
Abstract
Background and Objective
Daprodustat is a first-in-class hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI) approved in the USA for treatment of anemia owing to chronic kidney disease (CKD) in dialysis-dependent adults and in Japan for treatment of CKD in dialysis- and non-dialysis dependent adults. This analysis characterized the population pharmacokinetics (PopPK) of daprodustat in adults with CKD and evaluated the influence of intrinsic and extrinsic factors.
Methods
This PopPK analysis included data from one phase 2B and four phase 3 studies comprising 707 CKD subjects dose titrated to prespecified target hemoglobin levels with daprodustat doses ranging from 1 to 24 mg once daily and 2 to 48 mg given three times a week (TIW). Model development leveraged a previous phase 1/2 PopPK model. Stepwise covariate analysis included 20 extrinsic and intrinsic factors. Model evaluation used standard goodness-of-fit and visual predictive checks.
Results
Daprodustat PopPK was adequately characterized using a three-compartment distribution model with first-order elimination. The absorption phase was described using five transit compartments. Oral clearance and volume of distribution was 24.6 L/h and 26.9 L, respectively. Body weight dependence (with fixed allometric coefficients) of clearance and volume terms was a statistically significant covariate. Concomitant use of clopidogrel (moderate CYP2C8 inhibitor) decreased oral clearance, resulting in higher area under the plasma concentration-time curve (AUC) ratio of 1.59 (90% CI: 1.39–1.82), subjects’ dialysis status (non-dialysis versus dialysis) had an effect on absorption, with Cmax ratio of 1.19 (90% CI: 1.09–1.30). None of the other investigated intrinsic or extrinsic covariates, including concomitant administration with phosphate binders, oral iron and acid reducing agents resulted in a significant change in daprodustat systemic exposure.
Conclusion
The PopPK of daprodustat in the CKD population with anemia was adequately characterized. Allometrically-scaled body weight on clearance and volume, dialysis status on absorption and clopidogrel on clearance were statistically significant covariates.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Daprodustat pharmacokinetics in chronic kidney disease was adequately characterized by a three-compartment distribution and first-order elimination with five transit absorption compartments describing the variable absorption of the drug. |
Of the 20 intrinsic and extrinsic patient covariates evaluated, body weight effect on clearance and volume of distribution, dialysis status on absorption rate and coadministration of clopidogrel (moderate CYP2C8 inhibitor) effect on clearance were statistically significant in patients with CKD with anemia. |
None of the other investigated intrinsic or extrinsic covariates, concomitant dosing with phosphate binders, oral iron and acid reducing agents resulted in a meaningful change in daprodustat systemic exposure requiring dose-adjustment. |
1 Introduction
Daprodustat (Jesduvroq) is the first orally bioavailable hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI) approved in the USA for treatment of anemia owing to chronic kidney disease (CKD) in adults who have been receiving dialysis for at least 4 months [1]. It is also approved in Japan (Duvroq) for the treatment of anemia owing to CKD in dialysis-dependent (DD-CKD) and non-dialysis dependent (NDD-CKD) adults [2]. Daprodustat stimulates endogenous erythropoiesis and modulates iron metabolism through inhibition of HIF-prolyl-4-hydroxylases. This activity results in the stabilization and nuclear accumulation of HIF-1α and HIF-2α transcription factors, leading to increased transcription of the HIF-responsive genes, including erythropoietin and transferrin [3]. Daprodustat mimics the effects of hypoxia to stimulate erythropoiesis by increasing erythropoietin (EPO), and subsequently hemoglobin (Hgb) levels, without exposing the patient to supraphysiologic EPO levels [4, 5].
During phase 3 clinical trials (ASCEND studies), an individualized dosing regimen of daprodustat was administered either once daily (QD 1–24 mg) in NDD-CKD and DD-CKD or three times a week (TIW 2–48 mg) in DD-CKD subjects to align with their dialysis schedule. A semi-mechanistic longitudinal dose–hemoglobin response model for daprodustat was used to support the phase 3 doses in anemic subjects with CKD [6]. Daprodustat starting doses were based on baseline Hgb or prior erythropoietin (ESA) doses and aimed to reach or maintain Hgb within the target range with a minimum number of dose adjustments. This titration dosing regimen was applied to ensure achievement and maintenance of a recommended target hemoglobin range of 10–12 g/dL [7]. Currently, the QD regimen is approved in the USA. In addition to noncompartmental pharmacokinetic (PK) evaluation, three sequential PopPK analyses were undertaken at the different stages of daprodustat development to inform on dosing strategy as well as characterize the intrinsic and extrinsic factors influencing the systemic exposure of daprodustat. Although this paper focuses predominantly on the integrated phase 2B/3 data analysis, salient reference is made to the companion PopPK models [8, 9] developed previously using phase 1 and 2 studies as a starting point for the current analysis. A brief description of the basic pharmacokinetics and pharmacodynamics of daprodustat is provided for context to the PopPK characterization.
