
Abstract
Background and Objective
Given a high pharmacokinetic inter-individual variability and a low exposure target achievement, ganciclovir (GCV) therapeutic drug monitoring is sometimes used in children. We aimed to develop and validate Bayesian estimators based on limited sampling strategies for the estimation of GCV area under the concentration–time curve from 0 to 24 h in pediatric transplant recipients treated with valganciclovir (VGCV) or GCV.
Methods
Solid organ transplant or stem-cell transplant recipients who received GCV or VGCV and had available GCV concentrations per standard of care were retrospectively included in this study for pharmacokinetic modeling and development of Bayesian estimators using the iterative two-stage Bayesian method. Validation datasets included additional child recipients of a solid organ transplant or stem-cell transplant, and child recipients of a kidney or liver transplant enrolled in a previous study. Various combinations of three or two sampling times, applicable in clinical practice, were assessed based on the relative mean bias, standard deviation, and the root mean square error in a development dataset and three independent validation datasets.
Results
In the development dataset, the mean bias/standard deviation/root mean square error for the 1 h/2 h/3 h and 1 h/3 h limited sampling strategies were − 1.4%/9.3%/9.1% and − 3.5%/12.2%/12.3%, respectively for GCV, while for VGCV, the mean bias/standard deviation/root mean square error for the 1 h/2 h/6 h and 1 h/6 h limited sampling strategies were 0.7%/13.5%/13.3% and − 0.1%/12.1%/11.8%, respectively. In the independent validation datasets, seven (13%) and five (14%) children would have had misclassifications of their exposure using these Bayesian estimators and limited sampling strategies for VGCV and GCV, respectively.
Conclusions
Three plasma samples collected at 1 h/2 h/3 h and 1 h/2 h/6 h post-dose for GCV and VGCV respectively, are sufficient to accurately determine GCV area under the concentration–time curve from 0 to 24 h for pharmacokinetic-enhanced therapeutic drug monitoring.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Bayesian estimators and limited sampling strategies including samples at 1 h/2 h/3 h and 1 h/2 h/6 h were developed for intravenous GCV and enteral VGCV, respectively, in pediatric transplant recipients. |
The Bayesian estimators developed have been validated in three external datasets. |
These Bayesian estimators are available to the medical community at https://pharmaco.chu-limoges.fr/. |
1 Introduction
Ganciclovir (GCV) and its prodrug valganciclovir (VGCV) are the first-line drugs for the prophylaxis and treatment of cytomegalovirus (CMV) disease in solid organ transplant (SOT) and stem cell transplant (SCT) recipients [1,2,3]. No significant relationship has been observed between GCV trough concentration (C0) and GCV efficacy [4,5,6]. However, a GCV AUC0–24 h between 40 and 60 mg h/L is often referred to as a surrogate efficacy and safety target in adult transplant recipients. Indeed, a GCV area under the concentration–time curve from 0 to 24 h (AUC0–24 h) of 50 mg h/L was associated with an average incidence of CMV viremia of 1.3% during prophylaxis, whereas a GCV AUC0–24 h of 25 mg h/L was associated with an eight-fold higher incidence [7]. Moreover, the predicted incidence of neutropenia increased above 20% when GCV AUC0–24 h was > 60 mg h/L in adults with SOT [7, 8] and the predicted incidence of anemia increased from 26.6 to 51.9% when GCV AUC0–24 h exceeded 50 mg h/L [9]. These systemic exposure targets of GCV have been extrapolated to pediatric patients because of the lack of pharmacodynamic or exposure-effect studies in this population.
Several population pharmacokinetic (POPPK) analyses of GCV have been published in children, mostly in recipients of a SOT. These studies highlighted frequent insufficient GCV exposure for both intravenous (IV) GCV and enteral VGCV, a large inter-individual variability in pharmacokinetic (PK) parameters and a low probability of target attainment [10,11,12]. The actual US Food and Drug Administration-recommended dosing regimen based on body surface area using the Mosteller formula [13] and the creatinine clearance (CrCL) using the modified Schwartz formula [14] leads to overexposure in younger children and underexposure in older children [10,11,12, 15]. As a result, many other dosing regimens have been proposed in children, mostly weight based [11, 12, 15]. However, the probability of patients achieving the exposure target range (AUC0–24 h = 40–60 mg h/L) is still low, with a range of 23–65% in simulation studies [10,11,12, 15].
Given the high PK interindividual variability and the low probability of target achievement in children, therapeutic drug monitoring is highly recommended to ensure systemic exposure that optimizes the benefit-risk balance [16]. Considering that no significant correlation has been observed between GCV C0 and AUC0–24 h, therapeutic drug monitoring is generally performed based on the AUC0–24 h [17, 18].
