
Abstract
Inhibitors of sodium–glucose co-transporter type 2 (SGLT2) are proposed as a novel approach for the management of type 2 diabetes mellitus (T2DM). Several compounds are already available in many countries (dapagliflozin, canagliflozin, empagliflozin and ipragliflozin) and some others are in a late phase of development. The available SGLT2 inhibitors share similar pharmacokinetic characteristics, with a rapid oral absorption, a long elimination half-life allowing once-daily administration, an extensive hepatic metabolism mainly via glucuronidation to inactive metabolites, the absence of clinically relevant drug–drug interactions and a low renal elimination as parent drug. SGLT2 co-transporters are responsible for reabsorption of most (90 %) of the glucose filtered by the kidneys. The pharmacological inhibition of SGLT2 co-transporters reduces hyperglycaemia by decreasing renal glucose threshold and thereby increasing urinary glucose excretion. The amount of glucose excreted in the urine depends on both the level of hyperglycaemia and the glomerular filtration rate. Results of numerous placebo-controlled randomised clinical trials of 12–104 weeks duration have shown significant reductions in glycated haemoglobin (HbA1c), resulting in a significant increase in the proportion of patients reaching HbA1c targets, and a significant lowering of fasting plasma glucose when SGLT2 inhibitors were administered as monotherapy or in addition to other glucose-lowering therapies including insulin in patients with T2DM. In head-to-head trials of up to 2 years, SGLT2 inhibitors exerted similar glucose-lowering activity to metformin, sulphonylureas or sitagliptin. The durability of the glucose-lowering effect of SGLT2 inhibitors appears to be better; however, this remains to be more extensively investigated. The risk of hypoglycaemia was much lower with SGLT2 inhibitors than with sulphonylureas and was similarly low as that reported with metformin, pioglitazone or sitagliptin. Increased renal glucose elimination also assists weight loss and could help to reduce blood pressure. Both effects were very consistent across the trials and they represent some advantages for SGLT2 inhibitors when compared with other oral glucose-lowering agents. The pharmacodynamic response to SGLT2 inhibitors declines with increasing severity of renal impairment, and prescribing information for each SGLT2 inhibitor should be consulted regarding dosage adjustments or restrictions in moderate to severe renal dysfunction. Caution is also recommended in the elderly population because of a higher risk of renal impairment, orthostatic hypotension and dehydration, even if the absence of hypoglycaemia represents an obvious advantage in this population. The overall effect of SGLT2 inhibitors on the risk of cardiovascular disease is unknown and will be evaluated in several ongoing prospective placebo-controlled trials with cardiovascular outcomes. The impact of SGLT2 inhibitors on renal function and their potential to influence the course of diabetic nephropathy also deserve more attention. SGLT2 inhibitors are generally well-tolerated. The most frequently reported adverse events are female genital mycotic infections, while urinary tract infections are less commonly observed and generally benign. In conclusion, with their unique mechanism of action that is independent of insulin secretion and action, SGLT2 inhibitors are a useful addition to the therapeutic options available for the management of T2DM at any stage in the natural history of the disease. Although SGLT2 inhibitors have already been extensively investigated, further studies should even better delineate the best place of these new glucose-lowering agents in the already rich armamentarium for the management of T2DM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
By inhibiting renal glucose reabsorption, sodium–glucose co-transporter type 2 (SGLT2) inhibitors increase glucosuria, reduce hyperglycaemia (without inducing hypoglycaemia), promote weight loss and exert a modest diuretic effect with blood pressure reduction. They act primarily independently of insulin, although secondary indirect effects on insulin secretion and action may occur due to reduced glucose toxicity. |
SGLT2 inhibitors have proven their efficacy in placebo-controlled trials in patients with type 2 diabetes mellitus (T2DM) treated with diet and exercise, as add-on to another glucose-lowering agent [metformin, sulphonylurea, pioglitazone, dipeptidyl peptidase-4 (DPP-4) inhibitor], in triple oral therapy and in combination with insulin; they are as active or even more active than other glucose-lowering agents (sulphonylureas or sitagliptin). |
The glucose-lowering efficacy is reduced in patients with renal impairment and some adverse events may occur that are directly linked to the unique mechanism of action of this pharmacological class: an increase in the incidence of genital mycotic infections, mild urinary tract infections and hypotension episodes (mainly in elderly patients with volume depletion). |
Large prospective placebo-controlled trials with cardiovascular outcomes are ongoing, in patients with T2DM and high cardiovascular risk, with canagliflozin, dapagliflozin and empagliflozin, the results of which should help the clinician in positioning SGLT2 inhibitors in the treatment algorithm of T2DM. |
1 Introduction
Type 2 diabetes mellitus (T2DM) affects more than 350 million people worldwide, and its prevalence is increasing. Despite a large armamentarium already being available for the management of hyperglycaemia in T2DM, glucose-lowering agents are not adequately effective in maintaining long-term glycaemic control in a majority of patients, even when used in combination [1]. Furthermore, most antihyperglycaemic agents are associated with adverse events such as hypoglycaemia and/or weight gain, which exert counterproductive effects and hamper adherence to treatment. Thus, there remains a medical need for improving pharmacological therapy of T2DM [2].
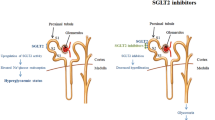
Inhibitors of sodium–glucose co-transporter type 2 (SGLT2) are new glucose-lowering agents with an original insulin-independent mode of action [3, 4]. They specifically target the kidney by blocking the reabsorption of filtered glucose, thus leading to increased urinary glucose excretion (UGE), especially when hyperglycaemia is present [5, 6]. This mechanism of action holds promise for patients with T2DM not only in terms of improvements in glycaemic control, with a limited risk of hypoglycaemia, but also considering the potential benefits of weight loss resulting from increased glucosuria and arterial blood pressure reduction associated with the osmotic effect [5, 6]. SGLT2 inhibitors may be used as monotherapy in diet-treated patients or in combination with any other glucose-lowering agent [7, 8]. The pharmacokinetic characteristics of SGLT2 inhibitors show an excellent oral bioavailability, a rather long elimination half-life (t ½) allowing once-daily administration, a low accumulation index, no active metabolites and a limited renal excretion [9]. Furthermore, these agents share a negligible risk of drug–drug interactions [10].
A leading article on SGLT2 inhibitors was published in 2011 discussing the progress and therapeutic potential of this drug class in T2DM, at a time when development was advanced (especially for dapagliflozin) but no agents had yet been approved for use [11]. Currently, there are three SGLT-2 inhibitors marketed in Europe and the USA (dapagliflozin [12, 13], canagliflozin [14–16] and empagliflozin [17–19]). A few others are commercialised or approved by the regulatory agency in Japan (ipragliflozin [20], luseogliflozin [21] and tofogliflozin [22]) and several others are in a late phase of development (ertugliflozin, remogliflozin, etc.) [23]. The aim of this review is to provide an updated analysis of the pharmacodynamics, efficacy and safety profile of these different SGLT2 inhibitors, with a special focus on dapagliflozin [24], canagliflozin [25], empagliflozin [26] and ipragliflozin [20] (Fig. 1).

Chemical structures of four sodium–glucose co-transporter type 2 inhibitors (dapagliflozin, canagliflozin, empagliflozin and ipragliflozin)
To identify relevant studies, an extensive literature search in MEDLINE was performed from 2008 to July 2014, with the following MeSH terms: SGLT2 inhibitor, canagliflozin, dapagliflozin, empagliflozin, ipragliflozin. No language restrictions were imposed but only studies reported as full papers (not as abstracts) were included in this review article. Reference lists of original studies, narrative reviews and previous systematic reviews were also carefully examined.
2 Dapagliflozin
2.1 Pharmacokinetic/Pharmacodynamic Analysis
2.1.1 Normal Kidney/Liver Function
The clinical pharmacokinetics and pharmacodynamics of dapagliflozin have recently been extensively reviewed [13]. Orally administered dapagliflozin is rapidly absorbed, generally achieving maximum (peak) plasma concentrations (C max) within 1–2 h. Dose-proportional systemic exposure to dapagliflozin has been observed over a wide dose range (0.1–500 mg) with an oral bioavailability of 78 %. Dapagliflozin has extensive extravascular distribution, as shown by a mean volume of distribution averaging 118 L (Table 1). Dapagliflozin metabolism occurs predominantly in the liver and kidneys by uridine diphosphate-glucuronosyltransferase-1A9 (UGT1A9) to the major inactive metabolite dapagliflozin 3-O-glucuronide (D3OG). Dapagliflozin is not appreciably cleared by renal excretion (<2 % of dose is recovered in urine as parent), in contrast to its major metabolite which is mainly eliminated via renal excretion. Almost no or only modest drug–drug interactions (without obvious clinical significance) were observed between dapagliflozin and other oral antidiabetic agents, cardiovascular (CV) medications or various drugs of potential interest because of a low therapeutic index [10].
Maximal increases in UGE were seen at doses ≥20 mg/day in patients with T2DM. Pharmacodynamic changes are dependent on plasma glucose and renal function, and decreases in UGE were observed due to the lower filtered load [plasma glucose × glomerular filtration rate (GFR)] in healthy volunteers than in subjects with T2DM. After multiple doses of dapagliflozin, UGE was associated with dose-related decreases in plasma glucose parameters in subjects with T2DM [13].
Besides increasing UGE, dapagliflozin exerts indirect metabolic effects [27]. It improved muscle insulin sensitivity due to a reduced glucotoxicity. However, surprisingly, following dapagliflozin treatment, endogenous glucose production increased substantially and was accompanied by an increase in fasting plasma glucagon concentration. Thus, glucosuria induction following SGLT2 inhibition is associated with a paradoxical increase in glucose production [28].
2.1.2 Impaired Kidney Function
Following a single 50 mg dose of dapagliflozin, plasma concentrations of dapagliflozin and D3OG were incrementally increased with declining kidney function [29]. Steady-state C max values for dapagliflozin were 4, 6 and 9 % higher and for D3OG were 20, 37 and 52 % higher in patients with mild, moderate and severe renal impairment (RI), respectively, than in individuals with normal function. Total exposure [area under the concentration–time curve (AUC)] was likewise higher in patients with RI. These results indicate that the kidney, besides the liver, significantly contributes to dapagliflozin metabolism, resulting in higher systemic exposure with declining kidney function.
Compared with patients with normal renal function, steady-state renal glucose clearance was reduced by 42, 83 and 84 % in patients with mild, moderate or severe RI, respectively, leading to a progressive attenuation of the glucose-lowering effect. These findings are consistent with the observation of reduced efficacy in terms of glycated haemoglobin (HbA1c) diminution in this patient population (see Sect. 7.4) [29].
2.1.3 Impaired Liver Function
Compared with healthy subjects, systemic exposure to dapagliflozin in subjects with chronic liver disease was correlated with the degree of hepatic impairment. Due to the higher dapagliflozin exposures in cases of severe hepatic failure, a reduced starting dose of dapagliflozin 5 mg instead of 10 mg is recommended in patients with severe hepatic impairment. However, the benefit:risk ratio should be individually assessed because the long-term safety profile and efficacy of dapagliflozin have not been specifically studied in this population with hepatic impairment [30].
2.2 Efficacy
2.2.1 Blood Glucose Control
The efficacy of dapagliflozin has been evaluated in placebo-controlled randomised clinical trials (RCTs) in T2DM patients treated with diet and exercise (monotherapy) [31–36], in combination with metformin [37–41], a sulphonylurea (glimepiride) [42, 43], a thiazolidinedione (pioglitazone) [44] or a dipeptidyl peptidase-4 (DPP-4) inhibitor (sitagliptin) [45], in triple therapy with metformin plus sitagliptin [45], as add-on therapy to usual care in patients with CV disease [46] or in patients with moderate (stage 3) chronic kidney disease (CKD) [47], and in combination with insulin (with or without metformin) [48, 49] (Table 2). The results are remarkably consistent regarding the reduction in HbA1c and fasting plasma glucose across the trials, independently of the background glucose-lowering therapy. In all conditions, dapagliflozin increased the proportion of T2DM patients reaching an HbA1c target below 7 %. Overall, dapagliflozin 10 mg once daily resulted in a slightly greater reduction in fasting plasma glucose and HbA1c than dapagliflozin 5 mg once daily, whatever the background therapy. The most important trials have already been described in previous reviews [12, 13] and pooled in a few recent meta-analyses [50–52].