After single oral dosing, daprodustat is readily absorbed with tmax observed from 1 to 4 h followed by rapid elimination with a terminal phase elimination half-life range of 1–4 h [10, 11]. There was no accumulation of daprodustat following repeated once daily dosing which is consistent with its short terminal phase elimination half-life [12]. Daprodustat exhibits linear pharmacokinetics over a dose range of 10–100 mg [11, 12]. Oral absolute bioavailability of daprodustat is 65%. Renal excretion of the parent drug is negligible [10]. No relevant differences were observed in daprodustat PK between subjects with CKD and healthy subjects. Food was reported to minimally delay and lower the maximal plasma concentration (Cmax), without significantly affecting the overall area under the plasma concentration-time curve (AUC), therefore, daprodustat can be given with or without food [13]. Daprodustat is extensively metabolized primarily via hydroxylation by CYP2C8 and a minor contribution of CYP3A4 [13]. At steady state, the systemic exposure of the three major circulating human metabolites, M2, M3, and M13 (>10% drug related material) is generally lower relative to parent drug in subjects with CKD [14]. Concomitant administration of daprodustat with gemfibrozil, a strong inhibitor of CYP2C8, substantially increased daprodustat AUC 18.6-fold and half-life 3.70-fold; therefore the coadministration of daprodustat with strong CYP2C8 inhibitors is contraindicated [13]. However, no clinically relevant change in rate or extent of systemic exposure of daprodustat was shown with concomitant administration of trimethoprim, a weak CYP2C8 inhibitor [15]. Daprodustat was not a perpetrator of drug–drug interactions with pioglitazone (a sensitive CYP2C8 substrate) or rosuvastatin (an OATP1B1 transporter) [15]. No clinically relevant change in exposure was observed in mild to moderate hepatic impairment [14]; however, daprodustat has not been studied in subjects with severe hepatic impairment. The steady-state exposure of daprodustat is similar in subjects with normal renal function and those with varying degrees of renal impairment [11].
The effect of daprodustat on change in hemoglobin (Hgb) is indirect and occurs through a downstream cascade where prolyl-4-hydroxylase (PHD) inhibition leads to erythropoietin (EPO) stimulation and a subsequent increase in Hgb. Other pharmacodynamic markers of PHD inhibition indirectly modulate iron utilization and metabolism thereby also contributing towards change in Hgb response. The effects of daprodustat on Hgb have been captured in a modeling framework supporting dose titration [16] and helped establish the dose titration schedule studied in the Phase 3 clinical trials.
The objective of the present analysis was to further characterize the PopPK of daprodustat and the influence of covariates in subjects with CKD based on data from one US phase 2B study and four global phase 3 studies. This integrated PopPK analysis in patients with CKD with anemia further supported the pharmacokinetics aspects of daprodustat during regulatory submissions.
2 Methods
2.1 Data
2.1.1 Ethics Approval
All studies were conducted in accordance with the US Code of Federal Regulations (CFR), Good Clinical Practice, 21 CFR Parts 50, 56, and 312, the ethical principles set forth in the Declaration of Helsinki, the International Conference on Harmonisation guideline regarding Good Clinical Practice (E6 Consolidated Guidance, April 1996), and the ethical requirements referred to in the European Union Directive 2001/20/EC. The trial protocol, amendments, and informed consent forms were reviewed and approved by institutional review boards and national authorities.
2.1.2 Summary of Source Data
A subset of subjects from each of the five studies provided plasma samples for PopPK assessments. Sparse plasma samples, dosing, demographic, and covariate data from subjects recruited in the five studies (one Phase 2B and four Phase 3) with PK assessments were combined. A total of 707 patients who received daprodustat were included in the PopPK analysis (Table 1). A summary of the studies and study characteristics used in this analysis is provided in Table 2.
2.1.3 Bioanalytical Method
Plasma samples of daprodustat were assayed using a high-performance liquid chromatography tandem mass spectrometric method. The lower limit of quantification (LLOQ) of the assay used to quantify the concentration of daprodustat was 0.1 ng/mL in all studies. The applicable analytical run for study samples met all predefined acceptance criteria, including bias (within ± 15%) and precision (< 15%) of quality control samples analyzed with study samples, as recommended by the FDA in the bioanalytical method validation guidelines [17].
2.2 Model Development
2.2.1 Previous Models
The PK of daprodustat in healthy subjects and target CKD adult population were characterized throughout the clinical development program. Overall, three PopPK models were developed sequentially as depicted in Fig. 1 of the Electronic Supplementary Material (ESM), which also includes the types of studies used in these models. The first phase 1/2 PopPK model was the starting point for providing PK parameter estimates during development of both the Japan phase 3 PopPK model and eventually the current global phase 2B/3 PopPK model in the target CKD population. Briefly, the phase 1/2 PK model was developed using rich sampled phase 1 data in healthy volunteers and patients with CKD and was described by a three-compartment distribution model with first-order elimination. The absorption part of the model contained a series of transit compartments, followed by a saturable absorption from the final transit compartment into the central compartment. Four transit absorption compartments were used under fasted conditions, and seven transit compartments were used under fed conditions. Additionally, the model contained allometric scaling of clearance (exponent = 0.75) and volume parameters (exponent = 1) with fixed exponents, and an effect of current hemodialysis (during PK sampling) on clearance. The PK parameters from this phase 1/2 PopPK model are presented in Table S1 of ESM to provide some context as the starting point for the model development in the current Phase 2B/3 analysis.
2.2.2 Starting Model
The starting model for the phase 2B/3 analysis differed in several aspects from the phase 1/2 PK model described in the previous section. Because daprodustat was dosed without regard for food in the phase 2B/3 studies, four transit compartments were used in the starting model for all subjects (as opposed to four or seven transits with fasted or fed, respectively, in the phase 1/2 model). Additionally, the saturable absorption term in the phase 1/2 model (Michaelis constant of 182 mg) was removed from the model, because this term destabilized the model and had limited relevance at the dose range for the phase 2B/3 studies (maximum dose 48 mg for TIW dosing) [18]. Finally, owing to the proportion of plasma data below the limit of quantification (BLQ) up to 20% in these studies (Table 1), the M3 method (likelihood-based) was used to simultaneously analyze the non-BLQ and BLQ data in NONMEM [19]. The non-BLQ data were modelled as log-transformed data with additive residual error (on log-scale). In the starting model, all parameter estimates were fixed to the estimates of the phase 1/2 PK model, with the exception of the absorption rate constant (owing to the changes in the absorption model).