Determining the AUC0–24 h using the reference trapezoidal method is even more challenging in pediatrics than in adults because it requires many blood samples. Few studies have developed maximum a posteriori Bayesian estimators (MAP-BEs) based on limited sampling strategies (LSSs). In pediatric transplant recipients, only two papers reported LSSs for GCV after VGCV administration [12, 18]. The first study developed and validated in an independent dataset a MAP-BE for kidney transplant recipients based on the three-point LSS 0 h/2 h/4 h [18]. The second only showed a correlation between the two-point trapezoidal AUC from 2 to 5 hours (AUC2–5 h) and the AUC0–24 h without any external validation [12]. Additionally, no BE has been reported so far for pediatric SCT, whether receiving enteral VGCV or IV GCV. The aims of the present study were to develop, validate, and make available POPPK models and BEs based on LSSs for the estimation of GCV AUC0–24 h after VGCV or IV GCV administration in pediatric SOT or SCT recipients.
2 Materials and Methods
2.1 Patient Population
For the development of the POPPK model and BEs (development dataset), we retrospectively included children from a tertiary pediatric hospital center (CHU Sainte-Justine, Montreal, QC, Canada). Children were included if: they had undergone a transplantation (SOT or SCT) between January 2007 and December 2015; they received VGCV or IV GCV for the prevention of CMV infection; and had a complete GCV PK profile (four or more PK samples) performed per standard of care. At CHU Sainte-Justine, the strategy to prevent CMV disease is a pre-emptive approach: CMV DNA in peripheral blood (CMV DNAemia) is monitored weekly during the first 100 days and then monthly until 6 months after transplantation. Antiviral therapy with IV GCV (5 mg/kg/every12 h) or enteral VGCV (10 mg/kg/every 12 h) is started whenever CMV DNAemia is detected above a significant threshold that is not standardized and depends on risk factors (CMV DNAemia value, time since transplant, CMV status). Treatment duration is based on CMV DNA clearance and risk factors. The Institutional Review Board of the CHU Sainte-Justine approved the protocol and since 2015, a clinical pharmacology database has been approved and implemented that prospectively collects data from children with available PK concentrations, as per standard of care.
2.2 Sample Collection and Analytical Method
As per the local standard of care, GCV therapeutic drug monitoring was performed after a minimum of six doses (steady state) with a AUC0–24h target of 40–60 mg h/L. Blood samples were routinely collected in EDTA microtainers (0.25 mL) at pre-dose; 0.5; 1; 1.5; 2; 3; 6; and 12 h for IV GCV, and pre-dose; 0.5; 0.75; 1; 1.5; 2; 4; 6; and 12 h for enteral VGCV. Samples were centrifuged at 3500 rpm for 10 min (2634 g) immediately after sampling, and plasma stored (− 30 °C) at the hospital clinical laboratory. Plasma concentrations were determined within 72 h of sampling, using high-performance liquid chromatography with diode array detection (Electronic Supplementary Material [ESM S1]) [19].
2.3 PK Modeling and Model Evaluation
Pharmacokinetic modeling was performed using the iterative two-stage Bayesian method with home-made software (ITSIM) [20]. Intravenous GCV and enteral VGCV were modeled separately. For both administration routes, one-compartment and two-compartment structural models with first-order elimination were investigated. For enteral VGCV, different models of absorption were investigated, including a lag time and one or two gamma distributions (because some PK profiles showed two absorption peaks). All PK profiles were considered as independents because it was not part of our objectives to explore the inter-occasion variability. A combined error model was used to describe the residual variability.
Associations between CrCL or bodyweight (WT) and PK parameters (central volume of distribution and clearance [CL]) were investigated using visual examination (scatterplots or boxplots). The covariates were included in the model using exponent allometric functions (Equations 1–3). Scaling was centralized to median WT or CrCL. Covariates were tested in the model using stepwise forward addition and backward elimination.
where CLstd and Vstd represent population estimates for a typical patient, CrCL is the creatinine clearance for the ith individual, WT is the bodyweight for the ith individual, MedCrCL and MedWT represent median CrCL and weight for our population, and x, y, and z are the estimated exponents.
The final model was selected based on a combination of a decrease of the Akaike information criterion and Bayesian information criterion and the visual examination of the individual and the goodness-of-fit plots: individual-predicted and population-predicted vs observed concentrations, weighted residuals vs time and weighted residuals vs individual-predicted concentrations. Internal evaluation of the model was performed using prediction-corrected visual predictive checks [21]. A total of 1000 replicates were simulated using the final model to generate expected concentrations and the 90% prediction intervals. The observed data and its 90% intervals were overlaid onto the prediction intervals and compared visually.
2.4 Correlation Between AUC0–12/24 h and C 0
The relationship between AUC0–12/24 h estimates and observed C0 (within 30 min before the administration) was investigated for IV GCV and enteral VGCV using linear regression and the coefficients of determination were calculated.
