
Abstract
We aimed to review the pharmacokinetics (PK) of intravenous busulfan in paediatric patients, identify covariate factors influencing exposure, investigate evidence of changes in PK behaviour over time, and correlate exposure with efficacy and toxicity outcomes. A literature review was undertaken of original research published between 2007 and 2019, investigating the PK and pharmacodynamics (PD) of intravenous busulfan in patients ≤ 18 years of age. The review identified 41 publications characterising the PK, and 45 publications describing the PD, of busulfan. Median typical clearance (CL) was 0.22 L/h/kg and median typical volume of distribution was 0.69 L/kg. Patient weight, age, glutathione-S-transferase A1 (GSTA1) genotype and busulfan dosing day/time were the most commonly identified factors affecting CL. Of nine studies investigating changes in CL, seven reported reduced CL over the 4-day course of treatment. Exposure monitoring methods and therapeutic targets were heterogeneous across studies. Relationships between busulfan exposure and patient outcomes were observed in five studies. One study observed a cumulative area under the concentration-time curve over all days of treatment of between 78 and 101 mg/L·h, and two studies observed an average concentration at first dose of < 600 ng/mL improved overall survival, transplant-related mortality, or relapse. One study observed increased sinusoidal obstructive syndrome with maximum busulfan concentration > 1.88 ng/mL. Patient weight, age and GSTA1 genotype are important covariates to consider when individualising busulfan therapy. Reduced busulfan CL over time may need to be accounted for, particularly in patients not receiving phenytoin co-therapy. Standardised monitoring of busulfan exposure over the entire course of treatment and further investigation of the role of busulfan metabolites and pharmacogenomics is warranted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A large proportion of studies have estimated cumulative busulfan exposure across all days of treatment from drug measurements taken on day 1 only, assuming linear pharmacokinetic (PK) behaviour. Clearance (CL) of busulfan decreases over the course of treatment, therefore exposure targets in the current literature may not accurately reflect actual exposure attained in patients. Harmonisation of exposure monitoring practices and further studies that either measure busulfan exposure over the entire course of treatment or validation of Bayesian forecasting programmes to estimate cumulative exposure from less intensive sampling are required to establish appropriate exposure–outcome relationships. |
It is imperative that clinicians are aware of the time-dependent PK (decreased CL over the course of treatment) exhibited by busulfan and the variability of exposure observed between occasions, and consider this when making recommendations for dose adjustments for patients. Exposure monitoring should be repeated following any dose change. Reducing future doses should be considered if exposure is close to the upper limit of the therapeutic target. |
Pharmacogenomic differences and busulfan metabolites may both play key roles in influencing the PK and pharmacodynamics (PD) of busulfan, and may affect patient outcomes, in particular the incidence of sinusoidal obstructive syndrome. Both could be further investigated through population PK/PD modelling. |
1 Introduction
Busulfan is a bifunctional alkylating agent commonly used in haematopoietic stem cell transplant (HSCT) conditioning regimens. Paediatric patients with both malignant and non-malignant conditions can benefit from HSCT where the recipient’s blood and marrow are replaced with donor haematopoietic stem cells [1]. Intravenous administration of busulfan is preferred over oral administration in children due to improved bioavailability and reduced pharmacokinetic (PK) variability. While product information in Europe and the US [2, 3] recommends that intravenous busulfan is administered 6-hourly over 4 consecutive days in paediatric patients, once-daily dosing is increasingly being used in practice [4,5,6,7,8,9].
The PK of busulfan are complex (Fig. 1) and are not completely understood. Busulfan binds reversibly and irreversibly to blood cells and plasma proteins. Unbound (free) busulfan diffuses passively into body cells [10] where it is hydrolysed [11] to produce highly reactive, positively charged carbonium ions that alkylate and damage DNA. Busulfan may be metabolised through conjugation with glutathione, primarily via glutathione-S-transferase A1-1 (GSTA1-1) [12]. This conjugate may then be converted via cystathionine gammalyase (CTH), to tetrahydrothiophene (THT) and γ-glutamyldehydroalanylglycine (EdAG). EdAG condenses with glutathione to form a non-reducible analogue of glutathione disulfide (GSG), essentially trapping a glutathione molecule since it cannot be recycled [13]. EdAG may contribute to the toxicity of busulfan as it competes with glutathione for binding with GST enzymes, contributes to non-reducible glutathionylation of proteins [14,15,16] and decreases glutathione levels within the cell [13]. Busulfan decreases hepatic glutathione by 60% in vivo and by 95% in a cell that is already depleted in glutathione, resulting in increased cellular toxicity [17]. Another busulfan sulfonium ion conjugate is metabolised through the mercaptopurate pathway and converted to THT via CTH [11]. THT is thought to be actively transported into liver cells via an ATP-binding cassette (ABC) transporter. In the liver, THT is largely oxidised by flavin-containing monooxygenase-3 (FMO3) or through multiple cytochrome P450 (CYP) pathways (e.g. CYP2C9, CYP2C19 and possibly CYP39A1) [12]. Approximately 30% of the busulfan dose is excreted into the urine within 48 h, mainly as metabolites, < 2% is excreted in the urine unchanged, and negligible amounts are recovered in faeces [18]. Busulfan rapidly distributes into the cerebrospinal fluid and similar exposure is achieved at this site as in plasma [19]. A number of coadministered medications may interfere with the metabolism of busulfan. Agents of primary interest include acetaminophen, phenytoin, metronidazole and azole antifungals, as they are commonly used during HSCT and are known substrates, inducers and/or inhibitors of enzymes involved in busulfan metabolism. Some suggest that busulfan clearance (CL) may decrease over a 4-day treatment course [20]. Glutathione depletion may explain the time-dependent decrease in busulfan CL, particularly in adults > 40 years of age; in children a decrease in clearance over time of 8–15% has been observed [21, 22], however the impact of glutathione depletion has not yet been explored [23].

Diagram illustrating the distribution, metabolism and elimination of intravenous busulfan. CTH cystathionine gamma lyase, GST glutathione-S-transferase, DPEP dipeptidase, GGT gamma-glutamyl transferase, NAT N-acetyltransferase, EdAG γ-glutamyldehydroalanylglycine, GSH glutathione, GSG EdAG gluthathione conjugate, FMO3 flavin-containing monooxygenase-3, CYP cytochrome P450, ABC ATP-binding cassette transporter, THT tetrahydrothiophene, --- oval (grey) indicates cytotoxic moiety, ··· rectangle indicates metabolite, ·-·-rectangle indicates co-factor, black arrow indicates transport into/out of cell, grey curved arrow indicates step in the metabolism process
Busulfan is a highly toxic drug with a narrow therapeutic window [24,25,26,27,28]. Exposure monitoring is routinely performed during busulfan treatment to optimise drug exposure and improve treatment outcomes [2]. In a prospective, randomised study, PK-guided dosing of intravenous busulfan was shown to be superior to fixed-dose body surface area (BSA) dosing for adults with acute myeloid leukaemia and myelodysplastic syndrome, resulting in a reduction in relapse (38% vs. 56%) and transplant-related mortality (TRM; 24% vs. 39%), and an overall hazard ratio (HR) of 0.64 (95% confidence interval [CI] 0.35–0.94) [29]. Busulfan exposure is estimated using various indices, including the area under the concentration-time curve (AUC) over the dosing interval (AUCdose interval), the AUC over a 24 h period (AUC24), cumulative exposure following all doses (AUCcum) and average concentration at steady-state (Cssavg) either over the course of treatment or for a particular dose (see Table 1 for comparison values). AUCdose interval, AUCcum and Cssavg targets were originally proposed for 6-hourly oral or intravenous busulfan administration and have been extrapolated for use with once-daily intravenous administration [21, 30, 31].