2.2.3 Structural Model Update
The starting model was applied to the phase 2B/3 data to assess if the data were adequately described by the model. It was then explored whether re-estimation of selected PK parameters based on the phase 2B/3 data would significantly (p < 0.001) improve model fit. This approach was selected to give the flexibility to characterize the phase 2B/3 data while allowing the analysis to leverage the information from the phase 1/2 PK model, which included mostly studies with intensive PK sampling. Thus, a stepwise inclusion of estimated factors associated with the phase 2B/3 data on the fixed PopPK parameters was undertaken to quantify the differences in relation to the starting model to achieve an adequate fit of the current phase 2B/3 dataset. Models that resulted in a decrease of at least 10.8 in the objective function value (OFV) value [ρ < 0.001, χ2, 1 degree of freedom (df)] after inclusion of a factor were ranked, starting with the factor that generated the largest drop in the minimum value of the objective function (MVOF). Subsequently, the ranked factors were included one by one. If inclusion of the additional factor was still statistically significant at the ρ < 0.001 level, the factor was kept and the next factor in rank was tested until all ranked factors were tested. At this point, the model was evaluated to consider if the structural parameter update resulted in an adequate fit of the data, and if not, additional modifications of the model were performed to improve the model fit including evaluation of the number of absorption transit compartments. The resulting model was considered the base model.
2.2.4 Statistical Model
The inter-individual variability (IIV) for PK parameters was modelled as log-normal distributions. The decision to reduce or expand the IIV model was based on improvements in the objective function value (OFV) and model performance assessed by visual predictive checks (VPCs). Various combinations of inter-individual variability (IIV) models were investigated with and without off-diagonal elements. The IIV parameters were removed if they were estimated with high uncertainty [> 50% relative standard error (RSE)], resulted in large condition numbers (e.g., > 1000) and shrinkage of random effects (η-shrinkage and ε-shrinkage were preferably < 30%).
2.2.5 Covariate Analysis
Predefined covariates for evaluation are listed in Table S2 of ESM and covariates were selected on the basis of physiologic plausibility and/or prior knowledge. It is noteworthy that phosphate binders, acid reducing agents, and oral iron were new covariates tested in this global phase 2B/3 model and were not available for inclusion in the previous PopPK models. In addition, clopidogrel was the most commonly administered moderate CYP2C8 inhibitor with only three subjects recorded as taking other CYP2C8 inhibitors.
Following development of the base model, covariate effects of interest were included in a model-based evaluation using a step forward inclusion and backward elimination approach [20]. As a first step, all preselected continuous and categorical covariates were added individually to the base model (parameterization of covariate effects are specified in the ESM). Covariates achieving a drop in the OFV less than 6.63 points (p < 0.01, χ2, 1 df) during the univariate implementation were considered non-significant and were not tested further in the forward inclusion. Covariate relationships were tested in a forward inclusion (ΔOFV of 6.63 p < 0.01, χ2, 1 for 1 df) and backward exclusion (ΔOFV of 10.83, p < 0.001, χ2, for 1 df) procedure. If the final covariate model contained a highly correlated covariate, the superior covariate was selected on the basis of statistical significance or clinical considerations. It was also evaluated whether estimation or removal of the allometric exponents was statistically, significantly better than fixing them.
2.2.6 Model Evaluation
In addition to inspection of parameter estimates and goodness-of-fit (GoF) plots [21], the predictive performance and robustness of the developed PK model was evaluated using simulation-based method(s), namely visual predictive checks (VPC) and normalized prediction distribution errors (NPDE). The VPCs illustrate the model’s ability to simulate the data that have been used for the model development. Percentiles (10th, 50th and 90th) of the observations were compared with percentiles (including confidence intervals) of the simulated values. These VPC plots were stratified by study. VPCs for the non-BLQ data were created using dose-normalized log concentrations for both observed and simulated data. Additionally, to evaluate whether the model adequately predicted the percentage of data points below LLOQ, the data were also split into three bins: “predose/trough,” “0.5 h post dose sample,” “other samples.” For each of these bins, the observed percentage of BLQ data was compared with the interval of the simulated percentage of BLQ data.
The normalized prediction distribution errors (NPDEs) were obtained from the NONMEM output, using a method, which allows for its use in PK models that use the M3 method to analyze normal and BLQ PK data [22]. Each observation was simulated 1000 times (ESAMPLE = 1000 option in NONMEM). For each of the observations, the prediction discrepancy was calculated as the percentile of the observation in the predicted distribution. A decorrelation step was performed for both the observed and simulated data (to correct for the fact that more than one sample/observation was available per subject resulting in correlations). The resulting prediction distribution errors were reported following a normal distribution with mean 0 and variance of 1. From the NONMEM output, population predictions and NPDEs were obtained for both the normal and BLQ samples. To evaluate the model, the NPDEs were plotted versus the population predictions and time after dose.
For PK parameters that were fixed in the final PK model (because re-estimating via a phase 3 factor did not significantly improve the fit), the informativeness of the phase 2B/3 data to these parameters was evaluated by calculating the uncertainty of the parameter when estimated.
2.3 Simulations to Illustrate Impact of Covariates
Both forest plots and typical profile simulations were applied to visualize the impact of covariates on the estimated systemic exposure of daprodustat from the final model. Individual dose-normalized model-predicted AUC and Cmax for each occasion with PK observations was created, along with baseline covariates of each subject. Cmax at steady state were determined from the simulated profiles and AUC over the steady state dosing interval was calculated as: AUC = \(\frac{\text{dose * F1}}{\text{apparent clearance}}\).