2.5 BE and LSS
Based on the POPPK models developed, the best combinations of three or two sampling times applicable in clinical practice were selected by calculating the mean relative prediction error (relative bias), standard deviation (SD) and range of bias, imprecision (relative root mean square error [RMSE]), and the number of AUC0–24 h estimates out of ± 10%, 15%, and 20% intervals with respect to the trapezoidal AUC0–24 h. The selection of the sampling times was made with a maximum deviation of ± 0.25 h (i.e., 15 min) from the selected time. Using the surrogate efficacy target range AUC0–24 h = 40–60 mg h/L, the MAP-BE AUC0–24 h estimates (AUCBE,0–24 h) were graphically compared to the trapezoidal AUC0–24 h (AUCtrap,0–24 h) and the number of exposure misclassifications potentially leading to inaccurate dose adjustment was calculated. For this purpose, GCV AUC0–24 h < 40 mg h/L was classified as underexposure and GCV AUC0–24 h > 60 mg h/L as overexposure.
2.6 Validation
The performances of the selected LSSs (2 and 3 points) were evaluated in three independent datasets and the relative mean bias, SD and range, RMSE, and number of AUC0–24 h out of ± 10, 15, and 20% the trapezoidal AUC0–24 h were calculated. The AUCBE,0–24 h were also graphically compared to the reference AUCtrap,0–24 h to estimate the number of inaccurate classifications.
2.6.1 Validation Dataset 1
The first validation dataset included pediatric recipients of SOT and SCT between August 2015 and February 2019 (subsequent patients) at CHU Sainte-Justine, who received GCV (5 mg/kg/every 12 h) or VGCV (10 mg/kg/every 12 h) as preemptive therapy, had available GCV concentrations per standard of care and were enrolled in the CHU Sainte-Justine Clinical Pharmacology Database. Blood samples were drawn and GCV concentrations determined in a similar manner to the development dataset.
2.6.2 Validation Dataset 2
The second validation dataset included children who underwent a SOT or a SCT between February and July 2019 (subsequent patients) at CHU Sainte-Justine with similar conditions to the previous validation dataset.
2.6.3 Validation Dataset 3
The third external validation dataset included child recipients of a kidney or a liver transplant and enrolled in a study previously published [10]. All the patients received 2 days of IV GCV followed by 2 days of VGCV. The GCV and VGCV daily doses of 260 mg/m2 and 520 mg/m2, respectively, were based on adult recommendations with dose adjustment for estimated CrCL by Schwartz. Pharmacokinetic plasma samples were collected on day 2 of GCV (pre-dose and 1; 2–3; 5–7; and 10–12 h) and on days 1 and 2 of VGCV (pre-dose; 0.25–0.75; 1–3, 5–7; and 10–12 h). Plasma concentrations were determined using another validated, high-performance liquid chromatography-tandem mass spectrometry method [22, 23].
3 Results
3.1 Patient Population
A total of 27 children treated with IV GCV (209 PK samples, 31 PK profiles, median of seven samples per profile) and 32 children treated with enteral VGCV (293 PK samples, 43 PK profiles, median of seven samples per profile) were included in the development dataset. The patient characteristics of the development and validation datasets are described in Table 1. No significant difference was observed between the development and validation datasets, except that children were younger in the second validation set and that there was no SCT patients in the third validation dataset.
3.2 PK Modeling and Model Evaluation
For both drugs, a two-compartment model with linear elimination best described the pharmacokinetics of ganciclovir. A double gamma distribution best described the absorption phase of VGCV for the enteral model. The parameters estimated by the model were: the apparent central volume of distribution, the apparent clearance, the intercompartmental transfer constants (k12, k21), and the absorption parameters: shape and scale of the two gamma distributions (a1, b1 and a2, b2 for the first and second, respectively) and the fraction of VGCV absorbed during the first gamma distribution (r) [20]. A combined analytical error model was used to describe the residual error (error = 0.02 + 0.05 × C for VGCV, error = 0.1 + 0.05 × C for GCV). The estimates of the individual PK parameters are presented in Table 2.
No association between covariates (WT, CrCL) and PK parameters was observed (no decrease in the Akaike information criterion and Bayesian information criterion; Table S2 of the ESM). The final models without covariate were therefore retained.
The diagnostic plots of the models are shown in Fig. 1. The prediction-corrected visual predictive checks show that the average prediction of the simulated data matched the observed concentration–time profiles and that the variability was reasonably estimated (Fig. 2).

Diagnostic plots of enteral (1) and intravenous models (2): A individual-predicted concentrations and B population-predicted concentrations vs observed concentrations; C weighted residuals error vs time after last dose; and D vs individual-predicted concentrations

Prediction-corrected visual predictive checks of the enteral (A) and intravenous (B) models. Percentiles (5%, 50%, and 95%) of observations (gray dashed lines) and predictions (gray solid lines) are overlaid with the observations (symbols). GCV ganciclovir
3.3 Correlation Between AUC0–12/24 h and C 0
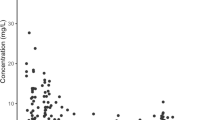
The relationships between BEs of GCV AUC0–12/24 h and observed C0 after enteral VGCV or IV GCV are shown in Fig. 3. Thirty-four and 30 GCV profiles after VGCV and GCV, respectively included C0 values. The coefficients of determination were 0.36 and 0.27 for VGCV and GCV, respectively.