Model-informed approaches, including the population PK/pharmacodynamic (PD) approach, provide a rationale to precise dosing for an individual and are increasingly being integrated into clinical decision-making tools [32]. Population models can assist with initial dose selection and can be applied in a Bayesian forecasting programme for subsequent dose adjustment [32]. The optimal therapeutic target of busulfan may depend on multiple factors, such as disease status, relapse risk, concomitant chemotherapeutic agents and the extent of the intended bone marrow suppression. There is a lack of consensus on how best to perform exposure monitoring to improve outcomes and reduce toxicity, therefore a review of the literature would be helpful.
Thus this review aims to (1) summarise the PK of intravenous busulfan in paediatric patients; (2) identify important covariate factors that influence busulfan exposure; (3) report evidence of time-dependent PK behaviour; and (4) summarise the PD of intravenous busulfan in paediatric patients by examining the relationship between exposure monitoring targets and efficacy and toxicity outcomes.
2 Methods
2.1 Structured Literature Review
A literature search was conducted in April 2020 using the MEDLINE, PUBMED, EMBASE and OVID databases based on the following search terms: ‘busulfan’ OR ‘busulphan’ [AND ‘paediatric OR pediatric’], [AND ‘pharmacodynamic’], [AND ‘Paediatric OR Pediatric’ AND ‘pharmacokinetics’], [AND ‘paediatric OR pediatric’, AND/OR ‘outcomes’, AND/OR ‘survival’, AND/OR ‘toxicity’, AND/OR ‘target’, AND/OR ‘exposure’, AND/OR ‘population pharmacokinetic model’, AND/OR ‘metabolism’]. The search was restricted to studies described in English and published since the last literature review in this area in 2007 [33]. Reference lists were reviewed to retrieve additional articles. Studies were included in the review if they fulfilled the following criteria: (1) > 50% of patients received intravenous busulfan, with a minimum of five patients involved; and (2) more than 50% of patients were ≤ 18 years of age, and (3) PK parameters and/or exposure monitoring target range with outcome data were adequately described. After removing duplicates, the titles and abstracts of articles were screened, and full-text versions of relevant articles were subsequently retrieved and reviewed to confirm eligibility.
2.2 Data Extraction
Information on study design, participant demographics, busulfan dosing regimens, PK sampling schedules, PK parameter estimates, and exposure targets aimed for and achieved, as well as patient outcomes associated with efficacy and toxicity, were extracted. Typical values for busulfan PK parameters, including CL and volume of distribution (Vd), as well as intraindividual variability (IIV) and interoccasion variability (IOV) associated with these parameters and any change in PK parameters over the course of treatment, were noted. Busulfan exposure targets were recorded, including Cssavg, AUCdose interval, AUC24 or AUCcum. Patient outcomes recorded included overall survival (OS), event-free survival (EFS), TRM, relapse, sinusoidal obstructive syndrome (SOS), acute graft-versus-host disease (aGVHD), seizures and lung toxicity. The definition of each outcome as provided in the individual study was carried forward. Data on the HR, odds ratio (OR) and relative risk (RR) associated with outcomes were also collected as available. Population PK models were examined for method of validation, predictive value and the final equation used for CL, and these were tabulated.
2.3 Analysis
PK and PD data were extracted from studies and tabulated. Descriptive statistics were used to summarise information on study features. Reported CL values were converted to L/h/kg and Vd values were converted to L/kg to aid comparison. Reported PK exposure targets were converted to AUCcum if reported as Cssavg, AUC24 or AUCdose interval using Eqs. 1, 2, or 3, respectively, and converted to mg/L·h for comparison; those studies reporting AUCcum required no conversion. Relationships between the exposure target, actual exposure attained, and patient outcomes were examined together with patient demographic and clinical features.
3 Results
3.1 Studies Reviewed
Forty-one publications were identified characterising the PK, and 45 publications characterising the PD, of busulfan. Electronic supplementary Fig. 1s describes the steps in study selection.
3.2 Pharmacokinetics (PK) of Intravenous Busulfan
The dosing protocol and study design associated with studies examining the PK of busulfan is provided in electronic supplementary Table 1s. Twenty-four studies (59%) used retrospective data, 12 studies (29%) used prospective data, 2 studies (5%) used both types of data, and the remaining 3 studies (7%) did not report their data source. Twenty-two studies (54%) were based on four-times-daily dosing of busulfan, eight studies (19.5%) were based on once-daily dosing, three studies (7%) were based on twice-daily dosing and the remaining studies (19.5%) involved a mixture of dosing regimens. Participant numbers varied from 5 to 1610 patients, with a median of 52 per study. Median patient age was 5.6 years and medians from studies ranged from 0.48 to 13 years. Two studies focused on young children and had a mean cohort age < 1 year [22, 34]. The majority of studies (57%) combined patients with and without malignant conditions. Benzodiazepines, phenytoin or fosphenytoin, levetiracetam and sodium valproate were used for seizure prophylaxis in 16, 13, 4 and 1 studies, respectively; 17 (41%) studies did not report the agent used, and some studies were included several times as they reported the use of a mixture of agents.
Busulfan dose was based on patient age in 13 (32%) studies, patient body size (either weight descriptor or BSA) in 10 (24%) studies, a mixture of both in 5 (12%) studies, a mixture of test dose or body size in 2 (5%) studies, a standard dose for all patients in 3 (8%) studies, according to the results of a test dose in 4 (9.5%) studies, and was not recorded in 4 (9.5%) studies. Busulfan dose was calculated based on patient weight (mg/kg) in 27 (66%) studies, BSA (mg/m2) in 5 (12%) studies, a mixture of both in 5 (12%) studies, and was not recorded in 4 (10%) studies. In terms of exposure monitoring 17 studies (41%), included samples that were mainly (>80% of patients) taken following the first dose only. Thirty-six studies (88%) reported the exposure target used for monitoring. Of these studies, AUCdose interval was reported in 18 (50%) studies, AUCcum was reported in 3 (8%) studies, Cssavg was reported in 11 (31%) studies, and a combination of AUCdose interval and AUCcum was reported in 4 (11%) studies. The majority of studies targeted exposure within a predefined range, however 7 studies [6, 9, 21, 34,35,36,37] utilised target concentration intervention and aimed for a single specific AUCdose interval or AUCcum value. Following conversion to AUC24 (electronic supplementary Table 1s), the median minimum AUC24 target reported across all the PK studies was 15 mg/L·h (range 12.5–22.5) and the median maximum AUC24 target was 21.6 mg/L·h (range 15–37). When converted to an AUCcum value, reported busulfan exposure targets ranged from 57.6 to 148 mg/L·h over a 4-day course (excluding reduced intensity conditioning regimens) [electronic supplementary Table 1s].