Forest plots were created to illustrate the impact of predefined intrinsic or extrinsic factors of interest (listed in Table 2 of ESM) on the individual dose-normalized model-predicted AUC and Cmax. Covariates identified in the model were also included in the forest plots, even if they were not prespecified for completeness.
To assess the difference in AUC and Cmax for the populations of interest, a linear model [analysis of variance (ANOVA)] was fitted to the log-transformed dose-normalized exposure metrics predicted for each subject using the final PK model. The forest plot included the estimate and 90% confidence interval of the ratio of the exposure metrics in the populations of interest relative to the reference population. Comparisons were only included if the respective populations had a sufficient number of subjects (> 5% of subjects in the final PK dataset).
Typical daprodustat profiles without inter-individual variability were simulated to show the isolated impact of covariates included in the model. These simulations were stratified by the discrete covariates. In each plot, the typical daprodustat PK profile at median bodyweight and at the 5th and 95th percentiles of the bodyweight distribution in the data were depicted. From these typical profiles, the estimated impact of covariates included in the model on AUC and Cmax were also calculated.
2.4 Software
The analysis was performed by means of non-linear mixed-effects modelling as implemented in the NONMEM software package (version 7.5.0; Icon Development Solutions, Ellicott City, Maryland USA) [23], in combination with PsN (version 5.0.0) [24, 25]. GFortran (version 9.3.0) was used as compiler. Diagnostic graphics, exploratory analyses, and post-processing of NONMEM output were performed using R (version 4.0.3; The R foundation for Statistical Computing) [26], and RStudio (version 1.3.1039; RStudio Inc, Boston, USA)
3 Results
3.1 Analysis Dataset
For the PopPK population subset, there were 707 patients (males, 378; females, 329) and a total of 4096 plasma concentration values included in the analysis dataset. Mean age, weight, and body mass index were 59 years, 77 kg and 26.95 kg/m2, respectively, in the pooled dataset. A summary of the patient key demographics by study is included in Table 3. The actual doses were titrated based on hemoglobin (Hgb) level ranging from 1 to 24 mg as once daily dosing and 2–48 mg when given as three times a week (TIW) dosing schedule. There were 571 subjects on dialysis (DD), 136 subjects not on dialysis (NDD), and 3 subjects on peritoneal dialysis (PDD). Apart from the estimated glomerular filtration rate (eGFR), which was lower in DD subjects, no other apparent differences were observed between NDD, DD and PDD subjects.
3.2 Daprodustat PK Model
3.2.1 Base Model
In the starting PK model, all PK model parameters were fixed to the phase 1/2 PopPK structural model parameter estimates. Subsequently, the inclusion of a “phase 3 factor” (quantifying the difference between the phase 1/2 estimates and the current phase 2B/3 dataset) was tested for all structural parameters in a stepwise manner as described in Sect. 2.2.3. The following three “phase 3 factor” parameters were found to significantly improve the model and were included in the base model: absorption rate constant (PH3KA), a shared factor for central compartment volume (PH3V2), and first peripheral compartment volume (PH3V3) as they had similar “phase 3 factor” estimates and a third factor on the second peripheral inter-compartmental clearance (PH3Q2). Two factors that were initially implemented (on V6 and F1) were removed owing to high (> 400%) RSE and collinearity issues, respectively. A five transit-compartment absorption model better characterized the phase 2B/3 data compared with the four transit compartments in the starting model. A sensitivity analysis showed the clearance (CL) parameter could be estimated with good precision (RSE = 3.0%) from the phase 2B/3 dataset without fixing this parameter in the PopPK model. Since this CL estimate was within 1% of the phase 1/2 estimate and was not significantly different, no “phase 3 factor” on CL was needed and the CL parameter was fixed to the phase 1/2 estimate. A schematic of the final PopPK phase 2B/3 model is provided in Fig. 1 with final PK parameters in Table 4.

Schematic of final PopPK model for daprodustat in phase 2B/3 studies. ka: absorption rate constant; Tr: transit absorption compartment; V2: central volume of distribution; V3: volume of distribution peripheral 1 compartment; V6: volume of distribution peripheral 2 compartment; CL: clearance
3.2.2 Covariate Effects
Bodyweight was incorporated in the structural part of the PopPK model with fixed allometric exponents for volume and clearance terms. Coadministration with clopidogrel showed a fractional decrease in CL [point estimate: − 0.42, 95% confidence interval (CI): − 0.54 to − 0.31]. In the non-dialysis population, there was a fractional increase in the rate of absorption constant (ka) with point estimate of 0.46 (95% CI 0.22–0.69). It is noteworthy that the phase 2 study 205665 was a significant covariate with effect on PK parameters, namely, inter-compartmental clearance (Q2), relative bioavailability (F1), and absorption rate constant (ka), when compared with the phase 3 studies. This observed disparity between the phase 2B and phase 3 studies could not be explained on the basis of any differences in patient characteristics between study 205665 and the other studies. All other tested covariates (Table 2 of ESM) were found not to be significant.