Association between trough concentration and area under the concentration–time curve from 0 to 12/24 h (AUC0–12/24 h) among pediatric solid organ transplant and stem cell transplant recipients receiving enteral valganciclovir (A) or intravenous ganciclovir (B). R2 coefficient of determination
3.4 BE and LSS
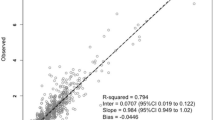
For both models, the mean relative bias, SD and range of bias, relative RMSE, and numbers of AUCBE,0–24 h estimates outside ± 10, 15, and 20% intervals of AUCtrap,0–24 h are summarized in Table 3 for each combination of two or three sampling times. The scatterplots of the AUCBE,0–24 h estimates vs the AUCtrap,0–24 h are shown in Fig. 4.

Scatterplots of the Bayesian area under the concentration–time curve from 0 to 24 h (AUC0–24 h) estimates vs trapezoidal AUC0–24 h for the combinations of two or three sampling times with the enteral (A) and intravenous (B) models. Gray dashed lines correspond to the AUC0–24 h threshold; gray shades correspond to the exposure area: underexposure: AUC0–24 h < 40 mg h/L, within the efficacy target: AUC0–24 h = 40–60 mg h/L, overexposure: AUC0–24 h >60 mg h/L. LSS limited sampling strategy
For enteral VGCV, the best LSS was LSS1,2,6, leading to a mean/SD [range] bias of 0.7%/13.5% [− 40 to 24%] and a RMSE of 13.3%. The Bland–Altman plots of the difference between AUCBE,0–24 h and AUCtrap,0–24 h vs the average of AUCBE,0–24 h and AUCtrap,0–24 h are shown in Fig. 5A. Three AUCBE,0–24 h estimates were outside the ± 20% interval. Among them, only one was misclassified as within the efficacy target range (AUCBE,0–24 h = 46.2 mg h/L) while AUCtrap,0–24 h would have classified it as “underexposure” (AUCtrap,0–24 h = 33.0 mg h/L) (Fig. 4A). The LSS1,6 showed a mean/SD bias of − 0.1%/12.1% [− 30% to 15%] and a RMSE of 12.1%. Two AUCBE,0–24 h estimates were outside the ± 20% interval, one of them being misclassified (AUCBE,0–24 h = 46.2 mg h/L vs AUCtrap,0–24 h = 33.0 mg h/L). Additionally, two other patients, while having bias below the ± 20% interval, would have had misclassified exposure (AUCBE,0–24 h = 37.9 mg h/L vs AUCtrap,0–24 h = 44.5 mg h/L and AUCBE,0–24 h = 39.7 mg h/L vs AUCtrap,0–24 h = 44.5 mg h/L).

Bland–Altman plots of the difference between Bayesian area under the concentration–time curve from 0 to 24 h (AUCBE,0–24 h) estimates and AUCtrap,0–24 h vs the average of AUCBE,0–24 h and AUCtrap,0–24 h for the selected three-point limited sampling strategy with the enteral (A) and intravenous (B) models. BE Bayesian estimator, trap trapezoidal
For IV GCV, the best LSS was LSS1,2,3 with a mean/SD bias of − 1.4%/9.5% [− 18% to 19%] and a RMSE of 9.1%. None of the AUCBE,0–24 h estimates were outside the ± 20% interval. The Bland–Altman plots of the difference between AUCBE,0–24 h and AUCtrap,0–24 h vs the average of AUCBE,0–24 h and AUCtrap,0–24 h are shown in Fig. 5B. Only one patient had a misclassified exposure (AUCBE,0–24 h = 39.3 mg h/L vs AUCtrap,0–24 h = 41.4 mg h/L) (Fig. 4B). The LSS1,3 showed a mean/SD bias of 3.5%/12.2% [− 35% to 19%] and a RMSE of 12.3%. Only one AUCBE,0–24 h estimate was outside the ± 20% interval but with a correct classification of exposure. However, one patient had inaccurate classification of exposure (AUCBE,0–24 h = 39.3 mg h/L vs AUCtrap,0–24 h = 41.4 mg h/L) while its absolute bias was less than 20%.
3.5 Validation
For the three validation datasets, the good performances were observed (Table 4). The scatterplots of the 3-point and 2-point AUCBE,0–24 h estimates vs the AUCtrap,0–24 h for each model are shown in Fig. 6. The performances of the BEs in the pooled validation sets overall and split by the type of transplantation (SOT or SCT) are presented in the ESM Table S3.