3.2.1 PK Parameters
To estimate PK parameters, a population modelling approach was applied in 21 studies (51%), 11 studies (27.5%) used a non-compartmental approach, 8 studies (19%) used a combination of approaches, and 1 study (2.5%) did not report the approach used. Of those using a population modelling approach, 16 studies (76%) characterised busulfan disposition using a one-compartmental model, and 5 studies (24%) used a two-compartment model. Seven population PK models were externally validated (Table 2) [20, 21, 38,39,40,41,42] and all were considered to have adequate predictive value. Key PK parameter values for each study are summarised in Table 2. Median typical CL reported using a population modelling approach was 0.22 L/h/kg (interquartile range [IQR] 0.20–0.24). Estimated IIV on CL ranged from 13.3 to 28% (estimated in 18 studies) and IOV on CL ranged from 3.9 to 16% (estimated in 14 studies). The median typical Vd when using a one-compartment model was 0.69 L/kg (IQR 0.64–0.71). Based on a two-compartment model, median typical Vd of the central compartment (V1) was 0.52 L/kg (range 0.22–0.73) and of the peripheral compartment (V2) was 0.33 L/kg (range 0.11–0.54). One study, that solely included young children [22] reported a median Vd of 0.8 L/kg and another observed a median Vd of 0.77 L/kg in children aged < 1 year [43]. Estimated IIV on Vd ranged from 7 to 65% (estimated in 18 studies) and IOV on Vd ranged from 6.1 to 26.3% (estimated in 5 studies).
3.2.2 Covariates that Affect the PK of Busulfan
Patient body weight, age, GSTA1 genotype and busulfan dosing day/time were most commonly found to have a significant influence on busulfan CL across multiple studies (Table 2, Fig. 2a). Other covariates identified as having a significant effect in at least one study were patient ideal body weight (IBW), postmenstrual age (PMA), fat-free mass (FFM), BSA, liver function tests (LFTs), and prior administration of fludarabine. Vd was commonly described allometrically in terms of patient body weight, however some studies used other body size descriptors, including BSA, IBW and FFM (Fig. 2b). Vd was reported to be larger in children < 1 year of age (0.77 ± 0.24 vs. 0.64 ± 0.11 L/kg, p = 0.04) and children < 4 years of age (0.73 ± 0.18 vs. 0.64 ± 0.11 L/kg, p = 0.001) compared with children older than 4 years of age in one study [43].

Covariate effects tested as having an influence on a busulfan CL (left hand panel) and b busulfan Vd (right hand panel) in population pharmacokinetic models. Covariates were either not included (dark shade) or included (light shade) in the final model, with the number of studies associated with each outcome shown in the figure. BSA body surface area, CrCl creatinine clearance, GST glutathione-S-transferase (A1, T1, M1, P1 variants)
Patient size, particularly actual body weight, was the most common covariate identified as influencing the CL of busulfan. Eleven studies applied allometric scaling for weight on CL using varying exponents. McCune et al. suggested that the biologically effective body size determining CL is proportional to FFM plus 51% of fat mass, whereas for Vd it is proportional to FFM plus 20% of fat mass [38]. A maturation function to describe changes in drug CL with age was applied in several studies [8, 20, 22, 38]. The function used and the estimated patient age at which busulfan metabolism reached adult levels varied across studies. Long-Boyle et al. reported a bell shape trend in CL, with it increasing up to the age of 12 years then decreasing to adult levels [20]. McCune et al. suggested that busulfan CL reaches 50% of adult levels at 46 weeks PMA and that busulfan CL, scaled to normal fat mass, is 95% of adult levels at 2.5 years postnatal age [38]. Savic et al. used an age-dependent nonlinear function to describe the maturation of CL and suggested that CL increases 1.7-fold between 6 weeks to 2 years of life [22].
One study described a decrease in busulfan CL with increasing alanine-S-transferase (AST) values [9], and another increased exposure on the first day of treatment when acylcarnitine, phenylacetylglutamine (both signs of liver damage) or deferoxamine were present in urine [44]; however, a further seven studies investigating varied liver function biomarkers found no significant impact [20, 21, 31, 40, 45,46,47]. Three studies reported that elevated ferritin concentrations (a sign of liver dysfunction) had no significant impact on busulfan CL [9, 40, 45], while another small non-population PK study reported that patients with ferritin concentrations ≥ 1000 ng/mL prior to HSCT had reduced busulfan CL compared with patients with lower ferritin levels (0.334 L/kg/h and 0.432 L/kg/h, respectively) [6].
Nine studies reported that polymorphic expression of metabolic enzymes influenced busulfan CL (Table 2) [8, 45, 48,49,50,51,52,53,54]; enzymes explored include GSTA1, GSTM1, GSTT1, GSTP1, CYP39A1, ABCB4, CYP2C219, SLC7A8, SLC22A4. Of four population PK models investigating glutathione-S-transferase A1 (GSTA1) genotype as a covariate, all but one [37] reported reduced busulfan CL in patients expressing the GSTA1*B allele compared with the wild-type genotype GSTA1*A/*A [8, 45, 48]. The reported reduction in CL in patients with the GSTA1*B allele versus wild-type genotype ranged from 10% to 30% across these studies. One study identified four GSTA1 diplotype groups that significantly affected CL [49]. Nava et al. reported that GSTA1 rapid metabolisers had a 7% increased CL and poor metabolisers a 12% reduced CL compared with normal diplotypes [8]. Ten Brink et al. reported that both GSTA1 and CYP39A1 genotypes were associated with busulfan CL and both haplotypes together could account for up to 17% of the variability in this parameter [50]. In this study, patients who were heterozygous for GSTA1*A/*B had an 8% lower busulfan CL compared with wild-type GSTA1 patients, and homozygous *B/*B patients had a 26% lower CL [50], while patients who were carriers of one of the variant CYP39A1*TC alleles had a 13% lower CL and homozygous patients had a 17% lower CL compared with CYP39A1*WT/*WT patients [50]. The effect of GSTA1 haplotype on CL may be dependent on age, with the GSTA1 haplotype having a larger influence in younger children in this study [50].
The impact of the condition being treated and associated concomitant medications on busulfan PK is still unclear. Six population PK studies found no significant difference in the PK of busulfan in children with malignant conditions compared with those without [7, 37, 38, 40, 55, 56] (Table 2, Fig. 2a). Coadministration of fludarabine was reported to result in a reduction in busulfan CL in two studies, ranging from 6% [46] to 30% [40]. However, a third study did not find any significant effect of the conditioning regimen on busulfan PK [56].
3.3 Evidence of Changes in Busulfan Clearance over the Course of Treatment
Nine studies examined whether there was any change in busulfan CL over the course of treatment (Table 3); seven (78%) documented a decrease in CL over 4 days of therapy ranging from 8.1 to 20% compared with CL on day 1 of therapy [9, 20, 38, 45, 51, 56, 57], whereas two (22%) studies did not identify any significant change [31, 39]. One study incorporated a Michaelis–Menten equation within their population PK model to explain the non-linearity observed [20]. Of the seven studies observing a decrease in CL over the course of treatment, four studies reported on patients using once-daily dosing schedules, while the two studies observing no change in CL over the course of treatment had smaller patient numbers and both were based on four-times-daily dosing [31, 39].
3.4 Pharmacodynamics of Intravenous Busulfan in Paediatric Patients
Table 4 provides a summary of the dosing protocol, study design and outcome data from the 45 studies that examined the PD of intravenous busulfan in paediatric patients. The number of participants varied, with a median of 47 (range 6–1781) per study. Median patient age was 7.4 years (range of median ages 0.5–12.8). Thirty-six (80%) studies solely utilised intravenous busulfan therapy, with the remaining nine (20%) studies including some patients who received busulfan orally. Twenty-one (47%) studies were based on four-times-daily dosing, eight (18%) were based on once-daily dosing, two (4%) were based on twice-daily dosing, eight (18%) were based on a mixture of dosing regimens, and six (13%) did not record this information.