3.2.3 Final Model
The final PopPK model adequately characterized the model parameter values and associated uncertainties (based on %RSE) relating to the influence of the phase 3 studies, as shown in Table 4. Low eta-shrinkage estimates of 14.6% and 7.6% for CL and absorption rate constant (ka), respectively, were obtained reflecting the validity of using post hoc individual parameter estimates for model diagnosis and creation of forest plots. Interindividual variability in volume parameters could not be estimated . The expected heterogeneity of the phase 2B/3 study populations is described by the parameters PH3KA, PH3V3/PH3V2, and PH3Q2, which reflect the estimates with corresponding 95%CIs (on log-scale) for the differences of these PK parameters in the current population relative to the corresponding parameters from phase 1/2 PopPK model. For ease of interpretation, after exponentiation, the phase 2B/3 data shows a fractional decrease (relative to the phase 1/2 PopPK model estimates) in the absorption rate constant (ka) [point estimate: −0.28 (95% CI −0.23 to −0.33], a fractional increase in peripheral volume of distribution (V2/V3) [point estimate: 0.54 (95% CI 0.43–0.66)] and fractional decrease in the inter-compartmental clearance (Q2) [point estimate: −0.76 (95% CI −0.66 to −0.83)]. For the covariate effect of phase 2B study 205665, the three parameters Q2, ka and F1 were significantly different from those of the four phase 3 studies in the present analysis (Table 4).
In the final model, three PK parameters remained fixed on the phase 1/2 estimates (CL, Q and V6). As a sensitivity analysis, a model was tested in which these were also estimated . The estimates for CL and Q had good precision (RSE of 3.0% and 19.9%, respectively) and were estimated within 1% of the phase 1/2 model parameter estimates (data not shown). In addition, this sensitivity analysis showed parameter V6 was estimated at 115L (324% higher than phase 1/2 estimate) with low precision (RSE = 53%), and this model was not significantly better than the final model (p > 0.001, df = 3).
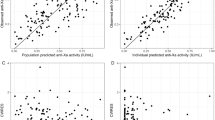
All parameters (structural and covariate) were estimated with good precision (RSE < 30%, Table 4). The condition number was 12.8, indicating that the final model was stable with acceptable collinearity of the parameter estimates. The final model adequately described the BLQ observations as shown by the VPC where the 95% confidence intervals (Fig. 2) of the model-predicted percentage of BLQ contain the observed mean percentage of BLQ data. Appropriateness of the final PopPK model was also evaluated using goodness-of-fit (Fig. 2 of ESM) plots as well as simulation-based diagnostics including VPC of the dose-normalized daprodustat concentration versus time stratified by study (Fig. 3). Additionally, normal prediction discrepancy error (NPDE) plots with pooled data with time and predicted concentrations, respectively (Fig. 3 of ESM) were also evaluated. NPDE plots stratified by study are shown in Fig. 4 of ESM. These plots indicate adequacy of the model to describe the observed data with no evidence of systematic bias. The apparent deviation from the identity line at low concentrations (particularly visible in the population prediction versus observation plots; Fig. 2 of ESM) can be explained by the lower limit of quantification of 0.1 ng/mL. While it is possible to have predicted concentrations below 0.1 ng/mL, there will, by definition, not be any non-BLQ observations below 0.1 ng/mL. The VPC plots demonstrate that there was good concordance of the 10th, 50th, and 90th percentiles of the simulations and corresponding percentiles of observed data. However, some bias was noted at 24 h in the 90th percentile, possibly owing to outliers.

Visual predictive check of percentage of data below lower limit of quantification (BLQ) for final phase 2B/3 PK model. Solid lines depict the observed percentage of BLQ data, and the grey area shows the intervals of the percentage BLQ in the simulations, which include variability (interindividual and residual error) but not parameter uncertainty

Visual predictive check of dose-normalized daprodustat concentration versus time for final model stratified by study. Solid lines depict the observed median concentration, dashed lines the observed 10th and 90th percentiles, and the grey areas show the intervals of these statistics in the simulations, which include variability (interindividual and residual error) but not parameter uncertainty. Both x-axis and y-axis are displayed on a log-scale
3.3 Model-Based Simulations for Covariate Effects
In addition to body weight already included in the starting model, the other significant covariates, namely, coadministration with clopidogrel, showed a fractional decrease in CL [point estimate: −0.42 (95% CI −0.54 to −0.31)]. This equates to an AUC ratio for subjects on clopidogrel to those not on clopidogrel of 1.59-fold (90% CI 1.39–1.82) as depicted in the forest plot (Fig. 4). In the non-dialysis population, there was a fractional increase in the rate of absorption constant (ka) with point estimate of 0.46 (95% CI 0.22–0.69); thus, the Cmax ratio estimate for subjects not on dialysis versus dialysis is 1.19 (90% CI 1.09–1.30). Forest plots show the impact of these significant covariates as well as other demographic/physiological characteristics in the analysis dataset with respect to individual model-predicted dose-normalized AUC (Fig. 4) and Cmax (Fig. 5). Except for weight and clopidogrel, the 90% CIs of the other covariates did not reveal marked changes in exposure.

Forest plot for model-predicted dose-normalized AUC at steady-state. AUC, area under the plasma concentration-time curve over a 24-h dosing interval at steady state; n, number of individuals in each category. Circles indicate estimated change relative to reference. The error bars indicate the 90% confidence interval

Forest plot for model-predicted dose-normalized Cmax at steady-state. n, number of individuals in each category. Circles indicate estimated change relative to reference. The error bars indicate the 90% confidence interval
Furthermore, as shown in Fig. 6, population (typical) profiles of dose-normalized concentrations of daprodustat were simulated to illustrate the estimated size of covariate effects in the model, namely, bodyweight, clopidogrel use, and dialysis status. As shown in Table 5, concomitant dosing with clopidogrel as independent covariate resulted in a 1.74-fold higher AUC of daprodustat relative to the typical (independent) effect of clopidogrel on daprodustat. Additionally, the fold-change in Cmax and AUC based on the other covariates bodyweight and dialysis relative to a reference subject (non-dialysis dependent, not using clopidogrel, bodyweight of 74.6 kg) is shown in Table 5.