Scatterplots of the Bayesian area under the concentration–time curve from 0 to 24 h (AUC0–24 h) estimates vs trapezoidal AUC0–24 h for the selected 3-point and 2-point limited sampling strategies with the enteral (A) and intravenous (B) models in the different validation datasets. Blue dots correspond to the 3-point estimates, orange dots to the 2-point estimates; gray dashed lines correspond to the clinical decision AUC0–24 h thresholds; gray shades correspond to the different exposure areas: underexposure: AUC0–24 h < 40 mg h/L, within the efficacy target: AUC0–24 h = 40–60 mg h/L, overexposure: AUC0–24 h >60 mg h/L
As a sensitivity analysis, data from the development, validation 1 and 2 sets (validation set 3 has only a few C4h available) were pooled to compare the performances of the LSS1,2,6 chosen to the LSS1,4,6 that exhibited better performances but a lower number of patients available in the development set (Table 3). The overall performances were largely better with LSS1,2,6 (mean bias/RMSE = 2.0%/14.6%) in comparison to LSS1,4,6 (− 14.4%/30%).
The individual profiles with the best and worst performances in each dataset are shown in Fig. 7. For enteral VGCV, 7/52 (13%) GCV AUCBE,0–24 h had exposure misclassification (Fig. 6A). In detail, 4 were classified as below instead of within the efficacy target range; 1 within instead of below; 1 above instead of within; and 1 within instead of above the efficacy target range (ESM Table S4). Among the misclassified patients, assuming the GCV pharmacokinetics is linear and the AUC0–24 h targeted for dose individualization is 50 mg h/L, only two patients would have had a dose proposal resulting in AUCtrap > 60 mg h/L (AUCBE,0–24 h/AUCtrap,0–24 h = 38.6/47.7 mg h/L, leading to 50.0/61.8 mg h/L; AUCBE,0–24 h/AUCtrap,0–24 h = 39.8/50.0 mg h/L, leading to 50.0/62.8 mg h/L). Conversely, two patients would have had an exposure within the target, resulting in no dose change.

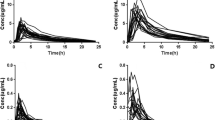
Examples of individual modeled pharmacokinetic (PK) profiles for enteral valganciclovir (best fits: 1; worst fits: 2) and intravenous ganciclovir (best fits: 3; worst fits: 4) in the development (A) and validation (B–D) datasets. Black lines and plots correspond to the observed concentrations and PK profiles; gray lines correspond to the modeled PK profiles using all available time points; blue and orange lines correspond to the modeled PK profiles using a 3-point and 2-point limited sampling strategy, respectively
For IV GCV, 5/35 GCV (14%) AUC0–24 h were misclassified, all from the third validation dataset (Fig. 6B), with three patients classified as below instead of within the efficacy target range and two patients within instead of above the range (ESM Table S4). Among the misclassified patients, two would have had a dose proposal resulting in AUCtrap,0–24 h > 60 mg h/L (AUCBE,0–24 h/AUCtrap,0–24 h = 35.0/42.0 mg h/L, leading to 50.0/61.1 mg h/L; AUCBE,0–24 h/AUCtrap,0–24 h = 35.7/43.6 mg h/L, leading to 50.0/62.8 mg h/L). Conversely, two patients would have had exposure within the target, resulting in no dose change.
4 Discussion
In this study, we developed POPPK models for enteral VGCV and IV GCV and MAP-BE based on 3-point or 2-point LSSs that accurately estimated GCV AUC0–24 h in pediatric transplant recipients. Afterwards, we thoroughly validated them in three independent external datasets. The determination of GCV AUC0–24 h is challenging in children because it requires many blood samples causing pain and stress, a prolonged hospital stay, or multiple visits. The main goal of this study was to develop MAP-BEs that can be used for routine care with LSSs including sampling times compatible with day hospital admissions, and not to perform a thorough POPPK study to characterize the sources of variability, for which a non-linear mixed-effect approach would have been preferred.
The size of the validation sets 1 and 2 were small and could have been grouped together. However, the data were available sequentially and gathering them would have led to the loss of the real design of the study. However, sensitivity analyses were performed in the pooled validation set (ESM Table S3) and the results were similar to those initially obtained.
To the best of our knowledge, only one MAP-BE based on a LSS for children has been reported, specifically for VGCV in kidney transplant recipients using three PK samples (0 h/2 h/4 h). It showed good performances in the development stage and an independent validation datasets (MPEdev = 2.6 ± 7.7%, MAPE = 5.5 ± 5.9%; MPEval = 3.1 ± 13.8%, MAPEval = 10.5 ± 9.1%) [18]. Another study assessed the correlation between single concentrations or 2-point trapezoidal AUC and AUC0–24 h and concluded that the 2-point AUC2–5 h was well correlated with the 4-point trapezoidal AUC0–24 h (coefficient of determination = 0.846). The authors developed a formula for the calculation of GCV AUC0–24 h based on trapezoidal AUC2–5 h but no validation was performed [12].