Twelve RCTs were eligible for quantitative synthesis and meta-analysis of dapagliflozin combined with conventional antidiabetic drugs. The overall effect size of HbA1c calculated from mean difference was −0.52 % with a 95 % confidence interval (CI) −0.60 to −0.45 (p < 0.001). The effect size of fasting plasma glucose was −1.13 mmol/L (95 % CI −1.33 to −0.93; p < 0.001) [51]. In another meta-analysis of ten RCTs, dapagliflozin treatment was associated with a reduction in HbA1c [weighted mean difference (WMD): −0.53 %; 95 % CI −0.58 to −0.47; p < 0.00001] and fasting plasma glucose (WMD: −1.06 mmol/L; 95 % CI −1.20 to −0.92; p < 0.00001). Overall, dapagliflozin monotherapy did not lead to hypoglycaemia [relative risk (RR) 1.44; 95 % CI 0.86–2.41; p = 0.17], although hypoglycaemic risk slightly increased (RR 1.16; 95 % CI 1.05–1.29; p = 0.005) when dapagliflozin was combined with other hypoglycaemic drugs [52].
Few clinical trials have compared the efficacy of dapagliflozin with that of another glucose-lowering agent, either metformin [31, 40] or glipizide (a sulphonylurea) [53, 54] (Table 3). A dose-ranging short-term monotherapy study compared various doses of dapagliflozin with placebo and as an exploratory investigation with metformin extended release (750–1,500 mg/day). The dapagliflozin dose of 5 mg/day exerted an HbA1c reduction similar to that of metformin, while the dose of 10 mg/day resulted in a greater HbA1c diminution [31]. These findings were confirmed in a longer double-blind study [40]. After 24 weeks, dapagliflozin 10 mg/day was non-inferior to metformin in reducing HbA1c in treatment-naive patients with T2DM and combination therapy was statistically superior to either monotherapy in controlling blood glucose [40]. The most important head-to-head trial compared dapaglifozin with the sulphonylurea glipizide as add-on therapies to metformin in a 52-week study [53], with an extension up to 104 weeks [54]. Despite similar 52-week glycaemic efficacy (HbA1c reduction of 0.52 % in both arms), dapagliflozin 5–10 mg/day produced much less hypoglycaemia than glipizide (3.5 % of patients vs. 40.8 %; p < 0.0001) in T2DM patients inadequately controlled with metformin [53]. Over 2 years, compared with glipizide, dapagliflozin demonstrated greater glycaemic durability, with a significantly decreased 18–104 week HbA1c coefficient of failure (0.13 %/year vs. 0.59 %/year; p = 0.0001), a significant difference in HbA1c reduction at 104 weeks (p = 0.0211) (Table 3) and again a low hypoglycaemia rate [54]. In a Bayesian network meta-analysis of RCTs involving anti-diabetes treatments added to metformin, dapagliflozin offers similar HbA1c control after 1 year, with similar or reduced risk of hypoglycaemia and the additional benefit of weight loss compared with DPP-4 inhibitors, thiazolidinediones and sulphonylureas [55].
2.2.2 Weight Loss
Due to the caloric loss associated with increased UGE, treatment with SGLT2 inhibitors offers the benefit of weight loss to overweight/obese patients with T2DM [56]. In a meta-analysis of 12 RCTs, the effect size of dapagliflozin on body weight was −2.10 kg with a 95 % CI −2.32 to −1.88 (p < 0.001) [51], while in another meta-analysis of ten RCTs, dapagliflozin treatment was also associated with a significant reduction in body weight (WMD: −1.63 kg; 95 % CI −1.83 to −1.43; p < 0.00001) [52]. Overall, dapagliflozin 10 mg provided a greater weight loss than dapagliflozin 5 mg, although this difference was rather small and not present in all studies (Table 2).
Because of the osmotic and possibly diuretic effect of dapagliflozin, it is of importance to differentiate between a weight reduction due to fluid loss and that due to fat loss [57]. At 24 weeks, dapagliflozin reduced total body weight, predominantly by reducing fat mass, visceral adipose tissue and subcutaneous adipose tissue in T2DM inadequately controlled with metformin [39].
These findings were confirmed over 102 weeks, when dapagliflozin improved glycaemic control, and reduced weight and fat mass [41]. Such dapagliflozin-induced weight loss was associated with improvement in overall health-related quality of life [58].
2.2.3 Blood Pressure Reduction
Dapagliflozin-induced SGLT2 inhibition for 12 weeks was associated with reductions in 24-h blood pressure, body weight, GFR and possibly plasma volume. Cumulatively, these effects suggest that dapagliflozin may have a diuretic-like capacity to lower blood pressure in addition to beneficial effects on glycaemic control [57]. In a pre-specified pooled analysis of 12 placebo-controlled studies for 24 weeks, dapagliflozin 10 mg was associated with a mean change from baseline in systolic blood pressure (SBP) of 4.4 mmHg and in diastolic blood pressure (DBP) of 2.1 versus 0.9 and 0.5 mmHg for SBP/DBP in the placebo group [59]. The blood pressure-lowering effects of dapagliflozin could be exploited in the clinical management of obese hypertensive patients with T2DM, particularly in patients with difficulty controlling arterial hypertension [60].
2.3 Safety
2.3.1 Urinary/Genital Infections
Safety data from 12 placebo-controlled RCTs with dapagliflozin were pooled to evaluate the relationship between glucosuria and urinary tract infections (UTIs) in patients with inadequately controlled diabetes [61]. Patients were treated with dapagliflozin (2.5, 5 or 10 mg, all once daily) or placebo once daily, either as monotherapy or as combined therapy for 12–24 weeks. Treatment of T2DM with dapagliflozin 5 or 10 mg was accompanied by a slightly increased risk of UTI (5.7 and 4.3 %, respectively, vs. 3.7 % with placebo). Infections were generally mild to moderate and clinically manageable. There was no definitive dose relationship between glucosuria and UTI [61].
Treatment with dapagliflozin was accompanied by an increased risk of vulvovaginitis or balanitis, related to the induction of glucosuria. For dapagliflozin 2.5 mg, 5 mg, 10 mg once daily and placebo, diagnosed infections were reported in 4.1, 5.7, 4.8 and 0.9 %, respectively. Events were generally mild to moderate, clinically manageable and rarely led to discontinuation of treatment [62].
2.3.2 Other Concerns
Dapagliflozin had no effect on markers of bone formation and resorption or bone mass densitometry after 50 weeks of treatment in both male and post-menopausal female patients whose T2DM was inadequately controlled on metformin [63]. Over 102 weeks, dapagliflozin did not affect markers of bone turnover or bone mass density in patients with T2DM inadequately controlled on metformin [41]. In a pooled analysis, there was no imbalance in fractures between dapagliflozin and comparator groups (overall incidence <1.6 %). However, in patients with moderate RI, the incidence of fractures was higher in dapagliflozin recipients than in placebo recipients [47].
Other possible concerns with dapagliflozin, especially the bladder and breast cancer issue, have been extensively discussed in previous reviews [7]. In the absence of new data, these concerns are not discussed further in the present paper.
3 Canagliflozin
3.1 Pharmacokinetic/Pharmacodynamic Analysis
3.1.1 Normal Kidney/Liver Function
The pharmacokinetics/pharmacodynamics and metabolism of canagliflozin have recently been reviewed [14, 15]. After oral administration, canagliflozin is rapidly absorbed in a dose-dependent manner across a dose range of 50–300 mg. The mean absolute oral bioavailability is approximately 65 %. After canagliflozin 100 or 300 mg, median time to C max (t max) values occur within 1–2 h, with steady-state levels attained after 4–5 days following multiple once-daily doses. Canagliflozin does not exhibit time-dependent pharmacokinetics and, following multiple 100 and 300 mg doses, accumulates in the plasma up to 36 %. The drug is extensively (99 %) bound to plasma proteins, mainly albumin (Table 1). Canagliflozin is mainly metabolised to two inactive O-glucuronide metabolites. Canagliflozin has only minor (≈7 % in humans) metabolism by cytochrome P450 (CYP) 3A4. In in vitro studies, canagliflozin did not induce or markedly inhibit most CYP enzyme expression [14]. Almost no or only modest (without clinical significance) drug–drug interactions were observed between canagliflozin and other oral antidiabetic agents, CV medications or various drugs with a low therapeutic index [10].
Canagliflozin dose-dependently reduced the calculated renal threshold for glucose excretion and increased UGE in healthy subjects [64]. In patients with T2DM, canagliflozin pharmacokinetics were dose dependent, and the t ½ ranged from 12 to 15 h. After 28 days, the renal threshold for glucose excretion was reduced, UGE was increased, and both HbA1c and fasting plasma glucose decreased in subjects administered canagliflozin [65].
Canagliflozin, besides its action on SGLT2, is also a low-potency sodium–glucose co-transporter type 1 (SGLT1) inhibitor, which may differentiate it from dapagliflozin and perhaps other SGLT2 inhibitors. A study tested the hypothesis that intestinal canagliflozin concentrations post-dose are sufficiently high to transiently inhibit intestinal SGLT1. Indeed, canagliflozin lowered postprandial glucose and insulin by delaying intestinal glucose absorption in addition to increasing UGE [66]. This additional effect may contribute to better control of postprandial glucose excursions in patients with T2DM and might at least partially explain why treatment with canagliflozin for 6–12 months improved model-based measures of β cell function in patients with T2DM [67].
3.1.2 Impaired Kidney Function
An open-label phase I single-dose study evaluated the pharmacokinetics and pharmacodynamics of canagliflozin in non-diabetic subjects with varying degrees of RI compared with healthy subjects [68, 69]. AUC and C max were slightly higher in subjects with mild RI and modestly higher in subjects with moderate to severe RI, but not end-stage renal disease (ESRD), than in those with normal function. Canagliflozin was negligibly removed by haemodialysis [69]. UGE after canagliflozin administration decreased as renal function decreased. Following canagliflozin treatment, the renal threshold for glucose was modestly higher in subjects with moderate to severe RI than in subjects with normal function and mild RI, and the pharmacodynamic response to canagliflozin declined with increasing severity of RI [68].
3.1.3 Impaired Liver Function
Mild (Child-Pugh class A) and moderate (Child-Pugh class B) hepatic impairment had no clinically meaningful effect on the pharmacokinetics of canagliflozin [25]. There is no clinical experience with canagliflozin in patients with severe (Child-Pugh class C) hepatic impairment, and thus the drug is not recommended in this specific population [14].
3.2 Efficacy
3.2.1 Blood Glucose Control
The efficacy of canagliflozin has been evaluated in placebo-controlled RCTs in drug-naïve T2DM patients treated with diet and exercise [70, 71], in combination with metformin [72–74], in triple therapy added to metformin plus a sulphonylurea [75] or metformin plus pioglitazone [76], and as add-on to usual care including insulin, in an elderly population [77] or in patients with moderate (stage 3) CKD [78, 79]. Two doses have been investigated compared with placebo. In all studies, the dose of 300 mg once daily provided a slightly greater reduction in HbA1c and fasting plasma glucose than the lower dose of 100 mg once daily (Table 4). According to a meta-analysis of ten trials including 6,701 patients, compared with placebo, canagliflozin produced absolute reductions in HbA1c levels when used as monotherapy (WMD −1.08 %, 95 % CI −1.25 to −0.90; p < 0.00001) or add-on treatment (WMD −0.73 %, 95 % CI −0.84 to −0.61; p < 0.00001) [80].
Canagliflozin was also compared to other glucose-lowering agents in head-to-head trials, using either a sulphonylurea (glimepiride) [81] or a DPP-4 inhibitor (sitagliptin 100 mg) [73, 82] as active comparators (Table 3). Canagliflozin 100 mg/day exerted a similar reduction in HbA1c to glimepiride while canagliflozin 300 mg/day produced a greater HbA1c reduction (least-squares mean difference of −0.12 %; 95 % CI −0.22 to −0.02). The proportion of patients with documented hypoglycaemic episodes was significantly lower with canagliflozin 100 and 300 mg than with glimepiride (6 and 5 %, respectively, vs. 34 %; p < 0.0001 for both) [81]. As compared to sitagliptin 100 mg in patients treated with metformin [73] or a metformin–sulphonylurea combination [82], canagliflozin induced a greater reduction at a daily dose of 300 mg (−0.15 %, 95 % CI −0.27 to −0.03 and −0.37 %, 95 % CI −0.50 to −0.25, respectively), without increasing the risk of hypoglycaemia (Table 3). In a post hoc analysis of this latter study involving patients with T2DM on metformin plus sulphonylurea, after 52 weeks patients treated with canagliflozin 300 mg/day demonstrated better attainment of individual and composite diabetes-related quality measures than did patients treated with sitagliptin 100 mg [83].
3.2.2 Weight Loss
In a meta-analysis of ten trials, canagliflozin led to greater body weight loss than placebo (WMD −2.81 kg, 95 % CI −3.26 to −2.37) [80]. Canagliflozin 300 mg/day provided a greater weight loss than canagliflozin 100 mg/day in all trials where the two doses were compared, and both doses of canagliflozin resulted in a significantly greater weight reduction than placebo (Table 4). In contrast to glimepiride, which induced some weight gain, canagliflozin reduced body weight [81]. This canagliflozin-associated weight reduction also offers some advantage over the DPP-4 inhibitor sitagliptin, which was weight-neutral [73, 82]. In a 52-week trial comparing canagliflozin with glimepiride [81], a body composition substudy using dual-energy X-ray absorptiometry scans showed that in the canagliflozin 100 and 300 mg groups, roughly two-thirds of the reduction in body weight was from fat mass and a third from lean body mass, whereas the increase in body weight with glimepiride included both fat and lean body mass. Analysis of abdominal fat in the canagliflozin groups with computed tomography (CT) imaging showed a slightly greater reduction in visceral adipose tissue than in subcutaneous adipose tissue [81]. These data confirmed previous findings reported with dapagliflozin after 24 and 102 weeks [39, 41]. Finally, weight loss of an amount demonstrated in clinical trials of canagliflozin was associated with improvements in weight-related quality of life and satisfaction with physical and emotional health [84].
3.2.3 Blood Pressure Reduction
In addition to glucose lowering, SGLT2 inhibitors are associated with weight loss and act as osmotic diuretics, resulting in a lowering of blood pressure [59]. In a systematic review and meta-analysis, SGLT2 inhibitors significantly reduced both SBP and DBP from baseline. Only canagliflozin had a significant dose–response relationship with SBP (p = 0.008) [85]. In patients with T2DM on background therapy with metformin and ACE inhibitors or angiotensin receptor antagonists, canagliflozin 300 mg/day exerted a transient reduction in plasma volume at week 1 that was largely attenuated by week 12. Nevertheless, reductions in body weight and blood pressure were observed at weeks 1 and 12 [86]. The osmotic diuretic effect may lead to postural hypotension and dizziness in susceptible subjects [87]. However, in a systematic review and meta-analysis, SGLT2 inhibitors had no significant effect on the incidence of orthostatic hypotension [85].
3.3 Safety
3.3.1 Urinary/Genital Infections
In a short-term 12-week trial evaluating five doses of canagliflozin (50 mg, 100 mg, 200 mg, 300 mg daily, or 300 mg twice daily), when compared with control subjects (placebo or sitagliptin 100 mg/day), canagliflozin increased UGE but was not associated with increased bacteriuria or adverse event reports of UTIs [88]. Canagliflozin treatment was also associated with an increase in vaginal colonisation with Candida species and in symptomatic vulvovaginal adverse events in women with T2DM [89].
In RCTs lasting up to 52 weeks, the most common adverse effects were genital mycotic infections occurring in 11–15 % of women exposed to canagliflozin versus 2–4 % of those randomised to glimepiride or sitagliptin. In men, corresponding proportions were 8–9 versus 0.5–1 %. UTIs were only slightly increased (5–7 %) with the use of canagliflozin compared with placebo (4 %) [87]. In a pooled analysis of clinical studies, genital mycotic infection incidences were higher with canagliflozin than control in patients with T2DM; however, events were generally mild to moderate in intensity and responded to standard treatments [90].
3.3.2 Other Concerns
An updated safety analysis of canagliflozin trials noted a non-significant imbalance in fracture incidence in patients treated with canagliflozin compared with control patients [7].
Other possible concerns have been extensively discussed in previous reviews [7, 14–16].
4 Empagliflozin
4.1 Pharmacokinetic/Pharmacodynamic Analysis
4.1.1 Normal Kidney/Liver Function
Single oral doses of empagliflozin were rapidly absorbed, reaching C max after 1.0–2.0 h (Table 1). Increases in empagliflozin exposure were roughly dose-proportional and a dose-dependent increase in UGE was observed for empagliflozin doses up to 100 mg [18]. Pharmacokinetic/pharmacodynamic characteristics of empagliflozin have recently been reviewed in healthy volunteers and patients with T2DM [17]. However, detailed description of absorption, distribution, metabolism and excretion characteristics of empagliflozin in humans has not been reported yet [91]. Almost no or only modest drug–drug interactions (without clinical significance in most cases) were observed between empagliflozin and other oral antidiabetic agents, CV medications or various drugs with a low therapeutic index [10, 17].
Empagliflozin exposure increased dose proportionally over the dose range 10–100 mg and showed linear pharmacokinetics with respect to time. Oral administration of empagliflozin at doses of 10, 25 or 100 mg once daily over 28 days resulted in significant increases in UGE and reductions in blood glucose compared with placebo in patients with T2DM [92]. These pharmacokinetic/pharmacodynamic data obtained after a single dose were confirmed after multiple doses of empagliflozin (2.5–100 mg) for 8 days [93].
A paradoxical increase in endogenous glucose production has been reported with empagliflozin in patients with T2DM, in both fasting and postprandial states [94]. These findings, which confirmed similar findings reported with dapagliflozin [94], suggest a whole-body metabolic adaptation following SGLT2 inhibition [95]. Nevertheless, despite this increase in endogenous glucose production, fasting plasma glucose levels were significantly reduced by empagliflozin in all RCTs comparing the SGLT2 inhibitor with placebo.
4.1.2 Impaired Kidney Function
The effect of impaired kidney function on the pharmacokinetics of empagliflozin was investigated in subjects with varying degrees of RI, who received a single dose of empagliflozin 50 mg [96]. The rate of absorption was slightly slower in subjects with RI than in those with normal renal function, with a median t max of 2.0–2.5 and 1.0 h, respectively [96]. After reaching C max, plasma drug concentrations declined in a biphasic fashion, which is consistent with previous reports in healthy subjects and patients with T2DM with normal kidney function [96]. Empagliflozin AUC∞ values increased by approximately 18, 20, 66 and 48 % in subjects with mild, moderate and severe RI and ESRD, respectively, in comparison to healthy subjects, which was attributed to decreased renal clearance [96]. There were also decreases in the mean fraction of the dose excreted in urine following drug administration with increasing RI [96]. Because the increase in drug exposure remained rather limited, no dose adjustment of empagliflozin is required in patients with RI.
The cumulative UGE decreased with increasing RI. The UGE change from baseline over 24 h was 97.6 g in subjects with normal renal function, decreasing to approximately 61.6 g in subjects with mild RI, 55.6 g in subjects with moderate RI, 18.2 g in subjects with severe RI and 0.8 g in subjects with ESRD. The decrease in UGE followed the same pattern as the decreases in the empagliflozin renal clearance with increasing RI [96].
4.1.3 Impaired Liver Function
As the increase in empagliflozin exposure was less than twofold in patients with impaired liver function, no dose adjustment of empagliflozin is required in these patients [97]. However, the clinical experience of empagliflozin in patients with hepatic impairment is limited and thus caution is required.
4.2 Efficacy
4.2.1 Blood Glucose Control
The efficacy of empagliflozin has been evaluated in placebo-controlled RCTs in T2DM patients treated with diet and exercise (monotherapy) [98–100], in combination with metformin [101, 102], in combination with pioglitazone (± metformin) [103], in triple therapy added to metformin plus sulphonylurea [104], as add-on to usual care (in patients with CKD) [105] and in combination with insulin [106]. A significant reduction in HbA1c and fasting plasma glucose levels was observed with empagliflozin throughout the spectrum of T2DM, whatever the baseline glucose-lowering therapy [19]. In most studies, the reduction was slightly greater with empagliflozin 25 mg once daily than with empagliflozin 10 mg once daily (Table 5). A recent systematic review and meta-analysis included ten studies with 6,203 participants. Compared with placebo, mean changes in HbA1c were −0.62 % (95 % CI −0.68 to −0.57 %) for empagliflozin 10 mg and −0.66 % (95 % CI −0.76 to −0.57 %) for empagliflozin 25 mg. Despite the clinically relevant improvement of glucose control, the incidence of hypoglycaemia with empagliflozin was similar to placebo (odds ratio [OR] 1.10; 95 % CI 0.87–1.39) [107].
Empagliflozin is the only SGLT2 inhibitor that was compared with metformin [98, 108], glimepiride [109] and sitagliptin [99, 101, 108] (Table 3). Empagliflozin had glycaemic efficacy that appeared to be slightly lower than that of metformin, both at the 10 mg and the 25 mg doses (no statistical analysis available) [98, 108]. In contrast, empagliflozin appeared to be superior to glimepiride, a difference that increased from week 52 to week 104. At week 104, the adjusted mean difference in change from baseline in HbA1c with empagliflozin versus glimepiride was −0.11 % (95 % CI −0.19 to −0.02; p = 0.0153 for superiority) [109]. Confirmed hypoglycaemic adverse events at week 104 were reported in 2 % of patients treated with empagliflozin and 24 % of patients treated with glimepiride [109]. Compared to sitagliptin 100 mg once daily, empagliflozin induced similar HbA1c reduction at a dose of 10 mg/day but a greater HbA1c reduction at a dose of 25 mg once daily, with no difference regarding the risk of hypoglycaemia [107]. Again, the difference between empagliflozin and sitagliptin increased as the duration of the trials progressed from 12 to 90 weeks (no statistical analysis available) (Table 3) [99, 101, 108].
4.2.2 Weight Loss
In a meta-analysis of ten RCTs, empagliflozin was associated with modest but significant body weight loss (WMD −1.84 kg; 95 % CI −2.30 to −1.38 vs. placebo) [107]. No obvious differences in weight loss were observed between the doses of empagliflozin 10 and 25 mg in the various trials when the SGLT2 inhibitor was added to various background antidiabetic therapies (Table 5). Again, the empagliflozin-associated weight loss contrasted with the weight gain induced by a sulphonylurea [109] and the weight neutrality observed with sitagliptin [99, 101, 108] (Table 3). No detailed body composition study has been published yet with the SGLT2 inhibitor empagliflozin.
4.2.3 Blood Pressure Reduction
Treatment with empagliflozin 25 mg once daily had a favourable effect on SBP (WMD −4.19 mmHg; 95 % CI −5.17 to −3.20), and to a lesser extent on DBP (WMD −1.88 mmHg; 95 %CI −2.71 to −1.04) compared with placebo. These findings were similar for the 10 mg dose. Of note, 24-h ambulatory blood pressure monitoring utilised in one of the included studies reported similar estimates [107].
4.3 Safety
4.3.1 Urinary/Genital Infections
No increase in UTIs but a modest increase in genital infections were noticed with empagliflozin 10 or 25 mg in an extension 78-week phase of two clinical trials in monotherapy (compared with metformin) or as add-on to metformin (compared with sitagliptin) [108]. However, in a meta-analysis of RCTs with more than 6,000 participants, an increased risk of genital tract infections (OR 3.31; 95 % CI 1.55–7.09) was observed with empagliflozin compared with placebo or other glucose-lowering agents [107].
4.3.2 Other Concerns
The effects of empagliflozin on bone metabolism or fracture risk have not been reported yet. According to the European assessment report, the frequency of bone fractures was 1.6 % in the placebo group, 1.6 % in the empagliflozin 10 mg group, 1.1 % in the empagliflozin 25 mg group, and 1.5 % in the ‘all comparators’ group. In subgroup analyses, no clinically meaningful changes in bone metabolism parameters or bone mineral density were observed with empagliflozin [26].
5 Ipragliflozin
Ipragliflozin [Suglat® (Japan)], a new orally active SGLT2 inhibitor, has been developed by Astellas Pharma and Kotobuki Pharmaceutical for the treatment of T2DM. Ipragliflozin has received its first global approval in this indication in Japan, for use as monotherapy or in combination with another antihyperglycaemic agent. Ipragliflozin is the first SGLT2 inhibitor to be approved in Japan [20]. The drug is available as tablets of 25 and 50 mg, the latter dose being that evaluated in a placebo-controlled RCT in Japanese T2DM patients [110]. In contrast, the doses used in two clinical trials performed in the USA and in Europe were much higher, i.e. 150 and 300 mg once daily, but in much heavier T2DM patients than Japanese subjects [111, 112]. The clinical pharmacokinetics/pharmacodynamics of ipragliflozin have been well-studied [113]. The efficacy/safety profile of ipragliflozin appears to be similar to that of other SGLT2 inhibitors, although the clinical experience with this new agent is more limited than that with dapagliflozin, canagliflozin and empagliflozin.
5.1 Pharmacokinetic/Pharmacodynamic Analysis
5.1.1 Normal Kidney/Liver Function
A multiple ascending-dose study assessed the pharmacokinetics/pharmacodynamics of ipragliflozin in healthy subjects after single doses and multiple once-daily doses for 10 days (dose levels 5–600 mg). Ipragliflozin was rapidly absorbed, with a median t max of 1.3 h after the last dose (Table 1). The AUC increased proportionally with increasing dose. The mean t ½ was 12 h following the last dose (Table 1). Ipragliflozin dose-dependently increased UGE up to a maximum of approximately 59 g of glucose excreted over 24 h following multiple doses, without affecting plasma glucose levels in healthy subjects [114]. Ipragliflozin did not affect the pharmacokinetics of sitagliptin, pioglitazone or glimepiride, and vice versa, suggesting that no dose adjustments are likely to be required when ipragliflozin is given in combination with other glucose-lowering drugs in patients with T2DM [113, 115].
These pharmacokinetic findings were confirmed in drug-naïve T2DM patients who were randomised to placebo or ipragliflozin once daily at doses of 50, 100, 200 or 300 mg for 28 days. Significant dose-dependent increases in UGE were observed in all ipragliflozin groups [116]. All ipragliflozin doses significantly reduced fasting plasma glucose, mean amplitude of glucose excursions and HbA1c levels compared with placebo. Combination treatment for 14 days with ipragliflozin and metformin was well-tolerated in patients with T2DM without hypoglycaemia. The addition of ipragliflozin (300 mg once daily) to metformin therapy did not result in a clinically relevant change in the pharmacokinetic properties of metformin [117].
5.1.2 Impaired Kidney Function
In a study designed to investigate the effects of RI on the pharmacokinetics of ipragliflozin in Japanese T2DM patients, C max and AUC were 1.17 and 1.21 times higher, respectively, in subjects with moderate RI than in subjects with normal renal function [113, 118]. In another study performed in European T2DM patients with moderate and severe RI, the AUC of ipragliflozin was, respectively, 40 and 47 % higher than in T2DM patients with normal renal function. In the latter study, ipragliflozin increased glucosuria in direct, linear proportion to GFR and degree of hyperglycaemia. Although absolute glucosuria decreased with declining GFR, the efficiency of ipragliflozin action (fractional glucose excretion) was maintained in patients with severe RI [118].
5.1.3 Impaired Liver Function
Moderate hepatic impairment (Child-Pugh score of 7–9) had no clinically relevant effects on the single-dose pharmacokinetics of ipragliflozin and its major inactive metabolite (M2) in a single-dose open-label study [119].
5.2 Efficacy/Safety Profile
The approval of ipragliflozin is based on a series of pivotal phase III trials in Japanese T2DM patients. Four trials tested ipragliflozin as monotherapy and six evaluated ipragliflozin in combination with another glucose-lowering agent in patients with insufficient glycaemic control on that agent alone. The results of these trials have been summarised in a recent review [20] but few of them have been reported in peer-reviewed journals to date.
After 12 weeks of treatment, ipragliflozin ≥50 mg/day in patients with T2DM dose-dependently decreased HbA1c and the effect appeared comparable with that of metformin [111] (Table 3). Compared to placebo, ipragliflozin treatment improved glycaemic control when added to metformin therapy and may be associated with weight loss and reductions in blood pressure (Table 6) [112].
A placebo-controlled RCT examined the efficacy and safety of ipragliflozin in combination with metformin in Japanese patients with T2DM. Patients were randomised in a 2:1 ratio to 50 mg ipragliflozin or placebo once daily for 24 weeks. HbA1c decreased significantly in the ipragliflozin group (−0.87 %; adjusted mean difference from placebo: −1.30 %; p < 0.001), without inducing hypoglycaemia and associated with weight loss (Table 6). The overall incidence of treatment-emergent adverse events was similar in both groups [110]. Overall, these efficacy/safety findings were in agreement with those previously reported with other SGLT2 inhibitors, dapagliflozin, canagliflozin and empagliflozin [20].
6 Other Sodium–Glucose Co-Transporter Type 2 Inhibitors in Development
Several other SGLT2 inhibitors, mostly developed by Japanese companies, have received first global approval in Japan [23, 120].
6.1 Luseogliflozin
Luseogliflozin [Lusefi® (Japan)] is an orally active second-generation SGLT2 inhibitor developed by Taisho Pharmaceutical for the treatment of patients with T2DM. The drug (presented as tablets of 2.5 and 5 mg) has received its first global approval for this indication in Japan, either as monotherapy or in combination with other antihyperglycaemic agents [21].
Randomised, single-blind, placebo-controlled, single ascending-dose (1–25 mg) and multiple ascending-dose (5 or 10 mg/day, 7 days) trials were conducted in healthy male Japanese subjects to investigate the safety, pharmacokinetics and pharmacodynamics of luseogliflozin [121]. After administration of a single oral dose of luseogliflozin, C max and AUC increased in a dose-dependent manner, and no food effects were observed on pharmacokinetic parameters. t max ranged from 0.667 to 2.25 h. The mean plasma t ½ of luseogliflozin after multiple dosing for 7 days ranged from 9.14 to 10.7 h, and no detectable accumulation of luseogliflozin was observed. UGE increased in a dose-dependent manner, ranging from 18.9 to 70.9 g (single-dose study) [121].
Three phase II–III 12- to 24-week placebo-controlled RCTs demonstrated the efficacy of luseogliflozin in Japanese patients with T2DM not well-controlled with diet and exercise (Table 6) [122–124]. No RCTs with luseogliflozin as add-on therapy to other glucose-lowering agents or compared with other glucose-lowering agents have been published yet.
6.2 Tofogliflozin
Tofogliflozin [Apleway®, Deberza® (Japan)], an orally active small molecule SGLT2 inhibitor, has been developed by Chugai Pharmaceutical for the treatment of T2DM, and a marketing authorisation application was filed in Japan in 2013 by licensees Sanofi K. K. and Kowa. In March 2014, tofogliflozin (20 mg once-daily dose) received its first global approval for this indication in Japan as either monotherapy or in combination with other antihyperglycaemic agents [22]. Tofogliflozin 10, 20 or 40 mg administered once daily as monotherapy significantly decreased HbA1c and body weight, and was generally well-tolerated in Japanese patients with T2DM (Table 6) [125]. Phase III studies were recently completed and support the findings of this combined phase II and III study.
6.3 Ertugliflozin
The pharmacokinetics, metabolism and excretion of the SGLT2 inhibitor ertugliflozin (PF-04971729) have been reported in healthy male subjects [126]. Ertugliflozin was well-absorbed and eliminated largely via glucuronidation, in a comparable manner to that previously reported with other SGLT2 inhibitors.
6.4 Remogliflozin
Remogliflozin etabonate is a SGLT2 inhibitor initially investigated by GlaxoSmithKline and now being developed in phase II by BHV Pharma for the treatment of T2DM [127, 128].
7 Discussion
7.1 Overall Efficacy/Safety Profile
Expanding therapy options for a complex patient population such as T2DM is critical, and SGLT2 inhibitors offer new options in both monotherapy and in combination with other glucose-lowering agents [129, 130]. SGLT2 inhibitors are the only oral antidiabetic agents that improve glycaemic control while reducing body weight, an interesting combined effect due to the increasing dual burden of obesity and T2DM [131]. Because of their original mechanism of action, SGLT2 inhibitors act independently of insulin. This means that these medications are effective in the entire spectrum of T2DM, whatever the residual insulin secretion capacity and the insulin resistance status of the patient [131]. Indeed, analysis of the various RCTs demonstrates a very consistent effect on reduction in HbA1c and body weight, whatever the background glucose-lowering therapy and the nature of the SGLT2 inhibitor used (Table 7). Because of the renal mechanism of action, two components may directly influence the efficacy of SGLT2 inhibitors: the level of hyperglycaemia (the higher the hyperglycaemia, the greater the UGE and the greater glucose-lowering effect) and the level of renal function (the lower the GFR, the lower the UGE and presumably the lower the glucose-lowering effect) [6].
The selectivity for SGLT2 may vary across the various SGLT2 inhibitors. For instance, whereas dapagliflozin is a highly selective SGLT2 inhibitor, canagliflozin also exerts some inhibition on SGLT1 [14, 15]. These SGLT1 transporters are present in the kidneys where they normally contribute to only 10 % of glucose reabsorption in the tubule. However, this contribution may be increased in special conditions, particularly when SGLT2 transporters are inhibited [132]. SGLT1 transporters are also present in the intestinal tract where they contribute to intestinal glucose absorption. Canagliflozin was shown to delay intestinal glucose absorption, in addition to increasing UGE, a mechanism that may contribute to lower postprandial glucose and insulin levels [66]. The clinical relevance of this effect remains unknown and only direct comparison of the clinical efficacy of different SGLT2 inhibitors (dapagliflozin vs. canagliflozin, for instance) may answer this question. Such head-to-head trials are lacking in the current literature.
The incidence of hypoglycaemia was low in most T2DM groups treated with SGLT2 inhibitors, except among patients receiving a sulphonylurea or insulin. The OR for any hypoglycaemia with SGLT2 inhibitors was 1.28 (95 % CI 0.99–1.65) compared with placebo and 0.44 (95 % CI 0.35–0.54) compared with other antidiabetic medications, including sulphonylureas [7]. However, after exclusion of sulphonylureas, a similar hypoglycaemic risk was observed with SGLT2 inhibitors and other glucose-lowering agents. Across all studies analysed, severe hypoglycaemia episodes (defined as requiring assistance from another person) were rare in all treatment groups and were seen primarily in participants already receiving a sulphonylurea [7]. These data obtained in selected patients participating in RCTs remain to be confirmed in real-life conditions.
The most prevalent adverse event associated to SGLT2 inhibitors consists of urinary (UTIs) and genital infections. These effects may be explained easily by the increased UGE, although no strict correlations could be found between these infections and the amount of glucose excreted in the urine [61]. According to a recent systematic review of studies comparing SGLT2 inhibitors with placebo (45 studies; n = 11,232) or with other glucose-lowering agents used as active comparators (13 studies; n = 5,175), UTIs were more common with SGLT2 inhibitors (OR = 1.42; 95 % CI 1.06–1.90) [7]. However, further studies enrolling larger numbers of subjects with longer-term exposure to SGLT2 inhibitors will be necessary to more fully understand the impact of these agents on the risk of developing UTI in real life. The risk of mycotic genital infections associated with SGLT2 inhibitors appears to be somewhat higher. In the same systematic review, an increased incidence of genital tract infections was reported with SGLT2 inhibitors compared with placebo (OR = 3.50; 95 % CI 2.46–4.99) and active comparators (OR = 5.06; 95 % CI 3.44–7.45) [7]. In general, the incidence was higher in female (vaginitis) than in male (balanitis) T2DM patients. These data are in agreement with those reported in separate analysis for dapagliflozin [62], canagliflozin [87, 90] and empagliflozin [107]. However, events were generally mild to moderate in intensity and responded to standard treatments and led to very few cases of drug discontinuation.
7.2 Indirect Metabolic Effects Beyond Glucosuria
The glucuretic effect resulting from SGLT2 inhibition leads to complex metabolic consequences [27, 95]. First, by the reduction of chronic hyperglycaemia and the attenuation of glucose toxicity, SGLT2 inhibitors can improve both insulin secretion by the β cells and peripheral tissue insulin sensitivity [27]. Second, chronic urinary glucose loss most probably leads to some compensatory mechanisms [28, 94]. One should involve increased energy intake. Indeed, in all clinical trials, the final weight loss induced by SGLT2 inhibition is lower than that calculated from energy loss associated with chronically increased UGE [27]. This conclusion was supported by results with empagliflozin 25 mg daily in a recent proof-of-concept 8-week trial in patients with type 1 diabetes, which has compared UGE increase and weight loss. In that study, despite stable prandial insulin, daily carbohydrate intake increased by almost 50 g/day (p = 0.0007) [133]. This effect may contribute to avoid hypoglycaemia. Another compensatory mechanism concerns endogenous glucose production, which is enhanced and most probably driven by an increased glucagon secretion [28, 94]. The risk of deleterious consequences on muscle metabolism (possible muscle atrophy) during SGLT2 inhibition appears negligible, and weight loss concerns mainly fat mass rather than lean body mass [39, 41, 81]. However, caution is recommended in elderly fragile patients with some degree of sarcopenia. Overall, despite these complex compensatory mechanisms, SGLT2 inhibitors exert a clinically relevant glucose-lowering activity while promoting some weight loss, a unique dual effect among oral antidiabetic agents.
7.3 Clinical Use in Elderly Patients
The elderly population with T2DM is increasing and deserves much attention. Older patients are more fragile and the risk of hypoglycaemia must be minimised to avoid severe adverse events. When added to a usual background regimen in an older population (almost half of the recruited patients were 65 years or older) with advanced T2DM and pre-existing CV disease, dapagliflozin improved glycaemic control without an increase in hypoglycaemic risk, promoted weight loss and was generally well-tolerated [46]. Similarly, in a large 26-week RCT recruiting 716 T2DM patients aged 55–80 years (mean 63.6 years), treatment with canagliflozin improved HbA1c levels, reduced body weight and SBP, and again was overall well-tolerated [77]. Pooled data from four randomised, placebo-controlled, 26-week phase III studies (n = 2,313) evaluating canagliflozin 100 and 300 mg were analysed by age: <65 years (n = 1,868; mean age 52.8 years) or ≥65 years (n = 445; mean age 69.3 years). Canagliflozin improved glycaemic control, body weight and systolic BP, and was generally well-tolerated in older patients with T2DM, with similar findings to those recorded in younger patients [134].
Thus, safety of SGLT2 inhibitors in elderly patients was consistent with that in the general population [135]. However, risks and benefits of treatment with SGLT2 inhibitors should be assessed in geriatric patients on a case-by-case basis. The osmotic diuretic effect may lead to postural hypotension and dizziness in susceptible older subjects [87]. Dose adjustment and special caution may be recommended in patients taking loop diuretics, especially in elderly people, if there are concerns or symptoms of volume-related adverse effects [15]. Finally, renal function should be assessed before initiating therapy and more carefully monitored in the elderly population with T2DM because of the rather high incidence of CKD.
7.4 Clinical Use in Patients with Chronic Kidney Disease
In contrast to all other glucose-lowering medications, SGLT2 inhibitors specifically act in the kidney [6]. Therefore, special attention should be paid in patients with CKD or who are susceptible to developing diabetic nephropathy [136]. Impaired renal function may interfere with the pharmacokinetic/pharmacodynamic parameters of SGLT2 inhibitors and is susceptible to altering their glucose-lowering efficacy/safety profile. Another interesting question concerns the potential effects of SGLT2 inhibitors on renal function itself and whether this novel pharmacological effect may influence the development of diabetic/hypertensive nephropathy in at-risk patients [137].
Because the renal clearance of available SGLT2 inhibitors is low, RI only marginally affects SGLT2 inhibitor pharmacokinetic parameters and exposure to parent drug; most metabolites eliminated in the urine are inactive and thus do not interfere with the pharmacological effects of the medications [9]. Specific pharmacokinetic studies showed heterogeneous results in patients with different degrees of CKD [9]: according to reported changes in systemic exposure, the daily dose of dapagliflozin [29] or empagliflozin [96] should not be reduced in patients with moderate CKD, whereas the maximum dose recommended for canagliflozin is 100 mg instead of 300 mg/day [68, 69].
In contrast, as already discussed (see Sects. 2.1.3, 3.1.3 and 4.1.3) [9, 138], pharmacodynamic changes are dependent on renal function and UGE progressively decreases with the degree of RI, leading to a progressive attenuation of the glucose-lowering effects in T2DM patients [29, 96, 118]. Nevertheless, despite the reduction in UGE in proportion to decreased GFR, the efficacy of SGLT2 inhibitors was demonstrated in patients with various degrees (mild to moderate) of CKD. In subjects with T2DM and stage 3 CKD, canagliflozin reduced HbA1c, body weight and blood pressure, and was generally well-tolerated (Table 4) [78, 79, 139]. The maximum recommended dosage is canagliflozin 100 mg once daily in patients with moderate RI, but the drug is not recommended in those with an estimated GFR (eGFR) of <45 mL/min/1.73 m2 [25]. In patients with T2DM and stage 2 or 3 CKD, empagliflozin reduced HbA1c and was well-tolerated (Table 5) [105]. The recommended maximum daily dose of empagliflozin in patients with stage 3 CKD is 10 mg and the drug should be discontinued when eGFR is persistently below 45 mL/min/1.73 m2 [26]. In another study in patients with moderate RI (stage 3 CKD), dapagliflozin did not improve glycaemic control, but nevertheless reduced body weight and blood pressure after 24 weeks (Table 2) [47]. Therefore, dapagliflozin is not recommended in T2DM patients with eGFR <60 mL/min/1.73 m2 (Table 7) [24]. It is noteworthy that all of these findings obtained in RCTs might not be applicable to the general population of patients with T2DM and CKD.
Even if SGLT2 inhibitors have been shown to be efficacious and safe in diabetic patients with mild to moderate CKD [78, 79], all drugs that may interfere with renal function, by decreasing GFR (for instance, NSAIDs), may reduce the glucose-lowering effects of SGLT2 inhibitors in fragile patients. In patients with normal renal function or mild RI, canagliflozin 300 mg was associated with slightly increased incidence of rare renal-related adverse events compared with placebo [7]. In clinical practice, renal function should be regularly monitored in diabetic patients treated with SGLT2 inhibitors, especially in patients with mild/moderate CKD, and all agents that may interfere with kidney function should be used with caution [138]. The approved SGLT2 inhibitors have limited use based on kidney function and should be used only in those with an eGFR >60 mL/min/1.73 m2 for dapagliflozin and ≥45 mL/min/1.73 m2 for canagliflozin and empagliflozin [136].
It has been suggested that early tubular cell proliferation and increased sodium–glucose co-transport, as triggered by the diabetic milieu, enhance proximal tubule reabsorption and make the GFR supranormal through the physiology of tubuloglomerular feedback. Thus, when investigating renal function in diabetic disease models, the tubular system may play a role in the pathophysiology of the diabetic kidney [140]. Clinical data for the nephroprotective effects of SGLT2 inhibitors currently are limited compared to the more extensive experimental literature [141]. Interestingly, short-term treatment with empagliflozin attenuated renal hyperfiltration in subjects with type 1 diabetes, likely by affecting tubular–glomerular feedback mechanisms [142]. These findings, although preliminary, suggest that SGLT2 inhibitors may have a role in diabetic kidney disease [137]. Further studies are required to investigate the long-term renal outcomes with SGLT2 inhibitors in patients with T2DM. This issue will be evaluated as a secondary endpoint in a ongoing 4-year trial comparing empagliflozin and glimepiride in patients with T2DM inadequately controlled with metformin and diet/exercise [143]. Two other more specific trials are currently investigating the effects of canagliflozin on renal endpoints in adult participants with T2DM [CANVAS-R (Study of the Effects of Canagliflozin on Renal Endpoints in Adult Subjects with T2DM)] (ClinicalTrials.gov identifier NCT01989754); [CREDENCE (Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation)] (ClinicalTrials.gov Identifier NCT02065791) [144]. Most probably, valuable information will also be drawn from several ongoing prospective studies with primary CV outcomes (see below).
7.5 Clinical Use in Patients with Cardiovascular Risk/Disease
Evidence concerning the importance of glucose lowering in the prevention of CV outcomes remains controversial [144]. Given the multi-faceted pathogenesis of atherosclerosis in diabetes, it is likely that any intervention to mitigate this risk must address CV risk factors beyond glycaemia alone. Phase II–III RCTs all together demonstrate that SGLT2 inhibitors improve glucose control, body weight and blood pressure when used as monotherapy or add-on to other antihyperglycaemic agents in patients with T2DM. Thus, SGLT2 inhibitors appear to have the potential to reduce CV risk in patients with T2DM through reductions in various risk markers/factors [145, 146]. In a systematic review and meta-analysis, SGLT2 inhibitors significantly reduced both SBP (WMD −4.0 mmHg; 95 % CI −4.4 to −3.5) and DBP (WMD −1.6 mmHg; 95 % CI −1.9 to −1.3) from baseline [85]. While not approved for blood pressure lowering, they may potentially aid blood pressure goal achievement in people within 7–10 mmHg of goal [59]. However, in another systematic review, a higher risk for hypotension was found with SGLT2 inhibitors than with other antidiabetic medications (OR = 2.68; 95 % CI 1.14–6.29) [7]. Orthostatic hypotension should be avoided in fragile elderly patients, especially in those receiving loop diuretics, even if it appears to be a rather rare event [85]. The effects of SGLT2 inhibitors on lipid profile appear limited [146], although controversial. Indeed, a concerning adverse effect of canagliflozin is an average 8 % increase in plasma levels of low-density lipoprotein cholesterol (LDL-C) compared with placebo [87]. However, some beneficial lipid effects (increased high-density lipoprotein cholesterol and decreased triglycerides) were also reported with canagliflozin [16]. Finally, SGLT2 inhibitors lower serum uric acid, an independent CV marker, through alteration of uric acid transport activity in renal tubule by increased glucosuria [147].
When added to a usual background regimen in an older population with advanced T2DM and pre-existing CV disease, dapagliflozin improved glycaemic control without an increase in hypoglycaemic risk, promoted weight loss and was well-tolerated [46]. However, this trial was not designed to investigate the effects of dapagliflozin on CV events in this high-risk population. A meta-analysis of CV outcomes for dapagliflozin, which was based on 14 trials (n ≈ 6,300), yielded an OR of 0.73 (95 % CI 0.46–1.16) compared with control [7]. Similarly, canagliflozin was not associated with an increased risk for the composite CV outcome compared with placebo or active comparator on the basis of data from ten trials that included a total of 10,474 patients (OR = 0.95; 95 % CI 0.71–1.26) [7].
No adequately powered trial has yet determined the effects of an SGLT2 inhibitor on either macrovascular or microvascular outcomes. However, a number of large-scale prospective trials are now ongoing to demonstrate the safety and possibly the efficacy of SGLT2 inhibitors on CV outcomes in T2DM patients at CV risk [148]. DECLARE (Dapagliflozin Effect on Cardiovascular Events)-TIMI (Thrombolysis In Myocardial Infarction) 58 (ClinicalTrials.gov identifier NCT01730534) is a multicentre, randomised, double-blind, placebo-controlled trial to evaluate the effect of dapagliflozin 10 mg once daily on the incidence of CV death, myocardial infarction or ischaemic stroke in patients with T2DM [149]. CANVAS (Canagliflozin Cardiovascular Assessment Study; ClinicalTrials.gov identifier NCT01032629 [149]) is a double-blind, placebo-controlled trial designed to evaluate the effects of canagliflozin (100 or 300 mg once daily) on the risk of CV disease and to assess safety and tolerability in patients with inadequately controlled T2DM and increased CV risk [149]. EMPA-REG OUTCOME™ (ClinicalTrials.gov identifier NCT01131676 [149]) is a double-blind placebo-controlled trial designed to determine the CV safety of empagliflozin (10 or 25 mg once daily) in a cohort of patients with T2DM and high CV risk, with the potential to show cardioprotection [150]. Results of all these CV outcome trials will be available within the next 4–5 years [148]. It will be of great interest to compare these results with those obtained in CV outcome studies performed with several DPP-4 inhibitors [151] to decide whether one or the other class offers a better cardioprotection in high-risk patients with T2DM. This comparison will help the physician in his/her choice between a SGLT2 inhibitor and a DPP-4 inhibitor after failure of monotherapy with metformin for the management of hyperglycaemia in T2DM [152].
7.6 Clinical Perspectives
Due to their complementary modes of action, there is a good rationale to combine a SGLT2 inhibitor with metformin or a SGLT2 inhibitor with a DPP-4 inhibitor to improve glycaemic control in patients with T2DM. Fixed-drug combinations are in current development, for instance a combination dapagliflozin–saxagliptin or empagliflozin–linagliptin. Findings of a pharmacokinetic study support the coadministration of empagliflozin and linagliptin without dose adjustments [153]. The combination of an SGLT2 inhibitor and an incretin mimetic/analogue such as liraglutide results in improved glycaemic control accompanied by significant weight loss [154]. This combination needs to be studied in a prospective RCT because the effect of each of the components of this combination is synergistically magnified by the addition of the partner drug [131].
Another promising perspective is the development of pharmacological agents that exert a dual inhibition of SGLT1 and SGLT2. SGLT1 is the primary co-transporter for glucose absorption from the gastrointestinal tract, and, as already discussed, SGLT2 is the primary co-transporter for glucose reabsorption in the kidney. In healthy subjects, SGLT1 inhibition reduced postprandial glucose levels and increased the release of gastrointestinal peptides such as glucagon-like peptide 1 (GLP-1) and peptide tyrosine tyrosine (PYY), whereas SGLT2 inhibition resulted in increased UGE [155]. LX4211, a first-in-class dual inhibitor of SGLT1 and SGLT2, was safe and well-tolerated and, due to its SGLT1 inhibition, produced strong postprandial glucose reductions and low UGE relative to selective SGLT2 inhibitors [155]. LX4211 may provide a promising new therapy for patients with T2DM, as suggested by the encouraging results of a short-term pilot study [156]. The potential long-term clinical benefits and safety of LX4211 treatment of T2DM will need to be confirmed in large clinical trials. Other dual SGLT1 and SGLT2 inhibitors (sotagliflozin or LX4211, Lexicon Pharmaceuticals; and LIK 066, Novartis) are in development and phase III trials are expected to begin in the near future [23].
SGLT2 inhibitors have a unique insulin-independent mechanism of action, which suggests that this new pharmacological approach could also be useful for the management of type 1 diabetes. Studies on adjunct therapeutic effects of SGLT2 inhibitors in individuals with type 1 diabetes are limited, but initial reports show favourable effects on reducing HbA1c, body weight, total daily insulin dose and hypoglycaemic events [157, 158]. Intriguingly, this drug may confer a degree of renal protection by reducing glomerular hyperfiltration that can arise in the diabetic state [142].
New glucose-lowering agents such as SGLT2 inhibitors are much more expensive than classical glucose-lowering agents (metformin, sulphonylureas), may have a price around that of DPP-4 inhibitors (gliptins), but are cheaper than injectable agents such as GLP-1 receptor agonists. No comparative pharmacoeconomic analysis of SGLT2 inhibitor therapy in T2DM has been published to date [159].
8 Conclusion
Multiple therapeutic classes of agents are available for the treatment of T2DM, and the armamentarium has expanded significantly in the past decade. Nevertheless, many patients with T2DM do not achieve and/or maintain glycaemic targets, despite therapy implementation and escalation. Most of the available antidiabetic agents aim at promoting insulin secretion or reducing insulin resistance. The kidney plays a vital role in maintaining blood glucose homeostasis by recovering glucose from glomerular filtrate, which is controlled by SGLT2 co-transporters expressed mainly in proximal tubule. In T2DM patients, inhibition of SGLT2 normalises glycaemic levels by preventing glucose from being reabsorbed through SGLT2 and re-entering the circulation. Thus, SGLT2 inhibition seems to be a logical approach and poses a novel insulin-independent mechanism of action for management of T2DM by promoting UGE in the body. This original mechanism results in decreased serum glucose, without hypoglycaemia, and offers the advantage of promoting weight loss and lowering blood pressure.
SGLT2 inhibitors have proven their efficacy in numerous placebo-controlled trials as monotherapy or in combination with various other glucose-lowering agents. They have been shown to be as effective as other antihyperglycaemic agents such as metformin, sulphonylureas or sitagliptin in head-to-head trials. SGLT2 inhibitors may offer the advantage of a better glucose-lowering durability, although this remains to be proven in long-term comparative studies. The overall safety profile of SGLT2 inhibitors is good, with a limited risk of hypoglycaemia, but a higher risk of benign UTIs and mycotic genital infections. No adequately powered trial has yet determined the effects of an SGLT2 inhibitor on either macrovascular or microvascular outcomes. However, a number of large-scale prospective trials are now ongoing to evaluate the clinical effects of SGLT2 inhibitors, especially on CV outcomes in high-risk patients with T2DM. The long-term effects on renal function also deserve further attention. If positive findings were obtained with SGLT2 inhibitors regarding glucose-lowering durability, CV outcomes or decreased risk of diabetic nephropathy, these results would be considered as major breakthroughs in the management of T2DM. Results from these clinical studies will help define the role for this new class of oral antidiabetic agents, with its unique mechanism of action, as a treatment option for reducing hyperglycaemia in patients with T2DM.
References
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2012;55(6):1577–96.
Tahrani AA, Bailey CJ, Del Prato S, et al. Management of type 2 diabetes: new and future developments in treatment. Lancet. 2011;378(9786):182–97.
Tahrani AA, Barnett AH, Bailey CJ. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol. 2013;1(2):140–51.
Hasan FM, Alsahli M, Gerich JE. SGLT2 inhibitors in the treatment of type 2 diabetes. Diabetes Res Clin Pract. 2014;104(3):297–322.
Bailey CJ. Renal glucose reabsorption inhibitors to treat diabetes. Trends Pharmacol Sci. 2011;32(2):63–71.
Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32(4):515–31.
Vasilakou D, Karagiannis T, Athanasiadou E, et al. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med. 2013;159(4):262–74.
Berhan A, Barker A. Sodium glucose co-transport 2 inhibitors in the treatment of type 2 diabetes mellitus: a meta-analysis of randomized double-blind controlled trials. BMC Endocr Disord. 2013;13(1):58.
Scheen AJ. Evaluating SGLT2 inhibitors for type 2 diabetes: pharmacokinetic and toxicological considerations. Expert Opin Drug Metab Toxicol. 2014;10(5):647–63.
Scheen AJ. Drug-drug interactions with SGLT-2 inhibitors, new oral glucose-lowering agents for the management of type 2 diabetes. Clin Pharmacokinet. 2014;53(4):295–304.
Neumiller JJ, White JR Jr, Campbell RK. Sodium-glucose co-transport inhibitors: progress and therapeutic potential in type 2 diabetes mellitus. Drugs. 2010;70(4):377–85.
Plosker GL. Dapagliflozin: a review of its use in type 2 diabetes mellitus. Drugs. 2012;72(17):2289–312.
Kasichayanula S, Liu X, Lacreta F, et al. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin Pharmacokinet. 2014;53(1):17–27.
Elkinson S, Scott LJ. Canagliflozin: first global approval. Drugs. 2013;73(9):979–88.
Lamos EM, Younk LM, Davis SN. Canagliflozin, an inhibitor of sodium-glucose cotransporter 2, for the treatment of type 2 diabetes mellitus. Expert Opin Drug Metab Toxicol. 2013;9(6):763–75.
Plosker GL. Canagliflozin: a review of its use in patients with type 2 diabetes mellitus. Drugs. 2014;74(7):807–24.
Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet. 2014;53(3):213–25.
Seman L, Macha S, Nehmiz G, et al. Empagliflozin (BI 10773), a potent and selective SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Drug Dev. 2013;2(2):152–61.
Scott LJ. Empagliflozin: a review of its use in patients with type 2 diabetes mellitus. Drugs. 2014;74(15):1769–84.
Poole RM, Dungo RT. Ipragliflozin: first global approval. Drugs. 2014;74(5):611–7.
Markham A, Elkinson S. Luseogliflozin: first global approval. Drugs. 2014;74(8):945–50.
Poole RM, Prossler JE. Tofogliflozin: first global approval. Drugs. 2014;74(8):939–44.
Nauck MA. Update on developments with SGLT2 inhibitors in the management of type 2 diabetes. Drug Des Devel Ther. 2014;8:1335–80.
European Medicines Agency. Assessment report. Forxiga (dapagliflozin). 2012. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002322/WC500136024.pdf. Accessed 20 Oct 2014.
European Medicines Agency. Assessment report. Invokana (canagliflozin). 2013. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/002649/WC500156455.pdf. Accessed 27 Oct 2014.
European Medicines Agency. Assessment report. Jardiance. International non-proprietary name: empagliflozin. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002677/WC500168594.pdf. Accessed 20 Oct 2014.
Scheen AJ, Paquot N. Metabolic effects SGLT2 inhibitors beyond increased glucosuria : a review of clinical evidence. Diabetes Metab. 2014;40(Suppl):S4–11.
Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124(2):509–14.
Kasichayanula S, Liu X, Pe Benito M, et al. The influence of kidney function on dapagliflozin exposure, metabolism and pharmacodynamics in healthy subjects and in patients with type 2 diabetes mellitus. Br J Clin Pharmacol. 2013;76(3):432–44.
Kasichayanula S, Liu X, Zhang W, et al. Influence of hepatic impairment on the pharmacokinetics and safety profile of dapagliflozin: an open-label, parallel-group, single-dose study. Clin Ther. 2011;33(11):1798–808.
List JF, Woo V, Morales E, et al. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32(4):650–7.
Ferrannini E, Ramos SJ, Salsali A, et al. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33(10):2217–24.
Bailey CJ, Iqbal N, T’Joen C, et al. Dapagliflozin monotherapy in drug-naive patients with diabetes: a randomized-controlled trial of low-dose range. Diabetes Obes Metab. 2012;14(10):951–9.
Kaku K, Inoue S, Matsuoka O, et al. Efficacy and safety of dapagliflozin as a monotherapy for type 2 diabetes mellitus in Japanese patients with inadequate glycaemic control: a phase II multicentre, randomized, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2013;15(5):432–40.
Ji L, Ma J, Li H, et al. Dapagliflozin as monotherapy in drug-naive Asian patients with type 2 diabetes mellitus: a randomized, blinded, prospective phase III study. Clin Ther. 2014;36(1):84–100.e9.
Kaku K, Kiyosue A, Inoue S, et al. Efficacy and safety of dapagliflozin monotherapy in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise. Diabetes Obes Metab. 2014;16(11):1102–10.
Bailey CJ, Gross JL, Pieters A, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375(9733):2223–33.
Bailey CJ, Gross JL, Hennicken D, et al. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med. 2013;11:43.
Bolinder J, Ljunggren O, Kullberg J, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab. 2012;97(3):1020–31.
Henry RR, Murray AV, Marmolejo MH, et al. Dapagliflozin, metformin XR, or both: initial pharmacotherapy for type 2 diabetes, a randomised controlled trial. Int J Clin Pract. 2012;66(5):446–56.
Bolinder J, Ljunggren O, Johansson L, et al. Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes Metab. 2014;16(2):159–69.
Strojek K, Yoon KH, Hruba V, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13(10):928–38.
Strojek K, Yoon KH, Hruba V, et al. Dapagliflozin added to glimepiride in patients with type 2 diabetes mellitus sustains glycemic control and weight loss over 48 weeks: a randomized, double-blind, parallel-group, placebo-controlled trial. Diabetes Ther. 2014;5(1):267–83.
Rosenstock J, Vico M, Wei L, et al. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA(1c), body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35(7):1473–8.
Jabbour SA, Hardy E, Sugg J, et al. Dapagliflozin is effective as add-on therapy to sitagliptin with or without metformin: a 24-week, multicenter, randomized, double-blind, placebo-controlled study. Diabetes Care. 2014;37(3):740–50.
Leiter LA, Cefalu WT, de Bruin TW, et al. Dapagliflozin added to usual care in individuals with type 2 diabetes mellitus with preexisting cardiovascular disease: a 24-week, multicenter, randomized, double-blind, placebo-controlled study with a 28-week extension. J Am Geriatr Soc. 2014;62(7):1252–62.
Kohan DE, Fioretto P, Tang W, et al. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int. 2014;85(4):962–71.
Wilding JP, Woo V, Soler NG, et al. Long-term efficacy of dapagliflozin in patients with type 2 diabetes mellitus receiving high doses of insulin: a randomized trial. Ann Intern Med. 2012;156(6):405–15.
Wilding JP, Woo V, Rohwedder K, et al.; Dapagliflozin 006 Study Group. Dapagliflozin in patients with type 2 diabetes receiving high doses of insulin: efficacy and safety over 2 years. Diabetes Obes Metab. 2014;16(2):124–36.
Musso G, Gambino R, Cassader M, et al. A novel approach to control hyperglycemia in type 2 diabetes: sodium glucose co-transport (SGLT) inhibitors: systematic review and meta-analysis of randomized trials. Ann Med. 2012;44(4):375–93.
Sun YN, Zhou Y, Chen X, et al. The efficacy of dapagliflozin combined with hypoglycaemic drugs in treating type 2 diabetes mellitus: meta-analysis of randomised controlled trials. BMJ Open. 2014;4(4):e004619.
Zhang M, Zhang L, Wu B, et al. Dapagliflozin treatment for type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. Diabetes Metab Res Rev. 2014;30(3):204–21.
Nauck MA, Del Prato S, Meier JJ, et al. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: a randomized, 52-week, double-blind, active-controlled noninferiority trial. Diabetes Care. 2011;34(9):2015–22.
Nauck MA, Del Prato S, Duran-Garcia S, et al. Durability of glycaemic efficacy over 2 years with dapagliflozin versus glipizide as add-on therapies in patients whose type 2 diabetes mellitus is inadequately controlled with metformin. Diabetes Obes Metab. 2014;16(11):1111–20.
Goring S, Hawkins N, Wygant G, et al. Dapagliflozin compared with other oral anti-diabetes treatments when added to metformin monotherapy: a systematic review and network meta-analysis. Diabetes Obes Metab. 2014;16(5):433–42.
Barnett AH. Impact of sodium glucose cotransporter 2 inhibitors on weight in patients with type 2 diabetes mellitus. Postgrad Med. 2013;125(5):92–100.
Lambers Heerspink HJ, de Zeeuw D, Wie L, et al. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab. 2013; 15 (9):853–62.
Grandy S, Hashemi M, Langkilde AM, et al. Changes in weight loss-related quality of life among type 2 diabetes mellitus patients treated with dapagliflozin. Diabetes Obes Metab. 2014;16(7):645–50.
Oliva RV, Bakris GL. Blood pressure effects of sodium-glucose co-transport 2 (SGLT2) inhibitors. J Am Soc Hypertens. 2014;8(5):330–9.
Engeli S, Jordan J. Novel metabolic drugs and blood pressure: implications for the treatment of obese hypertensive patients? Curr Hypertens Rep. 2013;15(5):470–4.
Johnsson KM, Ptaszynska A, Schmitz B, et al. Urinary tract infections in patients with diabetes treated with dapagliflozin. J Diabetes Complications. 2013;27(5):473–8.
Johnsson KM, Ptaszynska A, Schmitz B, et al. Vulvovaginitis and balanitis in patients with diabetes treated with dapagliflozin. J Diabetes Complications. 2013;27(5):479–84.
Ljunggren O, Bolinder J, Johansson L, et al. Dapagliflozin has no effect on markers of bone formation and resorption or bone mineral density in patients with inadequately controlled type 2 diabetes mellitus on metformin. Diabetes Obes Metab. 2012;14(11):990–9.
Sha S, Devineni D, Ghosh A, et al. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab. 2011;13(7):669–72.
Devineni D, Morrow L, Hompesch M, et al. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab. 2012;14(6):539–45.
Polidori D, Sha S, Mudaliar S, et al. Canagliflozin lowers postprandial glucose and insulin by delaying intestinal glucose absorption in addition to increasing urinary glucose excretion: results of a randomized, placebo-controlled study. Diabetes Care. 2013;36(8):2154–61.
Polidori D, Mari A, Ferrannini E. Canagliflozin, a sodium glucose co-transporter 2 inhibitor, improves model-based indices of beta cell function in patients with type 2 diabetes. Diabetologia. 2014;57(5):891–901.
Food and Drug Administration. Center for Drug Evaluation and Research report. Canagliflozin (Invokana). 2014. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204042Orig1s000ClinPharmR.pdf. Accessed 20 Oct 2014.
Devineni D, Marbury T, Curtin C, et al. Effects of renal function on canagliflozin (CANA) pharmacokinetics (PK) and pharmacodynamics (PD) in non-diabetic subjects (abstract no. PUB295). J Am Soc Nephrol. 2012;23:961A.
Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab. 2013;15(4):372–82.
Inagaki N, Kondo K, Yoshinari T, et al. Efficacy and safety of canagliflozin monotherapy in Japanese patients with type 2 diabetes inadequately controlled with diet and exercise: a 24-week, randomized, double-blind, placebo-controlled. Phase III study. Expert Opin Pharmacother. 2014;15(11):1501–15.
Rosenstock J, Aggarwal N, Polidori D, et al. Dose-ranging effects of canagliflozin, a sodium-glucose cotransporter 2 inhibitor, as add-on to metformin in subjects with type 2 diabetes. Diabetes Care. 2012;35(6):1232–8.
Lavalle-Gonzalez FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomised trial. Diabetologia. 2013;56(12):2582–92.
Qiu R, Capuano G, Meininger G. Efficacy and safety of twice-daily treatment with canagliflozin, a sodium glucose co-transporter 2 inhibitor, added on to metformin monotherapy in patients with type 2 diabetes mellitus. J Clin Translat Endocrinol. 2014;1:54–60.
Wilding JP, Charpentier G, Hollander P, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sulphonylurea: a randomised trial. Int J Clin Pract. 2013;67(12):1267–82.
Forst T, Guthrie R, Goldenberg R, et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes on background metformin and pioglitazone. Diabetes Obes Metab. 2014;16(5):467–77.
Bode B, Stenlöf K, Sullivan D, et al. Efficacy and safety of canagliflozin treatment in older subjects with type 2 diabetes mellitus: a randomized trial. Hosp Pract (1995). 2013;41(2):72–84.
Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab. 2013;15(5):463–73.
Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes mellitus and chronic kidney disease. Diabetes Obes Metab. 2014;16(10):1016–27.
Yang XP, Lai D, Zhong XY, et al. Efficacy and safety of canagliflozin in subjects with type 2 diabetes: systematic review and meta-analysis. Eur J Clin Pharmacol. 2014;70(10):1149–58.
Cefalu WT, Leiter LA, Yoon KH, et al. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet. 2013;382(9896):941–50.
Schernthaner G, Gross JL, Rosenstock J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care. 2013;36(9):2508–15.
Bailey RA, Damaraju CV, Martin SC, et al. Attainment of diabetes-related quality measures with canagliflozin versus sitagliptin. Am J Manag Care. 2014;20(1 Suppl):s16–24.
Traina S, Guthrie R, Slee A. The impact of weight loss on weight-related quality of life and health satisfaction: results from a trial comparing canagliflozin with sitagliptin in triple therapy among people with type 2 diabetes. Postgrad Med. 2014;126(3):7–15.
Baker WL, Smyth LR, Riche DM, et al. Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: a systematic review and meta-analysis. J Am Soc Hypertens. 2014;8(4):262–75.e9.
Sha S, Polidori D, Heise T, et al. Effect of the sodium glucose co-transporter 2 inhibitor canagliflozin on plasma volume in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2014;16(11):1087–95.
Mikhail N. Safety of canagliflozin in patients with type 2 diabetes. Curr Drug Saf. 2014;9(2):127–32.
Nicolle LE, Capuano G, Ways K, et al. Effect of canagliflozin, a sodium glucose co-transporter 2 (SGLT2) inhibitor, on bacteriuria and urinary tract infection in subjects with type 2 diabetes enrolled in a 12-week, phase 2 study. Curr Med Res Opin. 2012;28(7):1167–71.
Nyirjesy P, Zhao Y, Ways K, et al. Evaluation of vulvovaginal symptoms and Candida colonization in women with type 2 diabetes mellitus treated with canagliflozin, a sodium glucose co-transporter 2 inhibitor. Curr Med Res Opin. 2012;28(7):1173–8.
Nyirjesy P, Sobel JD, Fung A, et al. Genital mycotic infections with canagliflozin, a sodium glucose co-transporter 2 inhibitor, in patients with type 2 diabetes mellitus: a pooled analysis of clinical studies. Curr Med Res Opin. 2014;30(6):1109–19.
Grempler R, Thomas L, Eckhardt M, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14(1):83–90.
Heise T, Seewaldt-Becker E, Macha S, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics following 4 weeks’ treatment with empagliflozin once daily in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(7):613–21.
Heise T, Seman L, Macha S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple rising doses of empagliflozin in patients with type 2 diabetes mellitus. Diabetes Ther. 2013;4(2):331–45.
Ferrannini E, Muscelli E, Frascerra S, et al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest. 2014;124(2):499–508.
Cefalu WT. Paradoxical insights into whole body metabolic adaptations following SGLT2 inhibition. J Clin Invest. 2014;124(2):485–7.
Macha S, Mattheus M, Halabi A, et al. Pharmacokinetics, pharmacodynamics and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in subjects with renal impairment. Diabetes Obes Metab. 2014;16(3):215–22.
Macha S, Rose P, Mattheus M, et al. Pharmacokinetics, safety and tolerability of empagliflozin, a sodium glucose cotransporter 2 inhibitor, in patients with hepatic impairment. Diabetes Obes Metab. 2014;16(2):118–23.
Ferrannini E, Seman L, Seewaldt-Becker E, et al. A Phase IIb, randomized, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(8):721–8.
Roden M, Weng J, Eilbracht J, et al. Empagliflozin monotherapy in drug-naïve patients with type 2 diabetes: a randomised, 24-week, double-blind, placebo-controlled, parallel group, trial with sitagliptin as active comparator. Lancet Diabetes Endocrinol. 2013;1(3):208–19.
Kadowaki T, Haneda M, Inagaki N, et al. Empagliflozin monotherapy in Japanese patients with type 2 diabetes mellitus: a randomized, 12-week, double-blind, placebo-controlled. Phase II trial. Adv Ther. 2014;31(6):621–38.
Rosenstock J, Seman LJ, Jelaska A, et al. Efficacy and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, as add-on to metformin in type 2 diabetes with mild hyperglycaemia. Diabetes Obes Metab. 2013;15(12):1154–60.
Haring HU, Merker L, Seewaldt-Becker E, et al. Empagliflozin as add-on to metformin in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2014;37(6):1650–9.
Kovacs CS, Seshiah V, Swallow R, et al. Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: a 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab. 2014;16(2):147–58.
Haring HU, Merker L, Seewaldt-Becker E, et al. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2013;36(11):3396–404.
Barnett AH, Mithal A, Manassie J, et al. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2(5):369–84.
Rosenstock J, Jelaska A, Frappin G, et al. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. 2014;37(7):1815–23.
Liakos A, Karagiannis T, Athanasiadou E, et al. Efficacy and safety of empagliflozin for type 2 diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2014;16(10):984–93.
Ferrannini E, Berk A, Hantel S, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care. 2013;36(12):4015–21.
Ridderstrale M, Andersen KR, Zeller C, et al. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2(9):691–700.
Kashiwagi A, Kazuta K, Goto K, et al. Ipragliflozin in combination with metformin for the treatment of Japanese patients with type 2 diabetes: ILLUMINATE, a randomised, double-blind, placebo-controlled study. Diabetes Obes Metab. Epub 2014 Jun 12. doi: 10.1111/dom.12331.
Fonseca VA, Ferrannini E, Wilding JP, et al. Active- and placebo-controlled dose-finding study to assess the efficacy, safety, and tolerability of multiple doses of ipragliflozin in patients with type 2 diabetes mellitus. J Diabetes Complications. 2013;27(3):268–73.
Wilding JP, Ferrannini E, Fonseca VA, et al. Efficacy and safety of ipragliflozin in patients with type 2 diabetes inadequately controlled on metformin: a dose-finding study. Diabetes Obes Metab. 2013;15(5):403–9.
Kadokura T, Zhang W, Krauwinkel W, et al. Clinical pharmacokinetics and pharmacodynamics of the novel SGLT2 inhibitor ipragliflozin. Clin Pharmacokinet. 2014;53(11):975–88.
Veltkamp SA, Kadokura T, Krauwinkel WJ, et al. Effect of Ipragliflozin (ASP1941), a novel selective sodium-dependent glucose co-transporter 2 inhibitor, on urinary glucose excretion in healthy subjects. Clin Drug Investig. 2011;31(12):839–51.
Smulders RA, Zhang W, Veltkamp SA, et al. No pharmacokinetic interaction between ipragliflozin and sitagliptin, pioglitazone, or glimepiride in healthy subjects. Diabetes Obes Metab. 2012;14(10):937–43.
Schwartz SL, Akinlade B, Klasen S, et al. Safety, pharmacokinetic, and pharmacodynamic profiles of ipragliflozin (ASP1941), a novel and selective inhibitor of sodium-dependent glucose co-transporter 2, in patients with type 2 diabetes mellitus. Diabetes Technol Ther. 2011;13(12):1219–27.
Veltkamp SA, van Dijk J, Collins C, et al. Combination treatment with ipragliflozin and metformin: a randomized, double-blind, placebo-controlled study in patients with type 2 diabetes mellitus. Clin Ther. 2012;34(8):1761–71.
Ferrannini E, Veltkamp SA, Smulders RA, et al. Renal glucose handling: impact of chronic kidney disease and sodium-glucose cotransporter 2 inhibition in patients with type 2 diabetes. Diabetes Care. 2013;36(5):1260–5.
Zhang W, Krauwinkel WJ, Keirns J, et al. The effect of moderate hepatic impairment on the pharmacokinetics of ipragliflozin, a novel sodium glucose co-transporter 2 (SGLT2) inhibitor. Clin Drug Investig. 2013;33(7):489–96.
Kurosaki E, Ogasawara H. Ipragliflozin and other sodium-glucose cotransporter-2 (SGLT2) inhibitors in the treatment of type 2 diabetes: preclinical and clinical data. Pharmacol Ther. 2013;139(1):51–9.
Sasaki T, Seino Y, Fukatsu A, et al. Safety, pharmacokinetics, and pharmacodynamics of single and multiple luseogliflozin dosing in healthy Japanese males: a randomized, single-blind, placebo-controlled trial. Adv Ther. 2014;31(3):345–61.
Seino Y, Sasaki T, Fukatsu A, et al. Dose-finding study of luseogliflozin in Japanese patients with type 2 diabetes mellitus: a 12-week, randomized, double-blind, placebo-controlled, phase II study. Curr Med Res Opin. 2014;30(7):1231–44.
Seino Y, Sasaki T, Fukatsu A, et al. Efficacy and safety of luseogliflozin monotherapy in Japanese patients with type 2 diabetes mellitus: a 12-week, randomized, placebo-controlled, phase II study. Curr Med Res Opin. 2014;30(7):1219–30.
Seino Y, Sasaki T, Fukatsu A, et al. Efficacy and safety of luseogliflozin as monotherapy in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled, phase 3 study. Curr Med Res Opin. 2014;30(7):1245–55.
Kaku K, Watada H, Iwamoto Y, et al. Efficacy and safety of monotherapy with the novel sodium/glucose cotransporter-2 inhibitor tofogliflozin in Japanese patients with type 2 diabetes mellitus: a combined Phase 2 and 3 randomized, placebo-controlled, double-blind, parallel-group comparative study. Cardiovasc Diabetol. 2014;13:65.
Miao Z, Nucci G, Amin N, et al. Pharmacokinetics, metabolism, and excretion of the antidiabetic agent ertugliflozin (PF-04971729) in healthy male subjects. Drug Metab Dispos. 2013;41(2):445–56.
Dobbins RL, O’Connor-Semmes R, Kapur A, et al. Remogliflozin etabonate, a selective inhibitor of the sodium-dependent transporter 2 reduces serum glucose in type 2 diabetes mellitus patients. Diabetes Obes Metab. 2012;14(1):15–22.
Sykes AP, Kemp GL, Dobbins R, et al. Randomized efficacy and safety trial of once daily remogliflozin etabonate for the treatment of type 2 diabetes. Diabetes Obes Metab. Epub 2014 Sep 19. doi: 10.1111/dom.12393.
Salvo MC, Brooks AD, Thacker SM. Patient considerations in the management of type 2 diabetes—critical appraisal of dapagliflozin. Patient Prefer Adher. 2014;8:493–502.
Bonnet F, Scheen AJ. SGLT-2 receptor inhibitors : an opportunity to renew our therapeutic strategy for type 2 diabetes? Diabetes Metab 2014;40(Suppl):S1–3.
Scheen AJ, Van Gaal LF. Combatting the dual burden : therapeutic targeting of common pathways in obesity and type 2 diabetes. Lancet Diabetes Endocrinol. 2014;2:911–22.
Rieg T, Masuda T, Gerasimova M, et al. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol. 2014;306(2):F188–93.
Perkins BA, Cherney DZ, Partridge H, et al. Sodium-glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: results of an 8-week open-label proof-of-concept trial. Diabetes Care. 2014;37(5):1480–3.
Sinclair A, Bode B, Harris S, et al. Efficacy and safety of canagliflozin compared with placebo in older patients with type 2 diabetes mellitus: a pooled analysis of clinical studies. BMC Endocr Disord. 2014;14:37.
Elmore LK, Baggett S, Kyle JA, et al. A review of the efficacy and safety of canagliflozin in elderly patients with type 2 diabetes. Consult Pharm. 2014;29(5):335–46.
Scheen AJ. Pharmacokinetics, pharmacodynamics and clinical use of SGLT-2 inhibitors in patients with type 2 diabetes and chronic kidney disease. Clin Pharmacokinet. 2014 (unpublished data).
Stanton RC. Sodium glucose transport 2 (SGLT2) inhibition decreases glomerular hyperfiltration: is there a role for SGLT2 inhibitors in diabetic kidney disease? Circulation. 2014;129(5):542–4.
Scheen AJ. Pharmacokinetic considerations for the treatment of diabetes in patients with chronic kidney disease. Expert Opin Drug Metab Toxicol. 2013;9(5):529–50.
Yamout H, Perkovic V, Davies M, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes and stage 3 nephropathy. Am J Nephrol. 2014;40(1):64–74.
Vallon V, Thomson SC. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol. 2012;74:351–75.
De Nicola L, Gabbai FB, Liberti ME, et al. Sodium/glucose cotransporter 2 inhibitors and prevention of diabetic nephropathy: targeting the renal tubule in diabetes. Am J Kidney Dis. 2014;64(1):16–24.
Cherney DZ, Perkins BA, Soleymanlou N, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. 2014;129(5):587–97.
Ridderstrale M, Svaerd R, Zeller C, et al. Rationale, design and baseline characteristics of a 4-year (208-week) phase III trial of empagliflozin, an SGLT2 inhibitor, versus glimepiride as add-on to metformin in patients with type 2 diabetes mellitus with insufficient glycemic control. Cardiovasc Diabetol. 2013;12(1):129.
Scheen AJ, Charbonnel B. Effects of glucose-lowering agents on vascular outcomes in type 2 diabetes: a critical reappraisal. Diabetes Metab. 2014;40(3):176–85.
Basile JN. The potential of sodium glucose cotransporter 2 (SGLT2) inhibitors to reduce cardiovascular risk in patients with type 2 diabetes (T2DM). J Diabetes Complications 2013;27(3):280–6.
Ptaszynska A, Hardy E, Johnsson E, et al. Effects of dapagliflozin on cardiovascular risk factors. Postgrad Med. 2013;125(3):181–9.
Chino Y, Samukawa Y, Sakai S, et al. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos. 2014;35(7):391–404.
Foote C, Perkovic V, Neal B. Effects of SGLT2 inhibitors on cardiovascular outcomes. Diab Vasc Dis Res. 2012;9(2):117–23.
Neal B, Perkovic V, de Zeeuw D, et al. Rationale, design, and baseline characteristics of the Canagliflozin Cardiovascular Assessment Study (CANVAS)—a randomized placebo-controlled trial. Am Heart J. 2013;166(2):217–23.e11.
Zinman B, Inzucchi SE, Lachin JM, et al. Rationale, design, and baseline characteristics of a randomized, placebo-controlled cardiovascular outcome trial of empagliflozin (EMPA-REG OUTCOME). Cardiovasc Diabetol. 2014;13:102.
Scheen AJ. Cardiovascular effects of dipeptidyl peptidase-4 inhibitors: from risk factors to clinical outcomes. Postgrad Med. 2013;125(3):7–20.
Scheen AJ. SGLT2 versus DPP4 inhibitors for type 2 diabetes. Lancet Diabetes Endocrinol. 2013;1(3):168–70.
Friedrich C, Metzmann K, Rose P, et al. A randomized, open-label, crossover study to evaluate the pharmacokinetics of empagliflozin and linagliptin after coadministration in healthy male volunteers. Clin Ther. 2013;35(1):A33–42.
Bell DS. The potent synergistic effects of the combination of liraglutide and canagliflozin on glycemic control and weight loss. Am J Case Rep. 2014;15:152–4.
Zambrowicz B, Ogbaa I, Frazier K, et al. Effects of LX4211, a dual sodium-dependent glucose cotransporters 1 and 2 inhibitor, on postprandial glucose, insulin, glucagon-like peptide 1, and peptide tyrosine tyrosine in a dose-timing study in healthy subjects. Clin Ther. 2013;35(8):1162–73.e8.
Zambrowicz B, Freiman J, Brown PM, et al. LX4211, a dual SGLT1/SGLT2 inhibitor, improved glycemic control in patients with type 2 diabetes in a randomized, placebo-controlled trial. Clin Pharmacol Ther. 2012;92(2):158–69.
Lamos EM, Younk LM, Davis SN. Empagliflozin, a sodium glucose co-transporter 2 inhibitor, in the treatment of type 1 diabetes. Expert Opin Investig Drugs. 2014;23(6):875–82.
Henry RR, Rosenstock J, Edelman S, et al. Exploring the potential of the SGLT2 inhibitor dapagliflozin in type 1 diabetes: a randomized, double-blind, placebo-controlled pilot study. Diabetes Care. Epub 2014 Sep 30.
Asche CV, Hippler SE, Eurich DT. Review of models used in economic analyses of new oral treatments for type 2 diabetes mellitus. Pharmacoeconomics. 2014;32(1):15–27.
Janssen Research & Development, LLC. A study of the effects of canagliflozin (JNJ-28431754) on renal endpoints in adult participants with type 2 diabetes mellitus (CANVAS-R) [ClinicalTrials.gov identifier NCT01989754]. US National Institutes of Health, ClinicalTrials. gov. http://clinicaltrials.gov/show/NCT01989754. Accessed 1 Dec 2014.
AstraZeneca. Multicenter trial to evaluate the effect of dapagliflozin on the incidence of cardiovascular events (DECLARE-TIMI58) [ClinicalTrials.gov identifier NCT01730534]. US National Institutes of Health, ClinicalTrials.gov. http://clinicaltrials.gov/show/NCT01730534. Accessed 1 Dec 2014.
Janssen Research & Development, LLC. CANVAS – CANagliflozin cardioVascular Assessment Study [ClinicalTrials.gov identifier NCT01032629]. US National Institutes of Health, ClinicalTrials.gov. http://clinicaltrials.gov/show/NCT01032629. Accessed 1 Dec 2014.
Boehringer Ingelheim; Eli Lilly and Company. BI 10773 (Empagliflozin) cardiovascular outcome event trial in type 2 diabetes mellitus patients (EMPA-REG OUTCOME) [ClinicalTrials.gov identifier NCT01131676]. US National Institutes of Health, ClinicalTrials.gov. http://www.clinicaltrials.gov/show/NCT01131676. Accessed 1 Dec 2014.
Funding and conflict of interest
No sources of funding were used to assist in the preparation of this manuscript. No conflicts of interest are directly relevant to the content of this manuscript.
A. J. Scheen has received lecture/advisor fees from AstraZeneca/BMS, Boehringer Ingelheim, Eli Lilly, Janssen, Merck Sharp & Dohme, Novartis, NovoNordisk, Sanofi-Aventis and Takeda.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Scheen, A.J. Pharmacodynamics, Efficacy and Safety of Sodium–Glucose Co-Transporter Type 2 (SGLT2) Inhibitors for the Treatment of Type 2 Diabetes Mellitus. Drugs 75, 33–59 (2015). https://doi.org/10.1007/s40265-014-0337-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0337-y