Simulated typical dose-normalized PK profiles illustrating estimated covariate effects. Solid line indicates the profile of a subject with median bodyweight (74.6 kg), while the dashed lines indicate subjects with 5th and 95th percentile bodyweight. The exposure estimates (Cmax, AUC) in each panel are reported as: median [95th–5th] bodyweight. Ph3, phase 3; NDD, non-dialysis dependent; CLOPI, clopidogrel use; DD, dialysis dependent
4 Discussion
This is the first publication of an integrated population pharmacokinetics analysis of daprodustat in adults with CKD across the phase 2B and 3 studies in both NDD-CKD and DD-CKD indications and provided supportive evidence in the regulatory submissions of daprodustat. The previously developed PopPK models of daprodustat [9] in mostly healthy as well as patients with CKD were used as a starting point for the current analysis comprising a more heterogenous CKD population in terms of study designs, sampling variability, and patient characteristics (as well as new covariates). The final model in this analysis was a three-compartment distribution and first order elimination with a five-transit compartment absorption phase, which adequately described the pharmacokinetics of daprodustat. This final model also quantified the impact of the statistically significant covariates bodyweight (included as allometric exponent), dialysis status, and concomitant clopidogrel use on relevant PK parameters.
There were some changes relating to structural modifications and updates of parameter estimates that were needed to the final model compared with the previous phase 1/2 PopPK models based on the expected heterogeneity of the phase 3 population as mentioned above. The final model had five transit absorption compartments (N = 5) which can be explained by removal of the food restriction in the phase 2B/3 studies while the phase 1 PK model had four or seven transit absorption compartments when daprodustat was taken in fasted or fed state, respectively [18].
The updates of parameter estimates were explored by estimating “phase 3 factors” to quantify the difference in parameters in the current phase 2B/3 population relative to the phase 1/2 model parameter estimates. The fractional change in the absorption rate (ka), volume, and inter-compartmental clearance parameters were well estimated in the phase 2B/3 studies while keeping the remaining PK parameters fixed to those from the phase 1/2 model. Both the comprehensive model evaluation based on goodness-of-fit diagnostics and simulation-based assessments in addition to sensitivity analyses strongly support the characterization of the final model and suitability of the present analysis to investigate the impact of extrinsic and intrinsic factors on the exposure estimates of daprodustat.
Of the 20 intrinsic and extrinsic covariates evaluated, only body weight (included with fixed allometric exponents), and two statistically significant covariates were identified. Firstly, patients that were non-dialysis-dependent had 46% higher oral absorption rate constant compared to dialysis-dependent patients. Dialysis status in end stage renal failure (ESRD) is expected to decrease rate of absorption of drugs in chronic renal failure [27]. Secondly, concomitant clopidogrel (moderate CYP2C8 inhibitor) use reduced apparent clearance of daprodustat by 42%. As presented in Table 5, the typical fold change in Cmax between non-dialysis and dialysis dependent are not deemed to be clinically relevant. Although the 1.74-fold increase in AUC of daprodustat in a typical individual following concomitant dosing with clopidogrel (Table 5) would not be considered clinically important, further discussion during FDA regulatory review [1] recommended a reduction in the starting dose (2 mg once daily) of daprodustat by half in patients with CKD with anemia when initiating treatment with moderate CYP2C8 inhibitors such as clopidogrel. This recommendation was based on consideration of other data from drug-drug interaction studies evaluating administration of daprodustat with mild and strong CYP2C8 inhibitors, trimethoprim and gemfibrozil [13], respectively, as well as from physiologically-based pharmacokinetic (PBPK) modeling of daprodustat (manuscript in preparation) which predicted an average 4.3-fold increase in AUC for daprodustat when coadministered with clopidogrel. A recent publication evaluating the mechanism of CYP2C8-OATP1B1 drug–drug interaction with daprodustat predicted an average 3.6-fold increase in AUC when coadministered with repeated clinical doses of clopidogrel [28]. Possible reasons for the smaller effect size of the drug interaction of daprodustat with clopidogrel in the PopPK analysis may be owing to factors including lack of accurate timing of records for co-medication intake. However, since treatment with daprodustat requires a titration-based posology to achieve and maintain target hemoglobin levels in CKD patients with anemia, any clinical impact of clopidogrel coadministration on the PK of daprodustat would be mitigated based on this individualized dosing regimen. Moreover, following 4 weeks of daprodustat dosing, hemoglobin changes from baseline were similar in subjects with and without concomitant use of clopidogrel [1].
It is noteworthy that the phase 2B study 205665 was a significant covariate with effect on PK parameters, namely, inter-compartmental clearance (Q2), relative bioavailability (F1), and absorption rate constant (ka), resulting in much lower exposure when compared with the phase 3 studies. Moreover, the residual unexplained error in the model was higher for the phase 2B Study 205665 (standard deviation of additive error of 1.63 on log-scale) compared with the phase 3 studies in the analysis dataset. This difference could not be explained based on any deviations in either patient or study design characteristics. The estimation stabilization strategy used in the present analysis of phase 2B/3 data by fixing parameter estimates based on richly sampled PK data from early clinical studies in both healthy and target CKD population is a reasonable practical approach. Subsequent selective PK parameter updates based on the “phase 3 factors” stabilized the structural model as shown by the model evaluation tools and good model fit to the data. The fact that the CL parameter (which was fixed in the final model) could be estimated with good precision based on the phase 2B/3 data, supports the informativeness of the dataset for this parameter and the investigation of covariate effects for this parameter. However, it is acknowledged that there are alternative methods, such as combining study data across all phases of development for analysis [29] or using the Bayesian PRIOR subroutine in NONMEM [30, 31] to leverage the summary PK information (priors) to provide less biased parameter estimates compared to fixing PK parameters. Nevertheless, all these methods have their own well documented disadvantages and practical challenges [30, 31].
