancer.
Disclosure
Dr. Michiels declares no conflict of interest. Dr. Buyse report being employed by and holding stock in the International Drug Development Institute, and Dr. Saad reports being employed by the International Drug Development Institute.
Rights and permissions
About this article

Cite this article
Michiels, S., Saad, E.D. & Buyse, M. Progression-Free Survival as a Surrogate for Overall Survival in Clinical Trials of Targeted Therapy in Advanced Solid Tumors. Drugs 77, 713–719 (2017). https://doi.org/10.1007/s40265-017-0728-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0728-y