Additionally, no MAP-BE has been reported for enteral VGCV in pediatric SCT, or for IV GCV. The tools that we developed in the present work are innovative as they also allow the estimation of GCV AUC0–24 h in pediatric SCT recipients given VGCV and in either SOT or SCT pediatric recipients given IV GCV.
A two-compartment model with first-order elimination best described the GCV concentrations, consistent with previously published POPPK models in pediatric transplantation [10, 11, 22,23,24,25,26]. However, for the enteral formulation, we found that a double-gamma distribution better described the absorption of VGCV and its hydrolyzation to GCV by the intestinal esterases than a lagtime. Population PK models without covariates were retained for enteral VGCV and IV GCV as they showed the best performances in terms of likelihood (Akaike information criterion and Bayesian information criterion) and individual plots. Thus, despite renal elimination of GCV through glomerular filtration and tubular secretion and the inclusion of CrCL in several previous studies [10, 22,23,24,25,26], CrCL was not included as a covariate for GCV CL in our model. We think that covariates are useful in the case of simulation or for a priori estimation of the first dose [27]. In the case of BEs based on a LSS, the information is carried by the concentrations themselves (i.e., a patient with an altered renal function will have higher concentrations).
In any event, the VGCV and the GCV BEs were developed in patients with CrCL above 64 and 25 mL/min/1.73 m2, respectively, and should not be used for patients with lower CrCL. However, this limitation would have been the same if the CrCL had been included in the model as extrapolations require assumption of linearity that has to be evaluated. The influence on PK parameters of other covariates such as the type of transplant (SOT vs SCT) was not investigated because they were not significant in previously reported POPPK models.
The selection of the LSS times was based on clinical feasibility in addition to performance, to avoid patient hospital stay and to accurately describe GCV PK parameters. For VGCV, the LSS1,2,6 was selected even though the LSS1,4,6 showed better performances in term of relative bias and RMSE. This is because of the lower number of patients with a blood sample at 4 h than at 2 h after VGCV administration (16 vs 26 patients, respectively), leading us to select the latter. Moreover, on a theoretical viewpoint, the selection of C2h instead of C4h is more relevant to obtain information about the peak (i.e., the median time to maximum concentration is 2.18 h [1.7–3.0 h]) [8]. The selection of C6h in the VGCV LSS seems inconvenient for outpatients, but a late PK sample was required to accurately estimate GCV CL/F (which might have only been partly compensated by using CrCL as a covariate). Analysis on the pooled development and validation 1 and 2 datasets confirmed that the LSS1,2,6 was the best option. Similarly, Padullés Caldés et al. in their optimal LSS building found a higher bias and imprecision for GCV CL estimation with LSSs including only early time points (up to 5 h) as compared with those including a PK sample between 6 and 8 h [28].
While during the development of the BE phase, the sampling time selection was restricted to ± 0.25 h, during the validation phase, this cut-off would have led to only a small number of PK profiles available for validation (only five and three PK profiles in pooled datasets for VGCV and GCV, respectively). Thus, for each PK profile, the nearest available sampling time was used, with accurate determination of GCV AUC0–24 h.
The two-point LSS1,6 for VGCV showed very good performances as compared to the LSS1,2,6 in the development dataset but not in the validation datasets. Moreover, it does not decrease the length of hospital stay but has the advantage of decreasing the number of samples collected.
For GCV LSSs, the inclusion of t5h showed better performances but the low number of samples available led us to select t3h in the final LSS (12 and 16 patients, respectively). In the validation datasets, the time of the third PK sample ranged between 3.0 and 5.5 h and was associated with good performances, which confirms that BEs are flexible with respect to the actual sampling times. The two-point LSS1,3 for GCV showed comparable performances to the LSS1,2,3 in all the development and validation datasets, which is in favor of the two-point LSS1,3.
In addition to usual performance metrics, we aimed to select LSSs based on a minimum number of inaccurate exposure classifications that would have resulted in inaccurate dose adjustment in clinical practice. In the independent datasets, seven (13%) and five (14%) children would have had exposure misclassification using LSS1,2,6 for VGCV and LSS1,2,3 for IV GCV, respectively. Among them, two children for each LSS would have reached out-of-range exposure after dose adjustment. However, the surrogate efficacy target (40–60 mg h/L) is only correlated to a probability of breakthrough viremia, thus even if a GCV AUCtrap,0–24 h = 39.7 mg h/L is considering as underexposure (vs AUCtrap,0–24 h = 44.5 mg h/L, as presented above), the probability of breakthrough viremia is not very different than a GCV AUC0–24 h = 40 mg h/L. For both enteral VGCV and IV GCV, inaccurate classification of GCV exposure would only have led to an increase in the dose and increased AUCtrap,0–24 h up to 62.8 mg h/L, which is very close to the upper limit of the GCV target range. No dose adjustment based on exposure misclassification would have led to AUCtrap,0–24 h < 40 mg h/L.