3.4.1 Exposure Targets and Estimation
There was marked heterogeneity across the studies on how exposure monitoring was performed, how exposure was calculated, and which target was aimed for (Table 4). Busulfan exposure targets included Cssavg, AUC24, AUCcum and median AUC per dose over the course. Median reported observed results across all studies for each exposure matrix were Cssavg 652 ng/mL (n = 7; range 577–750); AUC24 19.8 mg/L·h (n = 6; range 16.0–21.9); AUCcum (excluding reduced intensity conditioning regimens) 74.8 mg/L·h (n = 6; range 74.0–91.0); and median AUCdose.interval for four-times-daily dosing 4.4 mg/L·h (n = 10; range 3.9–5.7) and twice-daily dosing 9.9 mg/L·h (n = 1). Of the 39 studies that performed exposure monitoring, 13 (33%) reported an exposure index that was attainable from the sampling method used; 12 (31%) reported an exposure result that could not have been obtained without assuming CL remained the same over time, as busulfan concentrations were not remeasured; and; 14 (36%) did not provide adequate information to determine how the reported exposure index was obtained relative to sampling.
3.4.2 Relationship between Exposure and Outcomes
A summary of studies that examined the relationship between busulfan exposure and patient outcomes is provided in Table 5.
3.4.2.1 Overall Survival and Event-Free Survival
OS was reported in 24 (53%) studies (from 0.3 to 5 years post-transplant), with a median value of 80% (range 40–100%), and EFS or disease-free survival (DFS) was reported in 20 studies (from 1 to 5 years post-transplant) and ranged from 29 to 100%. Only three studies described an association between busulfan exposure and OS [24, 58, 59] (Table 5) that included either an HR, RR or OR value. Bartelink et al. conducted the largest study, with 674 included patients. In that study, OS at 2 years was 80%, 72% and 64% in patients with a busulfan AUCcum between 78 and 101 mg/L·h, < 78 mg/L·h and > 101 mg/L·h, respectively [24]. Ansari et al. investigated 75 patients administered busulfan four times daily, with sampling performed after the first dose. Patients with a CssDose1 > 600 ng/mL (~ AUCcum > 57.6 mg/L·h) had a lower 5-year OS rate (50% vs. 90%, p = 0.001) and higher non-relapse mortality in multivariate analysis (HR 5.14, 95% CI 2.19–12.07; p = 0.001) compared with patients with a CssDose1 < 600 ng/mL [58]. Benadiba et al. also observed reduced OS in patients with a CssDose1 > 600 ng/mL compared with patients with a Cssavg < 600 ng/mL (37.5% vs. 81%, p = 0.006) [59]. Ansari and co-workers reported that a lower Cssavg is associated with improved EFS (p < 0.0005) [52] and a CssDose1 < 600 ng/mL is associated with improved EFS and OS (p < 0.0005) [49].
One publication examined CssDose1 together with Cssavg across the course of busulfan treatment [58]. The study (N = 75) involved four groups and included patients with CssDose1 < 600 ng/mL and Cssavg across the course < 750 ng/mL (~ AUCcum < 72 mg/L·h), CssDose1 < 600 ng/mL and Cssavg across the course > 750 ng/mL, CssDose1 > 600 ng/mL and Cssavg across the course < 750 ng/mL, and, lastly, CssDose1 > 600 ng/mL and Cssavg across the course > 750 ng/mL. This study reported that patients with CssDose1 < 600 ng/mL had significantly improved OS and EFS irrespective of the Cssavg across the course (OS 91%, 86%, 53% and 49%, respectively, p = 0.003; EFS 89%, 75%, 44% and 22%, respectively, p ≤ 0.001) [58].
3.4.2.2 Transplant-Related Mortality and Toxicity
TRM was reported in 18 studies with a median value of 9.1% (range 0–26%). One study reported increased TRM in patients with a busulfan AUCcum > 101 mg/L·h (HR 2.99, 95% CI 1.82–4.92, p ≤ 0.0001) (Table 5) and also increased transplant-related toxicity (TRT; HR 1.69, 95% CI 1.12–2.57, p =0.013) [24]. Ansari et al. identified that a CssDose1 > 900 ng/mL (~ AUCcum > 86.4 mg/L·h) correlated with increased TRT (p < 0.0005) [49]. CssDose1 > 900 ng/mL was associated with TRT irrespective of GSTA1 diplotype groups, whereas high risk of TRT for CssDose1 < 900 ng/mL was evident only for GSTA1 group IV carriers (slow metabolisers).
3.4.2.3 Engraftment and Relapse
Engraftment rate was reported in 26 studies with a median value of 93% (range 56–100%), and relapse rate was reported in 12 studies with a median value of 20% (range 10–35%). Bartelink et al. reported that graft failure or relapse occurred less in an optimum AUCcum group (78–101 mg/L·h) compared with a low AUCcum group (< 78 mg/L·h) [HR 0.57, 95% CI 0.39–0.84, p = 0.0041] [24]. Philippe et al. reported an increased risk of relapse for patients with an AUC6 < 3.7 mg/L·h (~ AUCcum < 59.2 mg/L·h) compared with > 3.7 mg/L·h (42.9% vs. 13.4%, p = 0.04) [28].
3.4.2.4 Sinusoidal Obstructive Syndrome
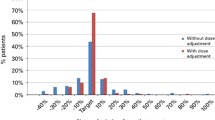
The incidence of SOS was reported in 31 studies with a median of 16% (range 0–34%). Two studies examined the incidence of SOS in relation to busulfan exposure [60, 61] and reported either an HR, RR or OR. An increased risk of SOS was observed in patients with a maximum drug concentration (Cmax) value ≥ 1880 ng/mL (63.3% vs. 21.3%), and in patients with increased time spent > 1300 ng/mL throughout the course of busulfan treatment, as estimated using Bayesian forecasting software [60]. A smaller study reported that decreased Cmax [43] correlated with an increased incidence of SOS. One study observed that patients administered busulfan 6-hourly with an AUC6 > 6.2 mg/L·h (~ AUCcum >99.2 mg/L·h) had an increased incidence of SOS compared with those with an AUC6 < 6.2 mg/L·h (33 vs. 15%, p < 0.05). However, this relationship was not evident in patients who received once-daily dosing of busulfan [61]. Interestingly, three studies that did not perform any exposure monitoring had a very low incidence of SOS [62,63,64], all below 5%. While the three studies with the highest incidence of SOS (24%, 25.6%, 31%, respectively) all performed exposure monitoring after the first dose only [60, 65, 66].
Eight studies reported other patient demographic and treatment factors that impacted on the risk of SOS (Table 6). Factors that increased the risk included the use of busulfan in combination with cyclophosphamide instead of fludarabine [4], GSTA1 genotypes, including GSTA1*B [67], GSTA1*B/*B, GSTA1*B1/*B1 [52], CTH c.1364 TT genotype [52], younger [27, 60, 65] or older age [66], weight < 9 kg [60], weight-based dosing as opposed to age-based dosing [68], and the use of once-daily intravenous busulfan with targeted concentration intervention [30]. One study suggested weight-based dosing was a predictor of SOS (HR 9.46, p = 0.009), with SOS at 42% compared with 5% for those receiving age-based dosing [68].
3.4.2.5 Acute Graft-versus-Host Disease
aGVHD incidence was reported in 20 studies with a median value of 30% (range 11–70%). Increased Cssavg correlated with higher cumulative incidence of aGVHD [52]. Ansari et al. reported that CssDose1 > 600 ng/mL (~ AUCcum > 57.6 mg/L·h) was associated with a higher incidence of aGVHD compared with a CssDose1 < 600 ng/mL (21 vs. 5%, p = 0.04) [58].
3.4.2.6 Other Toxicities
Neurotoxicity incidence was reported in eight studies with a median value of 1.6% (range 0–10%) and lung toxicity incidence was reported in seven studies with a median value of 5% (range 2–36%). One study reported that increased Cssavg correlated with higher cumulative incidence of lung toxicity and haemorrhagic cystitis, (p < 0.03) [52].
4 Discussion
This review summarises what is currently known about the PK and PD of intravenous busulfan in paediatric patients to assist clinicians in dosing of this agent for future patients. Heterogeneity was seen across the studies reviewed in terms of conditions being treated with HSCT, treatment regimens used, the way exposure monitoring was performed, the exposure target aimed for and obtained, outcomes assessed and the duration of follow-up. This marked heterogeneity limits opportunity for meta-analysis and comparison of results between studies since these factors impact on the inherent risk for certain toxicities and outcomes. Further prospective studies in particular patient groups, with specific conditioning regimens, would assist with validation of the current recommended therapeutic target. This would require large international, multicentre clinical trials, which was a key conclusion of a recently published meta-analysis that attempted to examine busulfan exposure and outcomes within paediatric patients [69]. Outcome data for EFS, aGVHD, seizures and lung toxicity were too sparse to analyse in this review.
IOV in the PK of busulfan was relatively low, highlighting the suitability of dosage adjustment based on exposure monitoring for this highly toxic chemotherapy agent. Both patient age and weight were found to influence busulfan CL and Vd in paediatrics. In childhood, age impacts on enzymatic maturation, plasma protein levels and body water:fat ratios, whereas weight relates to body size and impacts on liver blood flow. Both age and weight affect the liver-to-body-size ratio. CL of busulfan in younger children (> 1 to < 4 years) appeared to be higher than in older children. GST enzyme expression changes according to age [70]. Higher expression of GSTA1 and lower expression of GSTA2 have been reported in infants and young children compared with adults [70]. A higher liver-to-body-size ratio and increased liver blood flow largely explain the increased busulfan CL observed in young children (1–4 years of age), however it is possible that higher GSTA1 enzyme expression may also contribute. The ontogeny of alternative GST enzymes (GSTM and GSTP) and the effect of this on busulfan metabolism in different age groups requires further investigation. Lower expression of FMO3 and CYP450 enzymes has been reported in infants compared with older children and adults [70, 71], which could potentially lead to greater accumulation of THT and EdAG, resulting in increased inhibition of busulfan metabolism by GSTA1 in young infants compared with older children and adults. This may explain the reduced busulfan CL observed in young infants despite possible high GSTA1 expression [12].
Vd of busulfan may be higher in younger (< 4 years) compared with older children [22, 43], which could be due to differences in busulfan binding to plasma protein and changes in body composition with age. Busulfan covalently binds to plasma proteins, including albumin, the level of which is commonly reduced or primarily composed of fetal albumin (which has lower binding affinity) in infants [72].
Several studies reported that polymorphic expression of GSTA1 influences busulfan CL, with one study suggesting polymorphisms in CYP39A1 may also be important. Patients who are homozygous or heterozygous for the GSTA1*B allele have been shown to express less functional GSTA1 protein and thus have reduced CL of busulfan [11]. The incorporation of pharmacogenomic testing in clinical practice may be valuable for dosing of busulfan in children in the future. However, the high burden of concomitant medication interactions in HSCT and the cost of performing genomic testing may limit the utility of pharmacogenomic testing and be counterbalanced by therapeutic drug monitoring.
Several studies that investigated busulfan CL over the course of treatment reported that it decreased over time. It has been hypothesised that with repeated doses of busulfan, THT and EdAG may accumulate, resulting in a decrease in busulfan metabolism due to inhibition of the glutathione conjugation pathways [12, 13]. This may be more apparent with once-daily administration regimens that are associated with higher peak concentrations of busulfan, particularly if there is a step in the metabolism pathway that gets saturated, or glutathione depletion within the cell.
Phenytoin is a known inducer of several CYP450 enzymes, including CYP2B6 and CYP3A4/5, and a substrate of CYP2C9 and CYP2C19 [73]. Coadministration of phenytoin with busulfan may mask busulfan’s concentration or time-dependent non-linear PK behaviour due to the induction of enzymes responsible for the metabolism of THT, resulting in a reduction in the feedback inhibition of glutathione conjugation pathways. Nonlinear concentration- or time-dependent behaviour associated with busulfan therapy may be more apparent with the use of newer antiepileptics that are not metabolic inducers, and with once-daily administration of this agent. CYP2C9 genotype as a covariate on CL should be further investigated.
Studies suggest that too-high busulfan exposure can reduce OS and EFS. Data to date, while limited, tend to suggest targeting an AUCcum between 78 and 101 mg/L·h or CssDose1 < 600 ng/mL (~ AUCcum < 57.6 mg/L·h) may be most appropriate. While the current suggested target range for busulfan across all doses is a Cssavg of 600–900 ng/mL (~ AUCcum 57.6–86.4 mg/L·h) [2, 3], patients achieving a CssDose1 < 600 ng/mL may still be achieving adequate exposure over the course of treatment due to possible time-dependent PK behaviour and increased exposure with repeat dosing, while those with higher exposure initially may become overexposed with repeat dosing.
Busulfan may have a metabolite with activity, either cytotoxicity, toxic effects, or both. A few studies have demonstrated a relationship between busulfan exposure and certain adverse events. TRM and TRT appeared to be associated with higher busulfan exposure (i.e. an AUCcum > 101 mg/L·h [~ Cssavg > 1052 ng/mL] and CssDose1 > 900 ng/mL [~ AUCcum > 86.4 mg/L·h], respectively). Ansari et al. showed that GSTA1 group IV carriers, which are slow metabolisers, had increased TRT when CssDose1 was < 900 ng/mL. There is some evidence that the incidence of SOS is associated with higher busulfan exposure as well as patient genotype/haplotypes that result in slower busulfan metabolism [49, 52, 67]. These findings may be due to increased total cumulative busulfan intracellular exposure over the treatment course; increased time busulfan concentrations in plasma are above a certain concentration or potentially increased exposure to a toxic metabolite [49]. Free busulfan diffuses passively into cells, therefore more is likely to enter the cell with high concentrations or with prolonged exposure. If metabolism is slow, there is increased intracellular exposure to free busulfan, which could increase the amount hydrolysed to active moieties and therefore increase toxicity. Since busulfan metabolism is complex and involves multiple enzyme systems, the ontogeny of these enzymes and subsequent metabolite levels may play a role in the observed increased risk of SOS in younger patients [27, 60, 65], especially if a metabolite is involved in the development of this toxicity. EdAG is a product of busulfan metabolism (Fig. 1) and has been observed to react with protein nucleophiles, in particular glutaredoxins, resulting in inactive moieties [15]. Interactions with glutaredoxins by EdAG may cause flow-on effects to pathways that are dependent on glutaredoxins for either glutathionylation or deglutathionylation and therefore cause further cytotoxicity [15]. EdAG usually forms a conjugate with glutathione, however it is possible that with higher concentrations observed with once-daily dosing, there may be more potential for EdAG to interact with alternate proteins, potentially contributing to increased toxicity and/or efficacy.
EdAG also contributes to intracellular glutathione depletion through its metabolism to GSG. Glutathione depletion decreases the ability for cells to respond to endogenous oxidative stress, process further busulfan through GST enzymes, and deactivate further EdAG produced. Accumulation of EdAG may also help explain the time-dependent CL observed, since it competes with glutathione for binding sites on GST enzymes [13]. Blood glutathione concentrations before administration of busulfan were positively correlated with busulfan CL (adjusted R2 = 0.45, p = 0.009) [74]. Increasing intracellular glutathione levels reduces busulfan-related hepatotoxicity [75] without impacting on myeloablation [76]. N-acetyl-cysteine (NAC) administration during busulfan therapy may help prevent SOS in patients at high risk, however further investigation into the effects on cancer cells and relapse risk is necessary [75, 76]. Alternatively, it has been observed that endothelial cells that are involved in the evolution of SOS have little to no GSTA1 activity, and that other GST enzymes may take on the role of conjugating busulfan with glutathione [77]. Therefore, the pharmacogenomics and ontogeny of these alternative GST enzymes may be more important in the development of SOS than GSTA1 [77].
It has been suggested CYP2C9 may also play a role in busulfan metabolism based on case studies reporting that coadministration of metronidazole [78, 79] and ketobemidone [80, 81] (known substrates/inhibitors of CYP2C9) increases busulfan concentrations. Patients who are CYP2C9*2 and *3 allele carriers (decreased enzyme activity) have been shown to have a higher ratio of busulfan to its metabolite sulfolane [82], suggesting this enzyme plays a significant role in the metabolism of THT. Furthermore, increased graft failure and decreased EFS have been demonstrated in malignant patients with a busulfan:sulfolane metabolic ratio above five compared with below [82], further supporting the idea of a metabolite with activity. A low metabolic ratio, when busulfan levels are low compared with sulfolane levels, suggests fast transition of unbound busulfan through the metabolism process to the final products that are then excreted. Higher busulfan:sulfolane metabolic ratios suggest slower initial metabolism and potentially decreased or increased concentrations of intermediate metabolite products (including THT), depending on which particular enzyme activity is reduced. Cyclophosphamide is metabolised to active moieties through CYP enzymes, including CYP2C9, which could confound the results observed, since slow metabolism of busulfan to sulfolane may also mean slower or reduced production of the active cyclophosphamide metabolite. Further studies examining the impact of pharmacogenomics and metabolite concentrations on both the PK and PD of busulfan should be prioritised.
Many studies in this review performed dose adjustment based on exposure estimates obtained after the first dose, with no or limited further monitoring performed on subsequent days [55, 83], but then reported an average exposure per dose or a cumulative exposure estimate to link to outcomes. Cumulative exposure based on measures taken on the first day of treatment may underestimate that actually achieved on later days if busulfan displays non-linear or time-dependent PK behaviours. If unnecessary dosage increases are being made based on these underpredictions, this may explain suggestions by one group of investigators [84] that exposure monitoring is a risk factor for SOS compared with administration of intravenous busulfan without monitoring.
Bayesian forecasting methods have an advantage over other methods in terms of making dosage predictions, as they are based on PK models that can consider patient covariate values and time-dependent PK behaviour. Bartelink and colleagues have recommended increasing the historical busulfan AUCcum target of 58–86 mg/L·h (Cssavg 600–900 ng/mL) to a new recommended target of 78–101 mg/L·h (Cssavg 812.5–1052 ng/mL) using Bayesian forecasting modelling software [24]. Some caution should be used in applying this increased therapeutic target if cumulative exposure is being estimated in a different manner to that within the publication.
Four key published population PK models have been externally validated [20, 21, 38, 39]. For patients who are overweight or underweight, the population PK model by Bartelink and colleagues [21] may be predictive. For once-daily dosing, the population PK models by Nguyen [39] and McCune et al. [38] have both been externally validated. The population PK model published by Long-Boyle et al. [20] was not externally validated for obese patients or once-daily dosing and had limited numbers of patients in the external validation cohort above the age of 12 years.
Currently, there is much heterogeneity in how exposure monitoring is performed, including which day(s) sampling is performed. Standardisation of exposure monitoring would allow for improved analysis of results and for meta-analysis to be performed. Harmonisation to one unit of measure for exposure as mg/L·h has been recommended [85] for future publications and protocols to prevent misinterpretation. To be in line with this recommendation, harmonisation of the exposure index reported to AUC instead of Css is recommended. Estimated AUCcum would be helpful to allow comparisons between studies and ensure that dose adjustments can be incorporated in the final exposure estimate. Further prospective studies measuring busulfan exposure over the entire course of treatment, or validation of Bayesian forecasting programmes to estimate cumulative exposure from less intensive sampling and utilisation in studies, is required to establish appropriate exposure–outcome relationships. It is imperative that clinicians are aware of the time-dependent PK (decreased CL over the course of treatment) exhibited by busulfan, and the variability of exposure observed between occasions, and consider this when making recommendations for dose adjustments for patients. Exposure monitoring should be repeated following any dose change. Reducing future doses should be considered if exposure is close to the upper limit of the therapeutic target.
5 Limitations
Comparing exposure in the PK and PD analysis was complicated by considerable variety in the dosing of busulfan, exposure targets monitored (Cssavg, AUCdose interval, AUC24 and AUCcum), how exposure was calculated, how dose adjustments are made, and whether subsequent monitoring was pursued after the first dose. A number of publications did not contain the level of detail required to ensure accurate comparisons. Conversion of exposure to one index measure was necessary for comparisons even though values were not necessarily equivalent and that changes to exposure due to dose modifications may not have been taken into account. A complete description of covariate model building was rare, therefore it was not always possible to determine whether the covariates had been screened against both CL and Vd. Due to small patient numbers receiving this drug worldwide, publications commonly combined data from different centres and countries, therefore administering different regimens and supportive care. A considerable number of studies did not report the seizure prophylaxis used and if multiple agents were used in the patient group, the number of patients receiving each agent was not always available. Publications with a mixture of daily and four-times-daily dosing did not differentiate the PK values between the different dosing schemes. Comparison of outcomes was complicated by significant variations in the condition being treated and therefore inherent differences in the likelihood of certain outcomes, including survival and toxicity. In addition, a large proportion of studies had both malignant and non-malignant patients included and outcome results were often not separated. Outcomes were not consistently defined or measured in the same way and duration of follow-up across studies varied, making outcomes difficult to compare between studies.
6 Conclusions
Patient age, body weight and GSTA1 genotype are important covariates to consider when individualising busulfan therapy. Reduced busulfan CL across the dosing period may need to be considered, particularly in patients not receiving phenytoin co-therapy. Busulfan exposure above the current recommended target range appears to be associated with reduced OS and EFS, and increased TRM, TRT and some toxicities. Standardisation of the process for monitoring and harmonisation of the exposure index used should allow improved interpretation of results of studies and future meta-analysis to be performed. We would recommend performing exposure monitoring and subsequent dosage adjustment based on the use of Bayesian forecasting programmes. Repeated sample testing should be performed following any dose modification. Improving accuracy of estimates of cumulative busulfan exposure would facilitate improved investigation of exposure–outcome relationships and allow better characterisation of CL changes over the treatment course. Further investigation of the influence of busulfan metabolites on both the PK and PD of busulfan is warranted.
References
Duarte RF, Labopin M, Bader P, Basak GW, Bonini C, Chabannon C, et al. Indications for haematopoietic stem cell transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2019. Bone Marrow Transplant. 2019;54(10):1525–52. https://doi.org/10.1038/s41409-019-0516-2.
Otsuka America Pharmaceutical Inc. Busulfex (busulfan) for injection. Silver Spring: US FDA; 2015.
Pierre Fabre Medicament. Busilvex: EPAR—product information. Amsterdam: European Medicines Agency; 2017. p. 1–40.
Bartelink IH, van Reij EML, Gerhardt CE, van Maarseveen EM, de Wildt A, Versluys B, et al. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: maintaining efficacy with less toxicity. Biol Blood Marrow Transplant. 2014;20(3):345–53. https://doi.org/10.1016/j.bbmt.2013.11.027.
Hong KT, Kang HJ, Choi JY, Hong CR, Cheon J-E, Park JD, et al. Favorable outcome of post-transplantation cyclophosphamide haploidentical peripheral blood stem cell transplantation with targeted busulfan-based myeloablative conditioning using intensive pharmacokinetic monitoring in pediatric patients. Biol Blood Marrow Transplant. 2018;24(11):2239–44. https://doi.org/10.1016/j.bbmt.2018.06.034.
Kim B, Lee JW, Hong KT, Yu KS, Jang IJ, Park KD, et al. Pharmacometabolomics for predicting variable busulfan exposure in paediatric haematopoietic stem cell transplantation patients. Sci Rep. 2017a;7(1):1711. https://doi.org/10.1038/s41598-017-01861-7.
Lee JW, Kang HJ, Kim S, Lee SH, Yu K-S, Kim NH, et al. Favorable outcome of hematopoietic stem cell transplantation using a targeted once-daily intravenous busulfan-fludarabine-etoposide regimen in pediatric and infant acute lymphoblastic leukemia patients. Biol Blood Marrow Transplant. 2015;21(1):190–5. https://doi.org/10.1016/j.bbmt.2014.09.013.
Nava T, Kassir N, Rezgui MA, Uppugunduri CRS, Huezo-Diaz Curtis P, Duval M, et al. Incorporation of GSTA1 genetic variations into a population pharmacokinetic model for IV busulfan in paediatric hematopoietic stem cell transplantation. Br J Clin Pharmacol. 2018;84(7):1494–504. https://doi.org/10.1111/bcp.13566.
Rhee SJ, Lee JW, Yu KS, Hong KT, Choi JY, Hong CR, et al. Pediatric patients undergoing hematopoietic stem cell transplantation can greatly benefit from a novel once-daily intravenous busulfan dosing nomogram. Am J Hematol. 2017;92(7):607–13. https://doi.org/10.1002/ajh.24734.
Hassan M, Andersson BS. Role of pharmacogenetics in busulfan/cyclophosphamide conditioning therapy prior to hematopoietic stem cell transplantation. Pharmacogenomics. 2013;14(1):75–87. https://doi.org/10.2217/pgs.12.185.
Huezo-Diaz P, Uppugunduri CRS, Tyagi AK, Krajinovic M, Ansari M. Pharmacogenetic aspects of drug metabolizing enzymes in busulfan based conditioning prior to allogenic hematopoietic stem cell transplantation in children. Curr Drug Metab. 2014;15(3):251–64. https://doi.org/10.2174/1389200215666140202214012.
El-Serafi I, Terelius Y, Abedi-Valugerdi M, Naughton S, Saghafian M, Moshfegh A, et al. Flavin-containing monooxygenase 3 (FMO3) role in busulphan metabolic pathway. PLoS ONE. 2017;12(11):e0187294. https://doi.org/10.1371/journal.pone.0187294.
Younis IR, Elliott M, Peer CJ, Cooper AJL, Pinto JT, Konat GW, et al. Dehydroalanine analog of glutathione: an electrophilic busulfan metabolite that binds to human glutathione S-transferase A1–1. J Pharmacol Exp Ther. 2008;327(3):770–6. https://doi.org/10.1124/jpet.108.142208.
Scian M, Atkins WM. Supporting data for characterization of the busulfan metabolite EdAG and the Glutaredoxins that it adducts. Data Brief. 2015a;5:161–70. https://doi.org/10.1016/j.dib.2015.09.002.
Scian M, Atkins WM. The busulfan metabolite EdAG irreversibly glutathionylates glutaredoxins. Arch Biochem Biophys. 2015b;583:96–104. https://doi.org/10.1016/j.abb.2015.08.005.
Scian M, Guttman M, Bouldin SD, Outten CE, Atkins WM. The myeloablative drug busulfan converts cysteine to dehydroalanine and lanthionine in redoxins. Biochemistry. 2016;55(33):4720–30. https://doi.org/10.1021/acs.biochem.6b00622.
DeLeve LD, Wang XD. Role of oxidative stress and glutathione in busulfan toxicity in cultured murine hepatocytes. Pharmacology. 2000;60(3):143–54. https://doi.org/10.1159/000028359.
Hassan M, Oberg G, Ehrsson H, Ehrnebo M, Wallin I, Smedmyr B, et al. Pharmacokinetic and metabolic studies of high-dose busulphan in adults. Eur J Clin Pharmacol. 1989;36(5):525–30. https://doi.org/10.1007/bf00558081.
Hassan M, Ehrsson H, Smedmyr B, Totterman T, Wallin I, Oberg G, et al. Cerebrospinal fluid and plasma concentrations of busulfan during high-dose therapy. Bone Marrow Transplant. 1989;4(1):113–4.
Long-Boyle JR, Savic R, Yan S, Bartelink I, Musick L, French D, et al. Population pharmacokinetics of busulfan in pediatric and young adult patients undergoing hematopoietic cell transplant: a model-based dosing algorithm for personalized therapy and implementation into routine clinical use. Ther Drug Monit. 2015;37(2):236–45. https://doi.org/10.1097/ftd.0000000000000131.
Bartelink IH, Boelens JJ, Bredius RG, Egberts AC, Wang C, Bierings MB, et al. Body weight-dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients: towards individualized dosing. Clin Pharmacokinet. 2012;51(5):331–45. https://doi.org/10.2165/11598180-000000000-00000.
Savic RM, Cowan MJ, Dvorak CC, Pai SY, Pereira L, Bartelink IH, et al. Effect of weight and maturation on busulfan clearance in infants and small children undergoing hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2013;19(11):1608–14. https://doi.org/10.1016/j.bbmt.2013.08.014.
Langenhorst JB, Dorlo TPC, van Maarseveen EM, Nierkens S, Kuball J, Boelens JJ, et al. Population pharmacokinetics of fludarabine in children and adults during conditioning prior to allogeneic hematopoietic cell transplantation. Clin Pharmacokinet. 2019;58(5):627–37. https://doi.org/10.1007/s40262-018-0715-9.
Bartelink IH, Lalmohamed A, van Reij EM, Dvorak CC, Savic RM, Zwaveling J, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: a multicentre, retrospective cohort analysis. Lancet Haematol. 2016;3(11):e526–36. https://doi.org/10.1016/s2352-3026(16)30114-4.
Hamidieh AA, Alimoghaddam K, Jahani M, Bahar B, Mousavi SA, Iravani M, et al. Non-TBI hematopoietic stem cell transplantation in pediatric AML patients: a single-center experience. J Pediatr Hematol Oncol. 2013;35(6):e239–45. https://doi.org/10.1097/MPH.0b013e31827080fc.
Hamidieh AA, Monzavi SM, Kaboutari M, Behfar M, Esfandbod M. Outcome analysis of pediatric patients with acute lymphoblastic leukemia treated with total body irradiation-free allogeneic hematopoietic stem cell transplantation: comparison of patients with and without central nervous system involvement. Biol Blood Marrow Transplant. 2017;23(12):2110–7. https://doi.org/10.1016/j.bbmt.2017.08.036.
Malar R, Sjoo F, Rentsch K, Hassan M, Gungor T. Therapeutic drug monitoring is essential for intravenous busulfan therapy in pediatric hematopoietic stem cell recipients. Pediatr Transplant. 2011;15(6):580–8. https://doi.org/10.1111/j.1399-3046.2011.01529.x.
Philippe M, Goutelle S, Guitton J, Fonrose X, Bergeron C, Girard P, et al. Should busulfan therapeutic range be narrowed in pediatrics? Experience from a large cohort of hematopoietic stem cell transplant children. Bone Marrow Transplant. 2016;51(1):72–8. https://doi.org/10.1038/bmt.2015.218.
Andersson BS, Thall PF, Valdez BC, Milton DR, Al-Atrash G, Chen J, et al. Fludarabine with pharmacokinetically guided IV busulfan is superior to fixed-dose delivery in pretransplant conditioning of AML/MDS patients. Bone Marrow Transplant. 2017;52(4):580–7. https://doi.org/10.1038/bmt.2016.322.
Bartelink IH, Bredius RG, Ververs TT, Raphael MF, van Kesteren C, Bierings M, et al. Once-daily intravenous busulfan with therapeutic drug monitoring compared to conventional oral busulfan improves survival and engraftment in children undergoing allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2008;14(1):88–98. https://doi.org/10.1016/j.bbmt.2007.09.015.
Vassal G, Michel G, Esperou H, Gentet JC, Valteau-Couanet D, Doz F, et al. Prospective validation of a novel IV busulfan fixed dosing for paediatric patients to improve therapeutic AUC targeting without drug monitoring. Cancer Chemother Pharmacol. 2008;61(1):113–23. https://doi.org/10.1007/s00280-007-0455-2.
Darwich AS, Ogungbenro K, Vinks AA, Powell JR, Reny JL, Marsousi N, et al. Why has model-informed precision dosing not yet become common clinical reality? Lessons from the past and a roadmap for the future. Clin Pharmacol Ther. 2017;101(5):646–56. https://doi.org/10.1002/cpt.659.
Hoy SM, Lyseng-Williamson KA. Intravenous busulfan: in the conditioning treatment of pediatric patients prior to hematopoietic stem cell transplantation. Paediatr Drugs. 2007;9(4):271–8.
Ward J, Kletzel M, Duerst R, Fuleihan R, Chaudhury S, Schneiderman J, et al. Single daily busulfan dosing for infants with nonmalignant diseases undergoing reduced-intensity conditioning for allogeneic hematopoietic progenitor cell transplantation. Biol Blood Marrow Transplant. 2015;21(9):1612–21. https://doi.org/10.1016/j.bbmt.2015.05.017.
Browning B, Thormann K, Donaldson A, Halverson T, Shinkle M, Kletzel M. Busulfan dosing in children with BMIs ≥85% undergoing HSCT: a new optimal strategy. Biol Blood Marrow Transplant. 2011;17(9):1383–8. https://doi.org/10.1016/j.bbmt.2011.01.013.
Tse WT, Duerst R, Schneiderman J, Chaudhury S, Jacobsohn D, Kletzel M. Age-dependent pharmacokinetic profile of single daily dose i.v. busulfan in children undergoing reduced-intensity conditioning stem cell transplant. Bone Marrow Transplant. 2009;44(3):145–56. https://doi.org/10.1038/bmt.2008.437.
Zwaveling J, Press RR, Bredius RG, van Derstraaten TR, den Hartigh J, Bartelink IH, et al. Glutathione S-transferase polymorphisms are not associated with population pharmacokinetic parameters of busulfan in pediatric patients. Ther Drug Monit. 2008;30(4):504–10. https://doi.org/10.1097/FTD.0b013e3181817428.
McCune JS, Bemer MJ, Barrett JS, Scott Baker K, Gamis AS, Holford NH. Busulfan in infant to adult hematopoietic cell transplant recipients: a population pharmacokinetic model for initial and Bayesian dose personalization. Clin Cancer Res. 2014;20(3):754–63. https://doi.org/10.1158/1078-0432.Ccr-13-1960.
Nguyen L. Integration of modelling and simulation into the development of intravenous busulfan in paediatrics: an industrial experience. Fundam Clin Pharmacol. 2008;22(6):599–604. https://doi.org/10.1111/j.1472-8206.2008.00651.x.
Paci A, Vassal G, Moshous D, Dalle JH, Bleyzac N, Neven B, et al. Pharmacokinetic behavior and appraisal of intravenous busulfan dosing in infants and older children: the results of a population pharmacokinetic study from a large pediatric cohort undergoing hematopoietic stem-cell transplantation. Ther Drug Monit. 2012a;34(2):198–208. https://doi.org/10.1097/FTD.0b013e31824c2f60.
Diestelhorst C, Boos J, McCune JS, Hempel G. Population pharmacokinetics of intravenous busulfan in children: revised body weight-dependent NONMEM(R) model to optimize dosing. Eur J Clin Pharmacol. 2014;70(7):839–47. https://doi.org/10.1007/s00228-014-1692-z.
Neely M, Philippe M, Rushing T, Fu X, van Guilder M, Bayard D, et al. Accurately achieving target busulfan exposure in children and adolescents with very limited sampling and the BestDose software. Ther Drug Monit. 2016;38(3):332–42. https://doi.org/10.1097/ftd.0000000000000276.
Schechter T, Finkelstein Y, Doyle J, Verjee Z, Moretti M, Koren G, et al. Pharmacokinetic disposition and clinical outcomes in infants and children receiving intravenous busulfan for allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2007;13(3):307–14. https://doi.org/10.1016/j.bbmt.2006.10.026.
Kim B, Lee JW, Hong KT, Yu K-S, Jang I-J, Park KD, et al. Pharmacometabolomics for predicting variable busulfan exposure in paediatric haematopoietic stem cell transplantation patients. Sci Rep. 2017b;7(1):1711. https://doi.org/10.1038/s41598-017-01861-7.
Gaziev J, Nguyen L, Puozzo C, Mozzi AF, Casella M, Perrone Donnorso M, et al. Novel pharmacokinetic behavior of intravenous busulfan in children with thalassemia undergoing hematopoietic stem cell transplantation: a prospective evaluation of pharmacokinetic and pharmacodynamic profile with therapeutic drug monitoring. