
Abstract
Microsomal triglyceride transfer protein (MTP) is one of the promising targets for the therapy of dyslipidemia and MTP inhibition can lead to robust plasma low-density lipoprotein cholesterol (LDL-C) reduction. Lomitapide, a small-molecule MTP inhibitor, was recently approved by the US FDA as an additional treatment for homozygous familial hypercholesterolemia (hoFH). However, liver-related side effects, including hepatic fat accumulation and transaminase elevations, are the main safety concerns associated with MTP inhibitors. Here, we review recent knowledge on the mechanisms underlying liver toxicity of MTP inhibitors. The contribution of altered levels of intracellular triglycerides, cholesteryl esters, and free cholesterols toward cellular dysfunction is specifically addressed. On this basis, therapies targeted to attenuate cellular lipid accumulation, to reduce risk factors for non-alcoholic fatty liver disease (NAFLD) (i.e., insulin resistance and oxidative stress) and to specifically inhibit intestinal MTP may be useful for ameliorating liver damage induced by MTP inhibitors. In particular, weight loss through lifestyle interventions is expected to be the most effective and safest way to minimize the undesirable side effects. Specific dietary supplementation might also have protective effects against hepatosteatosis. Despite that, to date, few clinical data support these therapeutic options in MTP inhibition-related liver damage, such proposed approaches may be further explored in the future for their use in preventing unwanted effects of MTP inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Microsomal triglyceride transfer protein (MTP) inhibitors have prominent effects in lowering low-density lipoprotein cholesterol levels in patients with hypercholesterolemia. |
Intracellular lipid imbalance contributes to hepatic fat accumulation and transaminase elevations, which are main safety concerns associated with MTP inhibitors. |
Although several non-pharmacologic and pharmacologic strategies appear promising in theory to minimize MTP inhibition-related hepatotoxity, robust clinical evidence on these strategies is lacking. |
1 Introduction
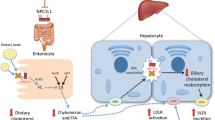
Microsomal triglyceride transfer protein (MTP), a key chaperone in the assembly and secretion of apolipoprotein B (apoB)-containing lipoproteins, is localized in the endoplasmic reticulum (ER) of hepatocytes and enterocytes [1]. Mutations in the gene encoding for MTP are the molecular basis of abetalipoproteinemia (ABL), a rare autosomal recessive disorder characterized by the absence of circulating apoB-containing lipoproteins of both intestinal and hepatic origin [2]. Following the discovery of the molecular cause of ABL in the early 1990s [3], MTP inhibition has emerged as a potential approach with well established efficacy in lowering plasma low-density lipoprotein cholesterol (LDL-C) in hypercholesterolemic individuals. Much data suggest that MTP inhibitors can be instrumental in combating homozygous familial hypercholesterolemia (hoFH) [4, 5]. Given the limited therapeutic options and the severe mortality and morbidity of hoFH, lomitapide, one of the systemic MTP inhibitors, was approved by the US FDA in December 2012 as an adjunct to a low-fat diet and other lipid-lowering treatments for hoFH patients [6]. However, substantial liver-related side effects, including hepatic steatosis and transaminase elevations, have been reported following this treatment in several clinical trials [4, 5, 7], thus hindering the wide application of such class of agents in clinic. Here, we review the liver toxicity and involved molecular mechanisms associated with MTP inhibition. Then we propose several potential therapeutic strategies, including pharmacologic and non-pharmacologic approaches, with the ultimate goal of minimizing the undesirable effects.
2 Hepatotoxicity of Microsomal Triglyceride Transfer Protein (MTP) Inhibition and Mechanistic Studies
In the human trials to date, lomitapide has always been associated with hepatic adverse issues. In the phase II study of lomitapide [4], all of the six patients with hoFH experienced increases in hepatic fat content, ranging from 10 % to 40 % at the highest dose of lomitapide (1.0 mg/kg per day), and elevations in liver aminotransferases were observed in four of the six patients. After suspension of the drug, liver fat and enzyme levels returned to baseline within 4 weeks in all patients except one, whose hepatic fat accumulation persisted for 14 weeks following active treatment. Recently, the results of a phase III trial of lomitapide (dose range, 5–60 mg/day; duration, 78 weeks) in 29 subjects with hoFH were published [5]. The mean liver fat content increased from 1 % at baseline to 8.6 % at week 26 when the dose of lomitapide was titrated up to 60 mg/day, and then remained at this level for the rest of the study (8.3 % at week 78) with stabilized drug dose. About one-third of the patients experienced an increase in transaminases more than three times the upper limit of normal (>3 × ULN), which appeared to be transient and manageable with temporary dose reduction.
2.1 MTP Inhibition and Intracellular Lipid Accumulation
In the liver and intestine, MTP is responsible for transferring neutral lipids (mainly triglycerides and cholesteryl esters) to nascent apoB-lipoproteins for secretion [8, 9]. Therefore, steatosis caused by MTP inhibition is expected to be explained by intracellular increases in both triglycerides and cholesteryl esters associated with impaired assembly and secretion of apoB-containing lipoprotein. Consistent with this speculation, studies with rodent models to investigate the effects of genetic ablation and chemical inhibition of MTP on tissue lipids showed high concentrations of triglycerides in hepatocytes and enterocytes. However, contrary to the expectations, deficiency of MTP triggered intracellular increases in free cholesterols and decreases in cholesteryl esters [10].
According to further mechanistic studies [9, 11–13], the effects that MTP deficiency exerts on cellular cholesterol homeostasis can be illustrated as follows. Under normal conditions, acyl-CoA:cholesterol acyltransferase (ACAT) synthesizes cholesteryl esters, and MTP transfers both free and esterified cholesterol to apoB-lipoproteins. When MTP is limiting, transfer of both free cholesterol and esterified cholesterol to apoB-lipoproteins is curtailed, leading to accumulation of both esterified and free cholesterol. Accumulation of esterified cholesterol inhibits esterification by ACAT enzymes, contributing to further accumulation of free cholesterol. Increased hepatic triglycerides and free cholesterol after MTP inhibition is consistent with previous studies [14–17].
2.2 MTP Inhibition and Elevated Plasma Transaminases
To understand the relationship between MTP inhibition, accumulation in liver lipids, and increases in plasma transaminases, Josekutty et al. conducted an experiment using mice fed a western diet and found that MTP inhibition enhances hepatic free cholesterol in the ER and mitochondria, which are associated with ER stress and oxidative stress, respectively, increasing transcription of the GPT/GOT1 genes through up-regulation of the IRE1α/cJun pathway [18]. GPT/GOT1 genes are responsible for encoding for the cytosolic isoforms of aspartate aminotransferase (AST)/alanine aminotransferase (ALT), namely ALT1/AST1. Thus, inhibition of MTP increases synthesis and release of ALT1/AST1 and ultimately leads to increases in plasma transaminases, without causing cell death. Results from this study also indicate the critical role played by cellular free cholesterol in the underlying mechanism of MTP inhibition-induced hepatotoxicity and therefore, lowering hepatic concentrations of free cholesterol may represent a significant way to circumvent liver-related side effects.
3 Lifestyle Interventions and Weight Loss
Nonalcoholic fatty liver disease (NAFLD) encompasses a broad spectrum of conditions, ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), cirrhosis, and hepato cellular carcinoma [19, 20]. Lifestyle interventions to induce weight loss, including dietary modification and increased physical activity, are recommended by recent guidelines for NASH treatment in adults and children [21, 22]. Reductions in body weight are accompanied by decreases in liver fat, which recently has been systematically reviewed [23, 24]. Weight loss is also associated with improvement in hepatic aminotransferases and insulin resistance (IR) [19, 24]. In addition, weight reduction through bariatric surgery markedly improved the major components of metabolic syndrome: glucose tolerance, hypertension, and hyperlipidemia [25].
Currently, the necessary degree of weight loss for improvement of NAFLD remains unknown. Studies in adults and children suggest that loss of 3–5 % of body weight reduces hepatic steatosis, while decreasing inflammation and progression of NASH may require as much as 10 % weight reduction [21]. In a 12-month trial of adults with type 2 diabetes, losing 1–5 % of body weight reduced hepatic steatosis by 33 % compared with a 65 % reduction in those who lost 5–10 % body weight and 80 % reduction in those who lost more than 10 % [26]. Similarly, in obese children with NAFLD, body mass index (BMI) reduction by 1–2 kg/m2 resolved NAFLD in 48 %, while a more than 2 kg/m2 reduction resolved NAFLD in 89–95 % [27]. Recent clinical studies demonstrated that body weight loss of more than 7 % resulting from a combination of diet and exercise, induced significant improvements in steatosis, inflammation, and hepatocyte ballooning [28]. Further evidence also supports the beneficial role of weight loss in histological improvements of NAFLD/NASH [29, 30]. Of note, aerobic and resistance exercise, even without weight loss, decreases hepatic steatosis and other markers of lipotoxicity [31, 32]. Pharmacologic agents that induce weight loss, such as orlistat, sibutramine, and rimonabant, have shown promising effects in the treatment of NAFLD/NASH [29, 33–35].
Collectively, weight reduction, primarily through restricted diet and the promoted physical activity, positively influences NAFLD/NASH. Therefore, we recommend this effective and safe strategy for both adults and children treated with MTP inhibitors, especially for those with co-existing obesity, metabolic syndrome, or diabetes. Further preclinical and clinical evidence is needed to validate the beneficial effects of weight loss on MTP inhibition-induced liver injury.
4 Dietary Supplements
Intake of specific dietary components may be particularly important in preventing and/or treating hepatic damage triggered by MTP inhibitors. Based on evidence from recent experiments, piperine (1-piperoylpiperidine), oxymatrine, and walnut (Juglans regia) have shown promise in this regard. Intake of these dietary supplements from natural sources may further enhance the beneficial effects of lifestyle modifications on MTP inhibition-related hepatic toxicity; however, this hypothesis is not supported by the clinical research so far.
Piperine is an alkaloid present in black pepper (Piper nigrum), long pepper (Piper longum), and other Piper species fruits (family: Piperaceae) [36]. Administration of piperine has been demonstrated to reverse high-fat diet-induced hepatic steatosis, as well as notably improve serum transaminase and lipid profiles in rats [37, 38]. The precise mechanisms and pathways explaining the liver-protective effects of piperine are unclear. It might be associated with reduced hepatic ER stress, improved IR, and suppression of lover X receptor (LXR)-α-mediated lipogenesis [37]. However, Choi et al. [38] proposed that the underlying mechanism is related to activation of adiponectin-AMP-activated protein kinase (AMPK) signaling, which plays an important role in mediating lipogenesis, fatty acid (FA) oxidation, and insulin signaling in the liver. Regardless of the exact mechanism, hepatic side effects of MTP inhibitors may benefit from dietary supplementation of piperine.
Oxymatrine, an active monomer isolated from the medicinal plant Sophora flavescens Ait [39], is found to ameliorate hyperlipidemia and hepatic lipid accumulation in rats with NAFLD [40]. Further observations indicate that the therapeutic effect of OMT on hepatic steatosis results partly from down-regulating sterol regulatory element-binding transcription factor 1 (SREBF1) and up-regulating peroxisome proliferator-activated receptor (PPAR)-α and MTP-mediated metabolic pathways simultaneously. These results imply that OMT may be used to attenuate MTP inhibition-induced hepatosteatosis.
Different components of the walnut have been used in folk medicine for protection against liver injury [41], and recent literature provides information about the hepatoprotective effects of walnuts. Walnut oil is capable of differentially modifying hepatic and systemic lipid homoeostasis in a rodent steatosis model. The drop in hepatic triglyceride content by high intake of walnut oil was paralleled by a significant elevation of fasting serum triglycerides, with increased very low-density lipoprotein (VLDL) secretion. These data share common features with the basic principle to lower ectopic lipid stores in the liver by increasing hepatic lipid disposal. With regard to the molecular mechanisms, walnut oil inhibits hepatic lipid accumulation probably by regulating hepatic gene expression of MTP and lipoprotein lipase (LPL) in obese Zucker rats [42]. Further, in vitro, walnut oil treatment significantly increased cholesterol efflux through decreasing the expression of the lipogenic enzyme stearoyl CoA desaturase 1 (SCD1) in macrophage-derived foam cells (MDFC) [43]. Based on both rodent and human data [42–46], dietary intervention with walnut oil also lowers plasma cholesterol levels as well as improving endothelial function and antioxidant potential. On the other hand, walnut leaf extract has shown promise in attenuating carbon tetrachloride (CCl4)-induced liver damage in rats [41]. In CCl4-treated rats, administration of walnut leaf extract significantly decreased serum transaminases and alkaline phosphatase levels, with increased antioxidant enzymes, including superoxide dismutase and catalase. Histopathological examination of livers showed reduced fatty degeneration, cytoplasmic vacuolization, and necrosis with walnut leaf extract treatment. The above beneficial effects of walnut intake suggest a novel way to protect against MTP inhibition-triggered hepatic injury.
5 Minimizing Cellular Lipid Accumulation
To avoid hepatic lipid accumulation and plasma transaminase elevation, adoption of an add-on therapy along with MTP antagonists has been proposed as one of the strategies to garner the full potential of MTP inhibition. In this respect, to inhibit intracellular lipid synthesis and to enhance lipid clearance appear to be two main therapeutic strategies.
5.1 Reducing Cellular Free Cholesterol Content
A viable means to attenuate free cholesterol buildup might be to inhibit cholesterol synthesis through HMG-CoA reductase. Statins inhibit HMG-CoA reductase, enhance hepatic low-density lipoprotein receptor (LDLR) expression, and lower plasma cholesterol [47]. Accumulating evidence from both in vitro and in vivo studies indicates that statins also have anti-inflammatory effects that are independent of their hypocholesterolemic activity [48–50]. Retrospective cohort data in 17 patients with NAFLD treated with statins for 10–16 years showed reduced liver steatosis but no effect on inflammation or fibrosis [51]. Furthermore, 5-year statin therapy has been associated with significant decreases or even normalization of serum aminotransferase activities in individuals affected with both coronary heart disease (CHD) and moderately elevated transaminases [52]. There is additional evidence for histological improvement in hepatic steatosis and reduction in serum transaminases of NAFLD/NASH after treatment with statins [53–59]. It has also been reported that atorvastatin therapy could prevent or delay the progression of liver steatosis into NASH [60]. Based on these findings, it is tempting to suggest that a combinatorial inhibition of MTP and HMG-CoA reductase may be effective in decreasing plasma cholesterol and preventing against hepatic fat accumulation. Similarly, potent inhibitors of squalene synthase might be used with MTP inhibitors to achieve these goals.
By comparison, ezetimibe, a sterol absorption inhibitor, exerts its lipid-lowering effect through inhibition of the intestinal Niemann-Pick C1-like 1 (NPC1L1) lipid transporter. This transporter is also expressed in the liver, where it facilitates cholesterol uptake and thus allows the retention of biliary cholesterol by hepatocytes [61, 62]. Ezetimibe could disrupt this process and thus might be useful in ameliorating liver fat accumulation. Evidence from human studies has validated a protective role of ezetimibe against NAFLD-related liver steatosis and elevated transaminases [7, 63]. Results from another clinical trial have also proved the usefulness of both combined ezetimibe/simvastatin (10/10 mg) treatment and simvastatin (20 mg/day) monotherapy to lower serum ALT and AST in patients with NAFLD [58]. Mechanistic studies indicated that ezetimibe not only reduced lipid synthesis in the liver, but also promoted lipid discharge from the liver by preventing post-translational degradation of MTP via a reduction of hepatic reactive oxygen species (ROS) generation, leading to inhibition of the development of NAFLD [64]. These findings raise the possibility of this drug being used for improving MTP inhibition-induced hepatic toxicity. Evidence from a phase II trial of lomitapide supports this potency of ezetimibe in a hyperlipidemic population [7]. In this trial, more patients receiving lomitapide alone than those receiving it in combination with ezetimibe discontinued study drugs due to transaminase elevations, with an incidence of 21.4 % (6/28) and 10.7 % (3/28), respectively. However, it is noteworthy that statin and ezetimibe have not shown promising results in avoiding hepatic fat accumulation and increases in liver enzymes in the phase III study of lomitapide [5]. Indeed, among the 29 lomitapide recipients with hoFH enrolled in the trial, 27 were also treated with statins and 22 with ezetimibe (all in combination with a statin). During the study, transaminase levels more than 3 × ULN were seen at least once in 10 of 29 patients. Mean hepatic fat in the 20 patients with evaluable nuclear magnetic resonance (NMR) scans increased from 1.0 % to 8.6 % with ascending dose of lomitapide. Further research, especially large prospective studies, should fully assess the efficacy and safety of statins or ezetimibe in dealing with MTP inhibition-induced liver abnormalities.
Another possible approach to lowering cellular cholesterol is to enhance its efflux mediated by LXR activation. LXRs belong to the nuclear receptor superfamily and carry on the function of controlling expression of genes involved in cholesterol efflux in macrophages, hepatic bile acid synthesis, and intestinal cholesterol absorption [65–68]. In apoe−/− and ldlr−/− mice, LXR agonists not only enhance cholesterol efflux and decrease atherosclerosis, but also increase hepatic bile acid synthesis and reduce hepatic cholesterol levels. These agonists reduce cholesterol absorption via ABCG5 and ABCG8 up-regulation in the intestine. Nevertheless, LXR agonists lead to high rates of hypertriglyceridemia [69], which can be alleviated by MTP antagonists. Therefore, it is worth examining whether LXR agonists and MTP inhibitors can be used in combination to avoid hepatic steatosis and the increases in plasma transaminases, without causing hypertriglyceridemia.
5.2 Avoiding Overt Accumulation of Triglycerides
Cellular triglyceride accumulation is one of the main characteristics of MTP inhibition. Triglyceride synthesis involves FA uptake, intracellular transport to microsomes by FA-binding proteins (FABPs), and acylation with glycerol by several monoacylglycerol acyltransferases and diacylglycerol acyltransferases (DGATs). Suppression of these steps is likely to reduce cellular triglyceride levels.
First, ablation of liver FABP (L-FABP) has been shown to lessen hepatic steatosis caused by treating mice with an MTP inhibitor [70, 71], supporting that co-repression of both L-FABP and MTP may be an effective means to reduce VLDL secretion without causing liver fat accumulation.
Second, joint inhibition of DGAT and MTP activity seems to be a promising way to avoid hepatic triglyceride accumulation based on several lines of evidence. DGAT, with two isozymes DGAT1 and DGAT2, catalyze the final reaction of triglyceride biosynthesis [72]. Absence of DGAT1 in mice shows a protective effect against a high-fat diet-induced hepatosteatosis [73, 74]. On the other hand, a lack of DGAT2 in mice reduces hepatic triglyceride synthesis and secretion, enhances FA oxidation, and lowers plasma triglyceride levels. Furthermore, a defect in DGAT2 in a rodent model causes a reduction in the messenger RNA (mRNA) levels of several hepatic lipogenic genes, including HMG-CoA reductase [75]. Recently, the target-specific silencing of DGAT2 using small interfering RNAs (siRNAs) has been found to decrease plasma levels of cholesterols as well as lessen hepatic triglyceride accumulation caused by MTP antagonism in mice [76]. Supporting evidence also comes from taxifolin, a plant flavonoid, which was shown to inhibit triglyceride synthesis and reducing apoB secretion by limiting triglyceride availability via DGAT and MTP activity in HepG2 cells, without increasing cellular lipids [77]. Thus, it is reconfirmed that joint inhibition of DGAT and MTP activity might avoid cellular triglyceride accumulation.
Third, cellular triglyceride levels can be reduced by up-regulating FA oxidation, in theory. PPARs are a superfamily of nuclear hormone receptors that are involved in the control of lipid metabolism. PPAR-α is the most expressed PPAR in hepatocytes and it acts as an efficient intracellular lipid sensor [78, 79]. Activation of PPAR-α promotes hepatic β-oxidation of FAs by augmenting expression of genes coding for enzymes of mitochondrial and peroxisomal β-oxidation [80]. Studies in fatty liver mice have confirmed the critical role of activated PPAR-α in maintaining sufficient clearance of lipids from the liver and preventing lipid accumulation and peroxidation in the liver [81, 82]. However, previous studies in humans and rats have shown inconsistent results that PPAR-α agonists decreased VLDL-triglyceride secretion [83–85], making it still far from clear what effects PPAR-α agonists would have on lipid accumulation in the liver.
Fibrates, pharmacological ligands of PPAR-α, have been demonstrated in animal models of steatosis and steatohepatitis to exert various protective effects against liver steatosis. Namely, fibrate-related PPARα activation may stimulate β-oxidation of FAs, reduce IR, and prevent inflammation [86–90]. The treatment for 11 obese patients with fatty liver with bezafibrate (400 mg/day) for 2–8 weeks effectively reduced macrovesicular steatosis and improved serum profiles of liver function and lipids [91, 92]. Administration of fenofibrate (200 mg/day) for 48 weeks among 16 patients with biopsy-confirmed NAFLD showed a significant improvement in biochemical parameters, including ALT, AST, alkaline phosphatase, and gamma-glutamyl transpeptidase (GGT). In regard to histological aspects, such treatment with fenofibrate only resulted in a decrease in the grade of hepatocellular ballooning degeneration, without marked changes in steatosis, lobular inflammation, and fibrosis [93]. There also exist some studies suggesting that fenofibrate clearly lowered liver triglyceride content in murine models of steatosis [82, 94, 95], despite an increased expression of genes involved in FA uptake and activation [94]. Besides, treatment with gemfibrozil decreased ALT levels in NASH patients [96], whereas beneficial effects of clofibrate on transaminase levels and liver histology were minimal in the treatment of NASH subjects [97]. Taken together, despite that the information currently available is not sufficient to draw conclusions on the benefits of PPAR-α activators in the therapy of NAFLD, PPAR-α offers a possible therapeutic target against steatotic liver caused by MTP antagonism. Still, experimental evidence is lacking in this aspect.
6 Suppressing Oxidative Stress
The mechanism of MTP inhibition at the molecular level has revealed that free cholesterol accumulation in mitochondria could cause an imbalance between excessive production of ROS and decreased antioxidant defenses, and finally induce oxidative stresses. Oxidative stress plays a pivotal role in the transition from simple steatosis to steatohepatitis [98, 99]. In this regard, antioxidants, including vitamin E (RRR-α-tocopherol), n-3 polyunsaturated FAs (PUFAs), and probucol, have exhibited a favorable effect on hepatic steatosis and NAFLD/NASH [19, 98].
Vitamin E is the most widely assessed antioxidant dealing with NAFLD and NASH [19, 98, 100–102], based on its activity as a free radical scavenger. Vitamin E is a chain-breaking antioxidant in free radical reactions, which is an important step in lipid peroxidation and membrane stabilization [98, 103]. Positive results of several in vivo animal studies reflect the potential therapeutic role of vitamin E in human NAFLD/NASH [104–108]. The beneficial effects of vitamin E in NAFLD and NASH also have been evidenced by accumulating clinical data [109–113]. In particular, results of two large multicenter randomized controlled trials (RCTs) of vitamin E were recently released. Vitamin E therapy (800 IU per day) for 96 weeks demonstrated a robust improvement in steatosis, inflammation, ballooning, and resolution of steatohepatitis in nondiabetic and noncirrhotic adults with aggressive NASH [114]. In children, the same dose of vitamin E showed a notable resolution of NASH, but without significant reduction in steatosis and inflammation [115]. All of the aforementioned studies have provided some evidence of a benefit of vitamin E in NAFLD/NASH; therefore, it is reasonable to assume that vitamin E administered simultaneously with MTP inhibitors might attenuate liver damage caused by MTP inhibition. In terms of this combinatorial strategy, validation from animal experiments and clinical trials is needed to assess the safety and therapeutic value.
n-3 PUFAs, naturally-occurring ligands of PPAR-α, are known to reduce VLDL triglyceride production and VLDL-apoB synthesis in the liver [116]. Intake of n-3 PUFAs appears to be beneficial for the treatment of NAFLD and NASH patients, with significant reduction in serum AST, ALT, and GGT. This medication also leads to amelioration in the extent of hepatic steatosis, fibrosis, and inflammation [117–121]. Several mechanisms may be involved in the anti-steatotic action of n-3 PUFAs, including preventing lipid peroxidation, positively influencing peripheral IR, activating PPAR-α, and suppressing lipogenic transcription factor sterol regulatory element-binding protein 1c (SREBP-1) [19]. Recent mechanistic studies in mice with parenteral nutrition-associated liver disease have shown that n-3 PUFAs exert their anti-inflammatory and insulin-sensitizing effects through a PPAR-γ action, independent from the PPAR-α pathway [122]. Of note, the value for n-3 PUFAs in improving NAFLD has been questioned by findings in a murine model of steatohepatitis in which n-3 PUFAs failed to prevent the development of steatohepatitis due to accumulation of hepatic lipoperoxides [123]. Therefore, in spite of some promising evidence with reduction in hepatic steatosis, the information currently available is not sufficient to draw conclusions on the benefits of n-3 PUFAs in the therapy of NAFLD. Further studies are required in order to explore benefits of n-3 PUFAs for attenuating liver damage caused by MTP inhibitors or other etiologies.
Probucol, a lipid-lowering agent with strong antioxidant properties, is reported to be both effective and safe for the treatment of NASH [98]. Among NASH patients with dyslipidemia, probucol therapy is capable of significantly reducing levels of serum aminotransferases, accompanied by an improvement in liver histology [124, 125]. These benefits can probably be attributed to its ability to reduce IR and oxidative stress [126]. Therefore, probucol is anticipated to have favorable effects on MTP inhibitor-induced steatotic liver. However, in spite of the possibly improved function of high-density lipoprotein (HDL), the decrease in plasma HDL cholesterol (HDL-C) and the prolongation of the QT interval remain major safety concerns with probucol treatment [127]. Thus, the efficacy and safety of probucol–MTP inhibitor combination therapy needs further validation.
7 Selective Intestinal MTP Inhibition
A mechanistic approach to circumvent the hepatotoxicity of MTP inhibition has been to develop enterocyte-specific inhibitors. As expected, such MTP inhibitors are associated with gastrointestinal adverse events, including nausea, flatulence, and diarrhea. Interestingly, these complications can be partially or completely avoided with a temporal separation of drug administration and food intake. Furthermore, the potential risk for intestinal steatosis is usually not considered a chronic problem because of the inherent capacity of the intestine for self-renewal. So far the most popular compounds of gut-selective MTP inhibitors, including dirlotapide, SLX-4090, and JTT-130, have shown good efficacy and minimal side effects in animal studies.
For example, dirlotapide was found to be clinically safe and effective in the reduction of lipid absorption and body weight in obese dogs, with only a few cases of temporary gastrointestinal discomfort and mildly elevated hepatic transaminases [128, 129]. This compound is approved by the FDA for the treatment of obesity in dogs. SLX-4090 has been demonstrated to reduce plasma LDL-C, triglycerides, and body weight in mice fed a high-fat diet, while showing no sign of hepatic complications [130]. Notably, treatment with JTT-130 in hyperlipidemic hamsters and guinea pigs resulted in a decrease in plasma triglyceride and LDL-C levels, without hepatic lipid accumulation [131, 132]. However, clinical development of JTT-130 was recently terminated. Given the above, the intestine-specific inhibition of MTP may be a promising way for future lipid-lowering interventions without inducing hepatotoxicity, and further clinical studies are warranted to substantiate their efficacy, safety, and tolerability.
8 Insulin-Sensitizing Agents
Recently, links between NAFLD and IR have been systematically reviewed by Gariani et al. [133] and Gaggini et al. [134]; both concluded that IR is one of the major triggering mechanisms for NAFLD progression. The key role that IR plays in NAFLD has led to numerous studies of insulin-sensitizing medications including metformin, thiazolidinediones (also known as glitazones), and incretin mimetics in NAFLD [135].
Metformin exerts anti-diabetic effects through increasing FA/glucose metabolism and improving insulin signaling in the liver and adipose tissue [136, 137]. Over the past decades, a growing number of clinical trials have been conducted to investigate the beneficial effects of metformin in NAFLD, in which the liver function, steatosis, and insulin sensitivity improved [138–144]. Furthermore, metformin treatment exhibited protective effects on metabolic abnormalities and cardiovascular risk, making it a promising therapeutic choice for NAFLD [145, 146]. Conversely, a recent meta-analysis found that metformin, when used in different doses and for different durations, did not result in histological improvement in liver [147]. In addition, metformin is not recommended as a specific treatment for adults with NASH, as this agent has no positive effect on liver histology [21].
Another clinically interesting class of insulin sensitizers are the thiazolidinediones (e.g. troglitazone, rosiglitazone, and pioglitazone) that act as agonists of PPAR-γ, improving glycemic control and insulin sensitivity, and by promoting the redistribution of triglycerides from the liver and muscle to the adipose tissue [148]. The original prototype compound troglitazone did improve IR and inflammation in NASH [149], but induced fulminant hepatitis in some individuals and was withdrawn. In early reports, rosiglitazone was demonstrated to normalize transaminases levels, inflammatory responses, and hepatic steatosis [150–152]. However, due to its obvious side effects (e.g. increased risk of heart attack), the use of rosiglitazone has been highly restricted in the USA and banned in Europe [153, 154]. Administration of pioglitazone, a PPAR α–γ agonist, led to significant improvement in insulin sensitivity, liver enzymes, hepatic steatosis, and inflammation in subjects with NAFLD/NASH in two placebo-controlled trials [114, 155]. Furthermore, according to a recent study, 4-month usage of metformin (1 g/day) and pioglitazone (30 mg/day) was safe and might have equally favorable effects on biomarkers of liver function, lipid profile, IR, and hepatic fat content in NAFLD patients [156]. However, pioglitazone was associated with weight gain and edema, which in turn aggravates heart failure. So the clinical usage of this drug is still under strict supervision in many countries [157].
All of the aforementioned data suggest that insulin sensitizers may suppress symptoms of NAFLD/NASH. Therefore, treatment of IR may be a therapeutic strategy in the treatment of MTP inhibition-induced hepatosteatosis and liver dysfunction, but this hypothesis remains unproven. Of note, thiazolidinediones should be administrated with caution because of the associated risk for severe adverse events.
9 Conclusion
As potential risk factors for progressive liver disease [158, 159], historically, hepatic steatosis and elevated liver enzymes remain major safety concerns associated with MTP inhibitors, thus restricting the availability of such agents to only hoFH patients. In order to address the delicate liver-related issues, appropriate measures should be taken promptly. Because hoFH is a rare disorder (1 per million births), small patient numbers limit the potential for carrying out clinical trials of therapeutic options. As a result, to date there exist no clinical data supporting the aforementioned therapeutic strategies in improving MTP-induced hepatic side effects, with the exception of statins and ezetimibe, both of which failed to show promising results in clinical studies [5].
Among those proposed treatment options, weight loss through lifestyle interventions is still the most effective and safest therapy for NAFLD. It also reduces several specific factors that facilitate the development of NAFLD (i.e., obesity, IR, dyslipidemia). Therefore, this approach can be recommended as a mainstay of therapies for all patients receiving MTP inhibition treatment. Special dietary supplements from natural sources, as well as a moderate intake of vitamin E and n-3 PUFAs, may further enhance the therapeutic effects of lifestyle interventions on the liver, with very few possible side effects. Metformin, with well established hypoglycemic and insulin sensitizing efficacy, may exert additional protective effects against MTP inhibition-induced hepatic steatosis, especially for diabetic and obese patients. Intestine-specific MTP inhibitors, despite that their supporting evidence is currently limited to animal studies, are anticipated to be both effective and safe lipid-lowering medications without unwanted hepatic side effects. Other pharmacologic strategies, including approaches to inhibit intracellular lipid accumulation and to reduce oxidative stress and IR, have also shown promise in this regard. However, they are associated with frustrating clinical data (statin and ezetimibe), inconclusive research evidence (LXR agonists, L-FABP inhibitors, DGAT inhibitors, PPAR-α agonists, and fibrates), or relatively high risks for severe adverse events (probucol and thiazolidinediones). With continuing efforts to further explore these potential non-pharmacologic and pharmacologic strategies, there is a possibility that safety of MTP inhibitors would be improved in hoFH patients.
References
Wetterau JR, Linand MC, Jamil H. Microsomal triglyceride transfer protein. Biochim Biophys Acta. 1997;1345:136–50.
Kane JP. Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins. In: Scriver CR, Sly WS, Childs B, et al, editors. The metabolic and molecular bases of inherited disease, 8th edition. New York: McGraw-Hill Professional, 2001.
Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, Hermier M, Schmitz J, Gay G, Rader DJ, Gregg RE. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001.
Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, Millar JS, Ikewaki K, Siegelman ES, Gregg RE, Rader DJ. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–56.
Cuchel M, Meagher EA, du Toit TH, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Du Plessis AM, Propert KJ, Sasiela WJ, Bloedon LT, Rader DJ. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6.
Norata GD, Tibollaand G, Catapano AL. Targeting PCSK9 for hypercholesterolemia. Annu Rev Pharmacol Toxicol. 2014;54:273–93.
Samaha FF, McKenney J, Bloedon LT, Sasiela WJ, Rader DJ. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2008;5:497–505.
Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, Wetterau JR. The role of the microsomal triglyceride transfer protein in abetalipoproteinemia. Annu Rev Nutr. 2000;20:663–97.
Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. Multiple functions of microsomal triglyceride transfer protein. Nutr Metab (Lond). 2012;9:14.
Iqbal J, Rudeland LL, Hussain MM. Microsomal triglyceride transfer protein enhances cellular cholesteryl esterification by relieving product inhibition. J Biol Chem. 2008;283:19967–80.
Rudel LL, Leeand RG, Cockman TL. Acyl coenzyme A: cholesterol acyltransferase types 1 and 2: structure and function in atherosclerosis. Curr Opin Lipidol. 2001;12:121–7.
Buhman KF, Accadand M, Farese RV. Mammalian acyl-CoA:cholesterol acyltransferases. Biochim Biophys Acta. 2000;1529:142–54.
Chang TY, Chang CC, Lin S, Yu C, Li BL, Miyazaki A. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and -2. Curr Opin Lipidol. 2001;12:289–96.
Sugimoto T, Yamashita S, Ishigami M, Sakai N, Hirano K, Tahara M, Matsumoto K, Nakamura T, Matsuzawa Y. Decreased microsomal triglyceride transfer protein activity contributes to initiation of alcoholic liver steatosis in rats. J Hepatol. 2002;36:157–62.
Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chretien Y, Koike K, Pessayre D, Chapman J, Barba G, Brechot C. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. Faseb J. 2002;16:185–94.
Mirandola S, Bowman D, Hussain MM, Alberti A. Hepatic steatosis in hepatitis C is a storage disease due to HCV interaction with microsomal triglyceride transfer protein (MTP). Nutr Metab (Lond). 2010;7:13.
Pan X, Hussain FN, Iqbal J, Feuerman MH, Hussain MM. Inhibiting proteasomal degradation of microsomal triglyceride transfer protein prevents CCl4-induced steatosis. J Biol Chem. 2007;282:17078–89.
Josekutty J, Iqbal J, Iwawaki T, Kohno K, Hussain MM. Microsomal triglyceride transfer protein inhibition induces endoplasmic reticulum stress and increases gene transcription via Ire1alpha/cJun to enhance plasma ALT/AST. J Biol Chem. 2013;288:14372–83.
Andersonand N, Borlak J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol Rev. 2008;60:311–57.
Corrado RL, Torresand DM, Harrison SA. Review of treatment options for nonalcoholic fatty liver disease. Med Clin N Am. 2014;98:55–72.
Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–23.
Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol. 2010;53:372–84.
Peng L, Wang J, Li F. Weight reduction for non-alcoholic fatty liver disease. Cochrane Database Syst Rev. 2011;(6):CD003619
Thoma C, Dayand CP, Trenell MI. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: a systematic review. J Hepatol. 2012;56:255–66.
De Ridder RJ, Schoon EJ, Smulders JF, van Hout GC, Stockbrugger RW, Koek GH. Review article: non-alcoholic fatty liver disease in morbidly obese patients and the effect of bariatric surgery. Aliment Pharmacol Ther. 2007;26(suppl 2):195–201.
Lazo M, Solga SF, Horska A, Bonekamp S, Diehl AM, Brancati FL, Wagenknecht LE, Pi-Sunyer FX, Kahn SE, Clark JM. Effect of a 12-month intensive lifestyle intervention on hepatic steatosis in adults with type 2 diabetes. Diabetes Care. 2010;33:2156–63.
Reinehr T, Schmidt C, Toschke AM, Andler W. Lifestyle intervention in obese children with non-alcoholic fatty liver disease: 2-year follow-up study. Arch Dis Child. 2009;94:437–42.
Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, Fava JL, Wing RR. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–9.
Harrison SA, Fecht W, Brunt EM, Neuschwander-Tetri BA. Orlistat for overweight subjects with nonalcoholic steatohepatitis: a randomized, prospective trial. Hepatology. 2009;49:80–6.
Nobili V, Manco M, Devito R, Di Ciommo V, Comparcola D, Sartorelli MR, Piemonte F, Marcellini M, Angulo P. Lifestyle intervention and antioxidant therapy in children with nonalcoholic fatty liver disease: a randomized, controlled trial. Hepatology. 2008;48:119–28.
Hallsworth K, Fattakhova G, Hollingsworth KG, Thoma C, Moore S, Taylor R, Day CP, Trenell MI. Resistance exercise reduces liver fat and its mediators in non-alcoholic fatty liver disease independent of weight loss. Gut. 2011;60:1278–83.
Johnson NA, Sachinwalla T, Walton DW, Smith K, Armstrong A, Thompson MW, George J. Aerobic exercise training reduces hepatic and visceral lipids in obese individuals without weight loss. Hepatology. 2009;50:1105–12.
Zelber-Sagi S, Kessler A, Brazowsky E, Webb M, Lurie Y, Santo M, Leshno M, Blendis L, Halpern Z, Oren R. A double-blind randomized placebo-controlled trial of orlistat for the treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:639–44.
Sabuncu T, Nazligul Y, Karaoglanoglu M, Ucar E, Kilic FB. The effects of sibutramine and orlistat on the ultrasonographic findings, insulin resistance and liver enzyme levels in obese patients with non-alcoholic steatohepatitis. Rom J Gastroenterol. 2003;12:189–92.
Gary-Bobo M, Elachouri G, Gallas JF, Janiak P, Marini P, Ravinet-Trillou C, Chabbert M, Cruccioli N, Pfersdorff C, Roque C, Arnone M, Croci T, Soubrie P, Oury-Donat F, Maffrand JP, Scatton B, Lacheretz F, Le Fur G, Herbert JM, Bensaid M. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology. 2007;46:122–9.
Wattanathorn J, Chonpathompikunlert P, Muchimapura S, Priprem A, Tankamnerdthai O. Piperine, the potential functional food for mood and cognitive disorders. Food Chem Toxicol. 2008;46:3106–10.
Jwa H, Choi Y, Park UH, Um SJ, Yoon SK, Park T. Piperine, an LXRalpha antagonist, protects against hepatic steatosis and improves insulin signaling in mice fed a high-fat diet. Biochem Pharmacol. 2012;84:1501–10.
Choi S, Choi Y, Choi Y, Kim S, Jang J, Park T. Piperine reverses high fat diet-induced hepatic steatosis and insulin resistance in mice. Food Chem. 2013;141:3627–35.
Wuand XN, Wang GJ. Experimental studies of oxymatrine and its mechanisms of action in hepatitis B and C viral infections. Chin J Dig Dis. 2004;5:12–6.
Shi LJ, Shi L, Song GY, Zhang HF, Hu ZJ, Wang C, Zhang DH. Oxymatrine attenuates hepatic steatosis in non-alcoholic fatty liver disease rats fed with high fructose diet through inhibition of sterol regulatory element binding transcription factor 1 (Srebf1) and activation of peroxisome proliferator activated receptor alpha (Pparalpha). Eur J Pharmacol. 2013;714:89–95.
Eidi A, Moghadam JZ, Mortazavi P, Rezazadeh S, Olamafar S. Hepatoprotective effects of Juglans regia extract against CCl4-induced oxidative damage in rats. Pharm Biol. 2013;51:558–65.
Fink A, Rufer CE, Le Grandois J, Roth A, Aoude-Werner D, Marchioni E, Bub A, Barth SW. Dietary walnut oil modulates liver steatosis in the obese Zucker rat. Eur J Nutr. 2014;53:645–60.
Zhang J, Grieger JA, Kris-Etherton PM, Thompson JT, Gillies PJ, Fleming JA, Vanden HJ. Walnut oil increases cholesterol efflux through inhibition of stearoyl CoA desaturase 1 in THP-1 macrophage-derived foam cells. Nutr Metab (Lond). 2011;8:61.
Berryman CE, Grieger JA, West SG, Chen CY, Blumberg JB, Rothblat GH, Sankaranarayanan S, Kris-Etherton PM. Acute consumption of walnuts and walnut components differentially affect postprandial lipemia, endothelial function, oxidative stress, and cholesterol efflux in humans with mild hypercholesterolemia. J Nutr. 2013;143:788–94.
Baneland DK, Hu FB. Effects of walnut consumption on blood lipids and other cardiovascular risk factors: a meta-analysis and systematic review. Am J Clin Nutr. 2009;90:56–63.
Ma Y, Njike VY, Millet J, Dutta S, Doughty K, Treu JA, Katz DL. Effects of walnut consumption on endothelial function in type 2 diabetic subjects: a randomized controlled crossover trial. Diabetes Care. 2010;33:227–32.
Nseir W, Mograbiand J, Ghali M. Lipid-lowering agents in nonalcoholic fatty liver disease and steatohepatitis: human studies. Dig Dis Sci. 2012;57:1773–81.
Weitz-Schmidt G. Statins as anti-inflammatory agents. Trends Pharmacol Sci. 2002;23:482–6.
Nseir W, Khateeb J, Tatour I, Haiek S, Samara M, Assy N. Long-term statin therapy affects the severity of chronic gastritis. Helicobacter. 2010;15:510–5.
Wierzbicki AS, Postonand R, Ferro A. The lipid and non-lipid effects of statins. Pharmacol Ther. 2003;99:95–112.
Ekstedt M, Franzen LE, Mathiesen UL, Holmqvist M, Bodemar G, Kechagias S. Statins in non-alcoholic fatty liver disease and chronically elevated liver enzymes: a histopathological follow-up study. J Hepatol. 2007;47:135–41.
Athyros VG, Tziomalos K, Gossios TD, Griva T, Anagnostis P, Kargiotis K, Pagourelias ED, Theocharidou E, Karagiannis A, Mikhailidis DP. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek atorvastatin and coronary heart disease evaluation (GREACE) study: a post-hoc analysis. Lancet. 2010;376:1916–22.
Rallidis LS, Drakoulisand CK, Parasi AS. Pravastatin in patients with nonalcoholic steatohepatitis: results of a pilot study. Atherosclerosis. 2004;174:193–6.
Hyogo H, Ikegami T, Tokushige K, Hashimoto E, Inui K, Matsuzaki Y, Tokumo H, Hino F, Tazuma S. Efficacy of pitavastatin for the treatment of non-alcoholic steatohepatitis with dyslipidemia: an open-label, pilot study. Hepatol Res. 2011;41:1057–65.
Mihaila RG, Nedelcu L, Fratila O, Rezi EC, Domnariu C, Deac M. Effects of lovastatin and pentoxyphyllin in nonalcoholic steatohepatitis. Hepatogastroenterology. 2009;56:1117–21.
Gomez-Dominguez E, Gisbert JP, Moreno-Monteagudo JA, Garcia-Buey L, Moreno-Otero R. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment Pharmacol Ther. 2006;23:1643–7.
Kiyici M, Gulten M, Gurel S, Nak SG, Dolar E, Savci G, Adim SB, Yerci O, Memik F. Ursodeoxycholic acid and atorvastatin in the treatment of nonalcoholic steatohepatitis. Can J Gastroenterol. 2003;17:713–8.
Abel T, Feher J, Dinya E, Eldin MG, Kovacs A. Safety and efficacy of combined ezetimibe/simvastatin treatment and simvastatin monotherapy in patients with non-alcoholic fatty liver disease. Med Sci Monit. 2009;15:S6–11.
Georgescuand EF, Georgescu M. Therapeutic options in non-alcoholic steatohepatitis (NASH). Are all agents alike? Results of a preliminary study. J Gastrointest Liver Dis. 2007;16:39–46.
Samyand W, Hassanian MA. Paraoxonase-1 activity, malondialdehyde and glutathione peroxidase in non-alcoholic fatty liver disease and the effect of atorvastatin. Arab J Gastroenterol. 2011;12:80–5.
Garcia-Calvo M, Lisnock J, Bull HG, Hawes BE, Burnett DA, Braun MP, Crona JH, Davis HJ, Dean DC, Detmers PA, Graziano MP, Hughes M, Macintyre DE, Ogawa A, O’Neill KA, Iyer SP, Shevell DE, Smith MM, Tang YS, Makarewicz AM, Ujjainwalla F, Altmann SW, Chapman KT, Thornberry NA. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc Natl Acad Sci USA. 2005;102:8132–7.
Jeuand L, Cheng JW. Pharmacology and therapeutics of ezetimibe (SCH 58235), a cholesterol-absorption inhibitor. Clin Ther. 2003;25:2352–87.
Yoneda M, Fujita K, Nozaki Y, Endo H, Takahashi H, Hosono K, Suzuki K, Mawatari H, Kirikoshi H, Inamori M, Saito S, Iwasaki T, Terauchi Y, Kubota K, Maeyama S, Nakajima A. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: an open-label, pilot study. Hepatol Res. 2010;40:566–73.
Wang X, Sugimoto K, Fujisawa T, Shindo N, Minato S, Kamada Y, Hamano M, Ohishi M, Ikegami H, Rakugi H. Novel effect of ezetimibe to inhibit the development of non-alcoholic fatty liver disease in fatty liver Shionogi mouse. Hepatol Res. 2014;44:102–13.
Ducheix S, Montagner A, Theodorou V, Ferrier L, Guillou H. The liver X receptor: a master regulator of the gut-liver axis and a target for non alcoholic fatty liver disease. Biochem Pharmacol. 2013;86:96–105.
Zelcerand N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–14.
Rader DJ. Liver X receptor and farnesoid X receptor as therapeutic targets. Am J Cardiol. 2007;100:n15–9.
Rigamonti E, Chinetti-Gbaguidiand G, Staels B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol. 2008;28:1050–9.
Zadelaar S, Kleemann R, Verschuren L, de Vries-Van DWJ, van der Hoorn J, Princen HM, Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27:1706–21.
Spann NJ, Kang S, Li AC, Chen AZ, Newberry EP, Davidson NO, Hui ST, Davis RA. Coordinate transcriptional repression of liver fatty acid-binding protein and microsomal triglyceride transfer protein blocks hepatic very low density lipoprotein secretion without hepatosteatosis. J Biol Chem. 2006;281:33066–77.
Higuchi N, Kato M, Tanaka M, Miyazaki M, Takao S, Kohjima M, Kotoh K, Enjoji M, Nakamuta M, Takayanagi R. Effects of insulin resistance and hepatic lipid accumulation on hepatic mRNA expression levels of apoB, MTP and L-FABP in non-alcoholic fatty liver disease. Exp Ther Med. 2011;2:1077–81.
Liu Q, Siloto RM, Lehner R, Stone SJ, Weselake RJ. Acyl-CoA:diacylglycerol acyltransferase: molecular biology, biochemistry and biotechnology. Prog Lipid Res. 2012;51:350–77.
Chenand HC, Farese RJ. Inhibition of triglyceride synthesis as a treatment strategy for obesity: lessons from DGAT1-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:482–6.
Chen HC. Enhancing energy and glucose metabolism by disrupting triglyceride synthesis: lessons from mice lacking DGAT1. Nutr Metab (Lond). 2006;3:10.
Yu XX, Murray SF, Pandey SK, Booten SL, Bao D, Song XZ, Kelly S, Chen S, McKay R, Monia BP, Bhanot S. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology. 2005;42:362–71.
Tep S, Mihaila R, Freeman A, Pickering V, Huynh F, Tadin-Strapps M, Stracks A, Hubbard B, Caldwell J, Flanagan WM, Kuklin NA, Ason B. Rescue of Mtp siRNA-induced hepatic steatosis by DGAT2 siRNA silencing. J Lipid Res. 2012;53:859–67.
Casaschi A, Rubio BK, Maiyoh GK, Theriault AG. Inhibitory activity of diacylglycerol acyltransferase (DGAT) and microsomal triglyceride transfer protein (MTP) by the flavonoid, taxifolin, in HepG2 cells: potential role in the regulation of apolipoprotein B secretion. Atherosclerosis. 2004;176:247–53.
Tailleux A, Woutersand K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 2012;1821:809–18.
Motojimaand K, Hirai T. Peroxisome proliferator-activated receptor alpha plays a vital role in inducing a detoxification system against plant compounds with crosstalk with other xenobiotic nuclear receptors. FEBS J. 2006;273:292–300.
Kane CD, Franconeand OL, Stevens KA. Differential regulation of the cynomolgus, human, and rat acyl-CoA oxidase promoters by PPARalpha. Gene. 2006;380:84–94.
Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38:123–32.
Harano Y, Yasui K, Toyama T, Nakajima T, Mitsuyoshi H, Mimani M, Hirasawa T, Itoh Y, Okanoue T. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int. 2006;26:613–20.
Edvardsson U, Ljungberg A, Linden D, William-Olsson L, Peilot-Sjogren H, Ahnmark A, Oscarsson J. PPARalpha activation increases triglyceride mass and adipose differentiation-related protein in hepatocytes. J Lipid Res. 2006;47:329–40.
Kesaniemiand YA, Grundy SM. Influence of gemfibrozil and clofibrate on metabolism of cholesterol and plasma triglycerides in man. JAMA. 1984;251:2241–6.
Petit D, Bonnefis MT, Rey C, Infante R. Effects of ciprofibrate and fenofibrate on liver lipids and lipoprotein synthesis in normo- and hyperlipidemic rats. Atherosclerosis. 1988;74:215–25.
Chou CJ, Haluzik M, Gregory C, Dietz KR, Vinson C, Gavrilova O, Reitman ML. WY14, 643, a peroxisome proliferator-activated receptor α (PPARα) agonist, improves hepatic and muscle steatosis and reverses insulin resistance in lipoatrophic A-ZIP/F-1 mice. J Biol Chem. 2002;277:24484–9.
Akbiyik F, Cinar K, Demirpence E, Ozsullu T, Tunca R, Haziroglu R, Yurdaydin C, Uzunalimoglu O, Bozkaya H. Ligand-induced expression of peroxisome proliferator-activated receptor alpha and activation of fatty acid oxidation enzymes in fatty liver. Eur J Clin Invest. 2004;34:429–35.
van Raalte DH, Li M, Pritchard PH, Wasan KM. Peroxisome proliferator-activated receptor (PPAR)-alpha: a pharmacological target with a promising future. Pharm Res. 2004;21:1531–8.
Haluzik MM, Lacinova Z, Dolinkova M, Haluzikova D, Housa D, Horinek A, Vernerova Z, Kumstyrova T, Haluzik M. Improvement of insulin sensitivity after peroxisome proliferator-activated receptor-alpha agonist treatment is accompanied by paradoxical increase of circulating resistin levels. Endocrinology. 2006;147:4517–24.
Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–91.
Nakamuta M, Morizono S, Soejima Y, Yoshizumi T, Aishima S, Takasugi S, Yoshimitsu K, Enjoji M, Kotoh K, Taketomi A, Uchiyama H, Shimada M, Nawata H, Maehara Y. Short-term intensive treatment for donors with hepatic steatosis in living-donor liver transplantation. Transplantation. 2005;80:608–12.
Perkins JD. Saying “Yes” to obese living liver donors: short-term intensive treatment for donors with hepatic steatosis in living-donor liver transplantation. Liver Transpl. 2006;12:1012–3.
Fernandez-Miranda C, Perez-Carreras M, Colina F, Lopez-Alonso G, Vargas C, Solis-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis. 2008;40:200–5.
Forcheron F, Abdallah P, Basset A, Del CP, Haffar G, Beylot M. Nonalcoholic hepatic steatosis in Zucker diabetic rats: spontaneous evolution and effects of metformin and fenofibrate. Obesity (Silver Spring). 2009;17:1381–9.
Srivastava RA, Jahagirdar R, Azhar S, Sharma S, Bisgaier CL. Peroxisome proliferator-activated receptor-alpha selective ligand reduces adiposity, improves insulin sensitivity and inhibits atherosclerosis in LDL receptor-deficient mice. Mol Cell Biochem. 2006;285:35–50.
Basaranoglu M, Acbayand O, Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J Hepatol. 1999;31:384.
Laurin J, Lindor KD, Crippin JS, Gossard A, Gores GJ, Ludwig J, Rakela J, McGill DB. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology. 1996;23:1464–7.
Al-Busafi SA, Bhat M, Wong P, Ghali P, Deschenes M. Antioxidant therapy in nonalcoholic steatohepatitis. Hepat Res Treat. 2012;2012:947575.
Albano E, Mottaran E, Occhino G, Reale E, Vidali M. Review article: role of oxidative stress in the progression of non-alcoholic steatosis. Aliment Pharmacol Ther. 2005;22(suppl 2):71–3.
Lomonaco R, Sunny NE, Bril F, Cusi K. Nonalcoholic fatty liver disease: current issues and novel treatment approaches. Drugs. 2013;73:1–14.
Pacanaand T, Sanyal AJ. Vitamin E and nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care. 2012;15:641–8.
Musso G, Antyand R, Petta S. Antioxidant therapy and drugs interfering with lipid metabolism: could they be effective in NAFLD patients? Curr Pharm Des. 2013;19:5297–313.
Chang CY, Argo CK, Al-Osaimi AM, Caldwell SH. Therapy of NAFLD: antioxidants and cytoprotective agents. J Clin Gastroenterol. 2006;40(suppl 1):S51–60.
Nan YM, Wu WJ, Fu N, Liang BL, Wang RQ, Li LX, Zhao SX, Zhao JM, Yu J. Antioxidants vitamin E and 1-aminobenzotriazole prevent experimental non-alcoholic steatohepatitis in mice. Scand J Gastroenterol. 2009;44:1121–31.
Phung N, Pera N, Farrell G, Leclercq I, Hou JY, George J. Pro-oxidant-mediated hepatic fibrosis and effects of antioxidant intervention in murine dietary steatohepatitis. Int J Mol Med. 2009;24:171–80.
Chung MY, Yeung SF, Park HJ, Volek JS, Bruno RS. Dietary alpha- and gamma-tocopherol supplementation attenuates lipopolysaccharide-induced oxidative stress and inflammatory-related responses in an obese mouse model of nonalcoholic steatohepatitis. J Nutr Biochem. 2010;21:1200–6.
Soden JS, Devereaux MW, Haas JE, Gumpricht E, Dahl R, Gralla J, Traber MG, Sokol RJ. Subcutaneous vitamin E ameliorates liver injury in an in vivo model of steatocholestasis. Hepatology. 2007;46:485–95.
Iida C, Fujii K, Koga E, Washino Y, Kitamura Y, Ichi I, Abe K, Matsura T, Kojo S. Effect of alpha-tocopherol on carbon tetrachloride intoxication in the rat liver. Arch Toxicol. 2009;83:477–83.
Yakaryilmaz F, Guliter S, Savas B, Erdem O, Ersoy R, Erden E, Akyol G, Bozkaya H, Ozenirler S. Effects of vitamin E treatment on peroxisome proliferator-activated receptor-alpha expression and insulin resistance in patients with non-alcoholic steatohepatitis: results of a pilot study. Intern Med J. 2007;37:229–35.
Vajro P, Mandato C, Franzese A, Ciccimarra E, Lucariello S, Savoia M, Capuano G, Migliaro F. Vitamin E treatment in pediatric obesity-related liver disease: a randomized study. J Pediatr Gastroenterol Nutr. 2004;38:48–55.
Hasegawa T, Yoneda M, Nakamura K, Makino I, Terano A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: a pilot study. Aliment Pharmacol Ther. 2001;15:1667–72.
Pietu F, Guillaud O, Walter T, Vallin M, Hervieu V, Scoazec JY, Dumortier J. Ursodeoxycholic acid with vitamin E in patients with nonalcoholic steatohepatitis: long-term results. Clin Res Hepatol Gastroenterol. 2012;36:146–55.
Dufour JF, Oneta CM, Gonvers JJ, Bihl F, Cerny A, Cereda JM, Zala JF, Helbling B, Steuerwald M, Zimmermann A. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin e in nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2006;4:1537–43.
Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85.
Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, Abrams SH, Scheimann AO, Sanyal AJ, Chalasani N, Tonascia J, Unalp A, Clark JM, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. 2011;305:1659–68.
Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n-6/n-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond). 2004;106:635–43.
Cappani M, Callelaand F, Biagini MR. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: a pilot study. Aliment Pharmacol. 2006;23:1143–51.
Spadaro L, Magliocco O, Spampinato D, Piro S, Oliveri C, Alagona C, Papa G, Rabuazzo AM, Purrello F. Effects of n-3 polyunsaturated fatty acids in subjects with nonalcoholic fatty liver disease. Dig Liver Dis. 2008;40:194–9.
Tanaka N, Sano K, Horiuchi A, Tanaka E, Kiyosawa K, Aoyama T. Highly purified eicosapentaenoic acid treatment improves nonalcoholic steatohepatitis. J Clin Gastroenterol. 2008;42:413–8.
Sofi F, Giangrandi I, Cesari F, Corsani I, Abbate R, Gensini GF, Casini A. Effects of a 1-year dietary intervention with n-3 polyunsaturated fatty acid-enriched olive oil on non-alcoholic fatty liver disease patients: a preliminary study. Int J Food Sci Nutr. 2010;61:792–802.
Zhu FS, Liu S, Chen XM, Huang ZG, Zhang DW. Effects of n-3 polyunsaturated fatty acids from seal oils on nonalcoholic fatty liver disease associated with hyperlipidemia. World J Gastroenterol. 2008;14:6395–400.
Prince E, Lazare FB, Treem WR, Xu J, Iqbal J, Pan X, Josekutty J, Walsh M, Anderson V, Hussain MM, Schwarz SM. ω-3 fatty acids prevent hepatic steatosis, independent of PPAR-α activity, in a murine model of parenteral nutrition-associated liver disease. JPEN J Parenter Enteral Nutr. 2013 Jun 11 [Epub ahead of print].
Larter CZ, Yeh MM, Cheng J, Williams J, Brown S, Dela PA, Bell-Anderson KS, Farrell GC. Activation of peroxisome proliferator-activated receptor alpha by dietary fish oil attenuates steatosis, but does not prevent experimental steatohepatitis because of hepatic lipoperoxide accumulation. J Gastroenterol Hepatol. 2008;23:267–75.
Merat S, Aduli M, Kazemi R, Sotoudeh M, Sedighi N, Sohrabi M, Malekzadeh R. Liver histology changes in nonalcoholic steatohepatitis after one year of treatment with probucol. Dig Dis Sci. 2008;53:2246–50.
Merat S, Malekzadeh R, Sohrabi MR, Sotoudeh M, Rakhshani N, Sohrabpour AA, Naserimoghadam S. Probucol in the treatment of non-alcoholic steatohepatitis: a double-blind randomized controlled study. J Hepatol. 2003;38:414–8.
Ishitobi T, Hyogo H, Tokumo H, Arihiro K, Chayama K. Efficacy of probucol for the treatment of non-alcoholic steatohepatitis with dyslipidemia: an open-label pilot study. Hepatol Res. 2013 Apr 17 [Epub ahead of print].
Yamashitaand S, Matsuzawa Y. Where are we with probucol: a new life for an old drug? Atherosclerosis. 2009;207:16–23.
Wren JA, Ramudo AA, Campbell SL, King VL, Eagleson JS, Gossellin J, Sunderland SJ. Efficacy and safety of dirlotapide in the management of obese dogs evaluated in two placebo-controlled, masked clinical studies in North America. J Vet Pharmacol Ther. 2007;30(suppl 1):81–9.
Gossellin J, McKelvie J, Sherington J, Wren JA, Eagleson JS, Rowan TG, Sunderland SJ. An evaluation of dirlotapide to reduce body weight of client-owned dogs in two placebo-controlled clinical studies in Europe. J Vet Pharmacol Ther. 2007;30(suppl 1):73–80.
Kim E, Campbell S, Schueller O, Wong E, Cole B, Kuo J, Ellis J, Ferkany J, Sweetnam P. A small-molecule inhibitor of enterocytic microsomal triglyceride transfer protein, SLx-4090: biochemical, pharmacodynamic, pharmacokinetic, and safety profile. J Pharmacol Exp Ther. 2011;337:775–85.
Mera Y, Odani N, Kawai T, Hata T, Suzuki M, Hagiwara A, Katsushima T, Kakutani M. Pharmacological characterization of diethyl-2-({3-dimethylcarbamoyl-4-[(4’-trifluoromethylbiphenyl-2-carbonyl)amino]p henyl}acetyloxymethyl)-2-phenylmalonate (JTT-130), an intestine-specific inhibitor of microsomal triglyceride transfer protein. J Pharmacol Exp Ther. 2011;336:321–7.
Aggarwal D, West KL, Zern TL, Shrestha S, Vergara-Jimenez M, Fernandez ML. JTT-130, a microsomal triglyceride transfer protein (MTP) inhibitor lowers plasma triglycerides and LDL cholesterol concentrations without increasing hepatic triglycerides in guinea pigs. BMC Cardiovasc Disord. 2005;5:30.
Gariani K, Philippeand J, Jornayvaz FR. Non-alcoholic fatty liver disease and insulin resistance: from bench to bedside. Diabetes Metab. 2013;39:16–26.
Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. 2013;5:1544–60.
Stein LL, Dongand MH, Loomba R. Insulin sensitizers in nonalcoholic fatty liver disease and steatohepatitis: current status. Adv Ther. 2009;26:893–907.
Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333:550–4.
Aliand S, Fonseca V. Overview of metformin: special focus on metformin extended release. Expert Opin Pharmacother. 2012;13:1797–805.
Uygun A, Kadayifci A, Isik AT, Ozgurtas T, Deveci S, Tuzun A, Yesilova Z, Gulsen M, Dagalp K. Metformin in the treatment of patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2004;19:537–44.
Nair S, Diehl AM, Wiseman M, Farr GJ, Perrillo RP. Metformin in the treatment of non-alcoholic steatohepatitis: a pilot open label trial. Aliment Pharmacol Ther. 2004;20:23–8.
Schwimmer JB, Behling C, Newbury R, Deutsch R, Nievergelt C, Schork NJ, Lavine JE. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology. 2005;42:641–9.
Nobili V, Marcellini M, Devito R, Ciampalini P, Piemonte F, Comparcola D, Sartorelli MR, Angulo P. NAFLD in children: a prospective clinical-pathological study and effect of lifestyle advice. Hepatology. 2006;44:458–65.
Loomba R, Lutchman G, Kleiner DE, Ricks M, Feld JJ, Borg BB, Modi A, Nagabhyru P, Sumner AE, Liang TJ, Hoofnagle JH. Clinical trial: pilot study of metformin for the treatment of non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2009;29:172–82.
de Oliveira CP, Stefano JT, de Siqueira ER, Silva LS, de Campos MD, Lima VM, Furuya CK, Mello ES, Souza FG, Rabello F, Santos TE, Nogueira MA, Caldwell SH, Alves VA, Carrilho FJ. Combination of N-acetylcysteine and metformin improves histological steatosis and fibrosis in patients with non-alcoholic steatohepatitis. Hepatol Res. 2008;38:159–65.
Garinis GA, Fruci B, Mazza A, De Siena M, Abenavoli S, Gulletta E, Ventura V, Greco M, Abenavoli L, Belfiore A. Metformin versus dietary treatment in nonalcoholic hepatic steatosis: a randomized study. Int J Obes (Lond). 2010;34:1255–64.
Landin K, Tengbornand L, Smith U. Treating insulin resistance in hypertension with metformin reduces both blood pressure and metabolic risk factors. J Intern Med. 1991;229:181–7.
Petersenand JS, DiBona GF. Acute sympathoinhibitory actions of metformin in spontaneously hypertensive rats. Hypertension. 1996;27:619–25.
Musso G, Cassader M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia. 2012;55:885–904.
Van Wagnerand LB, Rinella ME. The role of insulin-sensitizing agents in the treatment of nonalcoholic steatohepatitis. Ther Adv Gastroenterol. 2011;4:249–63.
Caldwell SH, Hespenheide EE, Redick JA, Iezzoni JC, Battle EH, Sheppard BL. A pilot study of a thiazolidinedione, troglitazone, in nonalcoholic steatohepatitis. Am J Gastroenterol. 2001;96:519–25.
Marlatt GA, Larimer ME, Mail PD, Hawkins EH, Cummins LH, Blume AW, Lonczak HS, Burns KM, Chan KK, Cronce JM, La Marr CJ, Radin S, Forquera R, Gonzales R, Tetrick C, Gallion S. Journeys of the circle: a culturally congruent life skills intervention for adolescent Indian drinking. Alcohol Clin Exp Res. 2003;27:1327–9.
Tiikkainen M, Hakkinen AM, Korsheninnikova E, Nyman T, Makimattila S, Yki-Jarvinen H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes. 2004;53:2169–76.
Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann-Heurtier A, Serfaty L, Podevin P, Lacorte JM, Bernhardt C, Bruckert E, Grimaldi A, Poynard T. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled fatty liver improvement with rosiglitazone therapy (FLIRT) trial. Gastroenterology. 2008;135:100–10.
Nissenand SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71.
Mazza A, Fruci B, Garinis GA, Giuliano S, Malaguarnera R, Belfiore A. The role of metformin in the management of NAFLD. Exp Diabetes Res. 2012;2012:716404.
Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, Berria R, Ma JZ, Dwivedi S, Havranek R, Fincke C, DeFronzo R, Bannayan GA, Schenker S, Cusi K. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–307.
Razavizade M, Jamali R, Arj A, Matini SM, Moraveji A, Taherkhani E. The effect of pioglitazone and metformin on liver function tests, insulin resistance, and liver fat content in nonalcoholic fatty liver disease: a randomized double blinded clinical trial. Hepat Mon. 2013;13:e9270.
Shahand P, Mudaliar S. Pioglitazone: side effect and safety profile. Expert Opin Drug Saf. 2010;9:347–54.
Farrelland GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–112.
Wierzbickiand AS, Oben J. Nonalcoholic fatty liver disease and lipids. Curr Opin Lipidol. 2012;23:345–52.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Project 81170262), National Program on Key Basic Research Project of China (973 Program) (No. 2012CB517504) and the Fundamental Research Funds for the Central Universities of Central South University (No.2013zzts325) (to L.M.J).
Conflict of interest
Minjie Lin, Shuiping Zhao, Li Shen, and Danyan Xu have no conflicts of interest that are directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lin, M., Zhao, S., Shen, L. et al. Potential Approaches to Ameliorate Hepatic Fat Accumulation Seen with MTP Inhibition. Drug Saf 37, 213–224 (2014). https://doi.org/10.1007/s40264-014-0147-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-014-0147-x