The main limitation of this study is its retrospective nature. Pharmacokinetic samples were collected during routine care, resulting in potential inaccuracies regarding dosing and/or sampling times, or associated clinical data. Another limitation is the use of the trapezoidal AUC0–24 h as the reference to evaluate BEs, particularly in patients with less than seven PK samples (median value for both drugs). Indeed, the number of PK samples was highly variable in the development dataset (four to ten samples per child). To test the robustness of our conclusions, the performances of the LSSs were calculated using as references the BEs obtained with all the individual time points available and showed better results than with the trapezoidal AUCs as references (ESM Table S5). Both routes were modeled separately while IV information can be of added value to model oral data. However, ITSIM used for PK modeling was not able to model simultaneously both dosing routes. Last, the analytical method of GCV determination was different between cohorts and could have resulted in inaccuracy in GCV AUC0–24 h as no cross-validation was performed. However, the goal of the third validation was to evaluate the BEs in a very different population to propose a model that can be used in routine care, which implies a wide range of different analytical methods.
5 Conclusions
In this study, we developed MAP-BEs based on three-point LSSs: 1 h/2 h/6 h and 1 h/3 h for VGCV and GCV, respectively, for both SOT and SCT pediatric recipients. The performances obtained in three independent datasets show that they accurately estimate GCV AUC0–24 h and can be used clinically for the therapeutic drug monitoring of these two drugs. They are now available to the medical community at https://pharmaco.chu-limoges.fr/.
References
Kotton CN, Kumar D, Caliendo AM, Huprikar S, Chou S, Danziger-Isakov L, et al. The Third International Consensus Guidelines on the management of cytomegalovirus in solid-organ transplantation. Transplantation. 2018;102:900–31.
Razonable RR, Humar A. Cytomegalovirus in solid organ transplant recipients: guidelines of the American Society of Transplantation Infectious Disease Community of Practice. Clin Transplant. 2019;33(9):e13512.
Boeckh M, Ljungman P. How we treat cytomegalovirus in hematopoietic cell transplant recipients. Blood. 2009;113(23):5711–9.
Giménez E, Solano C, Azanza JR, Amat P, Navarro D. Monitoring of trough plasma ganciclovir levels and peripheral blood cytomegalovirus (CMV)-specific CD8+ T cells to predict CMV DNAemia clearance in preemptively treated allogeneic stem cell transplant recipients. Antimicrob Agents Chemother. 2014;58:5602–5.
Gagermeier JP, Rusinak JD, Lurain NS, Alex CG, Dilling DF, Wigfield CH, et al. Subtherapeutic ganciclovir (GCV) levels and GCV-resistant cytomegalovirus in lung transplant recipients. Transpl Infect Dis. 2014;16:941–50.
Ritchie BM, Barreto JN, Barreto EF, Crow SA, Dierkhising RA, Jannetto PJ, et al. Relationship of ganciclovir therapeutic drug monitoring with clinical efficacy and patient safety. Antimicrob Agents Chemother. 2019;63:10.
Wiltshire H, Paya CV, Pescovitz MD, Humar A, Dominguez E, Washburn K, et al. Pharmacodynamics of oral ganciclovir and valganciclovir in solid organ transplant recipients. Transplantation. 2005;79:1477–83.
Genentech, Inc. Valcyte monography. 2010. https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021304s008,022257s003lbl.pdf. Accessed 5 May 2021.
Padullés A, Colom H, Bestard O, Melilli E, Sabé N, Rigo R, et al. Contribution of population pharmacokinetics to dose optimization of ganciclovir-valganciclovir in solid-organ transplant patients. Antimicrob Agents Chemother. 2016;60:1992–2002.
Åsberg A, Bjerre A, Neely M. New algorithm for valganciclovir dosing in pediatric solid organ transplant recipients. Pediatr Transplant. 2014;18:103–11.
Facchin A, Elie V, Benyoub N, Magreault S, Maisin A, Storme T, et al. Population pharmacokinetics of ganciclovir after valganciclovir treatment in children with renal transplant. Antimicrob Agents Chemother. 2019;63:e01192-e1219.
Villeneuve D, Brothers A, Harvey E, Kemna M, Law Y, Nemeth T, et al. Valganciclovir dosing using area under the curve calculations in pediatric solid organ transplant recipients. Pediatr Transplant. 2013;17:80–5.
Mosteller R. Simplified calculation of body-surface area. N Engl J Med. 1987;317:1098.
Schwartz GJ, Brion LP, Spitzer A. The use of plasma creatinine concentration for estimating glomerular filtration rate in infants, children, and adolescents. Pediatr Clin N Am. 1987;34:571–90.
Peled O, Berkovitch M, Rom E, Bilavsky E, Bernfeld Y, Dorfman L, et al. Valganciclovir dosing for cytomegalovirus prophylaxis in pediatric solid-organ transplant recipients: a prospective pharmacokinetic study. Pediatr Infect Dis J. 2017;36:745–50.
Stockmann C, Sherwin CMT, Knackstedt ED, Hersh AL, Pavia AT, Spigarelli MG. Therapeutic drug monitoring of ganciclovir treatment for cytomegalovirus infections among immunocompromised children. J Pediatric Infect Dis Soc. 2016;5:231–2.
Stockmann C, Roberts JK, Knackstedt ED, Spigarelli MG, Sherwin CM. Clinical pharmacokinetics and pharmacodynamics of ganciclovir and valganciclovir in children with cytomegalovirus infection. Expert Opin Drug Metab Toxicol. 2015;11:205–19.
Zhao W, Fakhoury M, Fila M, Baudouin V, Deschênes G, Jacqz-Aigrain E. Individualization of valganciclovir prophylaxis for cytomegalovirus infection in pediatric kidney transplant patients. Ther Drug Monit. 2012;34:326–30.
Kasiari M, Gikas E, Georgakakou S, Kazanis M, Panderi I. Selective and rapid liquid chromatography/negative-ion electrospray ionization mass spectrometry method for the quantification of valacyclovir and its metabolite in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;864:78–86.
Debord J, Risco E, Harel M, Le Meur Y, Büchler M, Lachâtre G, et al. Application of a gamma model of absorption to oral cyclosporin. Clin Pharmacokinet. 2001;40:375–82.
Keizer R. Vpc: create visual predictive checks. 2020. https://CRAN.R-project.org/package=vpc. Accessed 15 Jul 2020.
Pescovitz MD, Ettenger RB, Strife CF, Sherbotie JR, Thomas SE, McDiarmid S, et al. Pharmacokinetics of oral valganciclovir solution and intravenous ganciclovir in pediatric renal and liver transplant recipients. Transpl Infect Dis. 2010;12:195–203.
Vaudry W, Ettenger R, Jara P, Varela-Fascinetto G, Bouw MR, Ives J, et al. Valganciclovir dosing according to body surface area and renal function in pediatric solid organ transplant recipients. Am J Transplant. 2009;9:636–43.
Zhao W, Baudouin V, Zhang D, Deschênes G, Guellec CL, Jacqz-Aigrain E. Population pharmacokinetics of ganciclovir following administration of valganciclovir in paediatric renal transplant patients. Clin Pharmacokinet. 2009;48:321–8.
Vezina HE, Brundage RC, Balfour HH. Population pharmacokinetics of valganciclovir prophylaxis in paediatric and adult solid organ transplant recipients: population pharmacokinetics of valganciclovir prophylaxis after transplantation. Br J Clin Pharmacol. 2014;78:343–52.
Bradley D, Moreira S, Subramoney V, Chin C, Ives J, Wang K. Pharmacokinetics and safety of valganciclovir in pediatric heart transplant recipients 4 months of age and younger. Pediatr Infect Dis J. 2016;35:1324–8.
Sheiner L, Beal S, Rosenberg B, Marathe V. Forecasting individual pharmacokinetics. Clin Pharmacol Ther. 1979;26:294–305.
Padullés Caldés A, Colom H, Caldes A, Cerezo G, Torras J, Grinyó JM, et al. Optimal sparse sampling for estimating ganciclovir/valganciclovir AUC in solid organ transplant patients using NONMEN. Ther Drug Monit. 2014;36:371–7.
Acknowledgements
We thank Anémone Faivre-D’Arcier, nurse of the CHU Sainte-Justine Clinical Pharmacology Unit for the coordination of the Clinical Pharmacology Database.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by the internal funds from the CHU Sainte-Justine Research Center.
Conflicts of Interest/Competing Interests
Julie Autmizguine received salary support from the FRQS (Fonds de Recherche Santé Québec), and consults for Astellas Pharma Inc. Bénédicte Franck, Anders Åsberg, Yves Théorêt, Pierre Marquet, Philippe Ovetchkine, and Jean-Baptiste Woillard have no conflicts of interest that are directly relevant to the content of this article.
Ethics Approval
The Institutional Review Board of the CHU Sainte-Justine approved the protocol.
Availability of Data and Material
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code Availability
Code is available upon request (Rfile).
Authors’ Contributions
BF and JBW conceived, designed, and performed the analysis. BF, JA, and JBW wrote the paper. JA, AÅ, and PO conceived and collected the data. YT performed the analyses of samples. JA, AÅ, PM, and PO contributed to the data analysis and to manuscript writing.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Franck, B., Autmizguine, J., Åsberg, A. et al. Thoroughly Validated Bayesian Estimator and Limited Sampling Strategy for Dose Individualization of Ganciclovir and Valganciclovir in Pediatric Transplant Recipients. Clin Pharmacokinet 60, 1449–1462 (2021). https://doi.org/10.1007/s40262-021-01034-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01034-w