Because PK visits were scheduled on dialysis days for dialysis dependent subjects, the difference in PK between dialysis and non-dialysis days among subjects dependent on dialysis could not be investigated. However, in a phase 2 study, the steady-state pharmacokinetics of daprodustat in subjects with normal renal function, anemic non-dialysis (ND)-dependent CKD subjects (CKD stage 3/4) and anemic subjects on either hemodialysis (HD) or peritoneal dialysis (PDD) did not reveal any marked changes in the pharmacokinetics of daprodustat [11] which is expected given that daprodustat is highly protein bound (> 98%) [14]. Effects of various co-medications on daprodustat PK were investigated during covariate analysis. Only clopidogrel coadministration showed a statistically significant increase in systemic exposure, whilst other comedications, such as phosphate binders and oral iron, did not show relevant changes in AUC. However, since the timing of comedication intake was not recorded (and may therefore not have been simultaneous with daprodustat administration during PK visits), the possibility of a potential drug–drug interaction on (the extent of) absorption cannot be excluded.
5 Conclusions
The three-compartment distribution and first order elimination pharmacokinetic model with a five serial transit absorption compartments adequately described the pharmacokinetics of daprodustat in subjects with chronic kidney disease. While body weight, dialysis effect, and clopidogrel coadministration were statistically significant, of the 20 intrinsic and extrinsic patient covariates evaluated, the other covariates including phosphate binders, oral iron, and acid reducing agents did not show significant increases in systemic exposure.
References
JESDUVROQ (Daprodustat). [package insert] Durham, NC: GlaxoSmithKline. 2023 [cited 08 July 2024]; Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/216951s000lbl.pdf
Yap DYH, McMahon LP, Hao C. Recommendations by the Asian pacific society of nephrology (apsn) on the appropriate use of hif-ph inhibitors. Nephrology. 2020;26:105–18.
Gupta N, Wish JB. Hypoxia-inducible factor prolyl hydroxylase inhibitors: a potential new treatment for anemia in patients with CKD. Am J Kidney Dis. 2017;69(6):815–26.
Akizawa T, Nangaku M, Yonekawa T, Okuda N, Kawamatsu S, Onoue T, et al. Efficacy and safety of daprodustat compared with darbepoetin alfa in Japanese hemodialysis patients with anemia: a randomized, double-blind, phase 3 trial. Clin J Am Soc Nephrol. 2020;15(8):1155–65.
Meadowcroft AM, Cizman B, Holdstock L, Biswas N, Johnson BM, Jones D, et al. Daprodustat for anemia: a 24-week, open-label, randomized controlled trial in participants on hemodialysis. Clin Kidney J. 2019;12(1):139–48.
Mahar KM, Johnson BM. Population Kinetic/Pharmacodynamic (K/PD) Analyses of Daprodustat (Prolyl Hydroxylase Inhibitor) in Non-dialysis (ND) and Hemodialysis-Dependent (HDD) Anemia Patients with Chronic Kidney Disease (CKD). J Pharmacokinet Pharmacodyn. 2018;45(1):T-041.
Mactier R, Davies S, Dudley C, Harden P, Jones C, Kanagasundaram S, et al. Summary of the 5th edition of the renal association clinical practice guidelines (2009–2012). Nephron Clin Pract. 2011;118(1):c27–70.
Mahar K, Mills R, Goyal N. Population Pharmacokinetic (POP-PK) Analysis of Daprodustat (GSK1278863) and its Metabolites in Non-dialysis (ND) and Hemodialysis-Dependent (HDD) Anemia Patients with Chronic Kidney Disease (CKD). J Pharmacokinet Pharmacodyn. 2018;45(S64):T040.
Post T, Mahar K, Ashman M. Population pharmacokinetic analysis of daprodustat in healthy volunteers (HV), global phase 2 and Japanese phase 3 studies in chronic kidney disease patients with anemia following once daily (QD) or 3 times weekly (TIW) administration. Clin Pharm Ther. 2021;109:PI-031.
Mahar KM, Caltabiano S, Andrews S, Ramanjineyulu B, Chen L, Young G, et al. Clinical pharmacokinetics of daprodustat: results of an absorption, distribution, and excretion study with intravenous microtracer and concomitant oral doses for bioavailability determination. Clin Pharmacol Drug Dev. 2021;10(12):1419–31 (2021-12-01).
Caltabiano S, Cizman B, Burns O, Mahar KM, Johnson BM, Ramanjineyulu B, et al. Effect of renal function and dialysis modality on daprodustat and predominant metabolite exposure. Clin Kidney J. 2019;12(5):693–701.
Hara K, Takahashi N, Wakamatsu A, Caltabiano S. Pharmacokinetics, pharmacodynamics and safety of single, oral doses of GSK1278863, a novel HIF-prolyl hydroxylase inhibitor, in healthy Japanese and Caucasian subjects. Drug Metab Pharmacokinet. 2015;30(6):410–8.
Johnson BM, Stier BA, Caltabiano S. Effect of food and gemfibrozil on the pharmacokinetics of the novel prolyl hydroxylase inhibitor GSK1278863. Clin Pharmacol Drug Dev. 2014;3(2):109–17.
Mahar KM, Shaddinger BC, Ramanjineyulu B, Andrews S, Caltabiano S, Lindsay AC, Cobitz AR. Pharmacokinetics of daprodustat and metabolites in individuals with normal and impaired hepatic function. Clin Pharmacol Drug Dev. 2022;11(5):562–75.
Caltabiano S, Mahar KM, Lister K, Tenero D, Ravindranath R, Cizman B, Cobitz AR. The drug interaction potential of daprodustat when coadministered with pioglitazone, rosuvastatin, or trimethoprim in healthy subjects. Pharmacol Res Perspect. 2018;6(2):1–9.
Van den Berg P, Post T, Ahsman Mea. Population dose-response analysis of hemoglobin data from global phase 2 and Japanese phase 3 studies with daprodustat in chronic kidney disease patients with anemia following once daily (QD) or 3 times weekly (TIW) administration. Clin Pharmacol Ther. 2021;109.
Food and Drug Administration. Bioanalytic method validation guidance for industry. US Department of Health and Human Services; 2018.
Yamada M, Osamura M, Ogura H, Onoue T, Wakamatsu A, Numachi Y, et al. A single-dose, open-label, randomized, two-way crossover study in healthy Japanese participants to evaluate the bioequivalence and the food effect on the pharmacokinetics of daprodustat. Clin Pharmacol Drug Dev. 2020;9(8):978–84.
Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Pharmacodyn. 2008;35(4):401–21.
Lehr T, Staab A, Tillmann C, Trommeshauser D, Raschig A, Schaefer HG, Kloft C. Population pharmacokinetic modelling of NS2330 (tesofensine) and its major metabolite in patients with Alzheimer’s disease. Br J Clin Pharmacol. 2007;64(1):36–48.
Nguyen TH, Mouksassi MS, Holford N, Al-Huniti N, Freedman I, Hooker AC, et al. Model evaluation of continuous data pharmacometric models: metrics and graphics. CPT Pharmacometrics Syst Pharmacol. 2017;6(2):87–109.
Bauer RJ. NONMEM tutorial part ii: estimation methods and advanced examples. CPT Pharmacometrics Syst Pharmacol. 2019;8(8):538–56.
Beal S, Boeckmann L, Bauer R, Sheiner L. NONMEM User’s Guides. (1989–2009). 2009.
Lindbom L, Pihlgren P, Jonsson N. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57 (2005/09/01/).
Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85–94 (2004/08/01/).
R Core Team. R: A language and environment for statistical computing. Vienna, Austria. 2020.
Dreisbach AW, Lertora JJ. The effect of chronic renal failure on drug metabolism and transport. Expert Opin Drug Metab Toxicol. 2008;4(8):1065–74.
Bi YA, Jordan S, King-Ahmad A, West MA, Varma MVS. Mechanistic determinants of daprodustat drug-drug interactions and pharmacokinetics in hepatic dysfunction and chronic kidney disease: significance of OATP1B-CYP2C8 interplay. Clin Pharmacol Ther. 2024;115(6):1336–45.
Bruno R, Vivier N, Vergniol JC, De Phillips SL, Montay G, Sheiner LB. A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J Pharmacokinet Biopharm. 1996;24(2):153–72.
Gisleskog PO, Karlsson MO, Beal SL. Use of prior information to stabilize a population data analysis. J Pharmacokinet Pharmacodyn. 2002;29(5–6):473–505.
Chan Kwong AHP, Calvier EAM, Fabre D, Gattacceca F, Khier S. Prior information for population pharmacokinetic and pharmacokinetic/pharmacodynamic analysis: overview and guidance with a focus on the NONMEM PRIOR subroutine. J Pharmacokinet Pharmacodyn. 2020;47(5):431–46.
Acknowledgements
The authors thank all the patients (and their caregivers) who provided data for the PopPK model and support from the daprodustat development team at GSK. Medical writing support was provided by Misba Beerahee, PhD, of MBPharmaco Consulting Ltd., UK, and funded by GSK. The authors would also like to thank Lakshmi Vasist and Sandra Visser for influential conversations supporting the phase 1/2 and Japan modeling; Jan Vanderlinde, Priyanka Kocheta, and Yufei Du for programming the modeling datasets; and Italo Poggesi for his considerate review of the manuscript. All individuals and institutions mentioned in the acknowledgements have provided written permission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
All clinical studies mentioned in this manuscript were funded by GSK.
Conflicts of Interest/Competing
K.M.M. and S.Y. are employees of GSK and hold GSK shares. T.M.P., S.C.G., and E.M. are consultants at LAP&P, the Netherlands.
Ethics Approval
All studies were conducted in accordance with the US Code of Federal Regulations (CFR), Good Clinical Practice, 21 CFR Parts 50, 56, and 312, the ethical principles set forth in the Declaration of Helsinki, the International Conference on Harmonization guideline regarding Good Clinical Practice (E6 Consolidated Guidance, April 1996), and the ethical requirements referred to in the European Union Directive 2001/20/EC. The study protocols were approved by the institutional review boards or ethics committees of the study sites.
Consent to Participate
Informed consent was obtained from all individual participants included in the studies.
Consent for Publication
Not applicable.
Code Availability
Model code is available in Electronic Supplementary Materials
Author Contributions
All authors contributed to critically revising the manuscript for important intellectual content and approve the manuscript for publication.
Data availability statemen
Please refer to GSK weblink to access GSK’s data sharing policies and as applicable seek anonymised subject level data via the link https://www.gsk-studyregister.com/en/.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mahar, K.M., Yang, S., Mesic, E. et al. Integrated Population Pharmacokinetics of Daprodustat in Patients with Chronic Kidney Disease with Anemia. Clin Pharmacokinet (2024). https://doi.org/10.1007/s40262-024-01417-9
Accepted:
Published:
DOI: https://doi.org/10.1007/s40262-024-01417-9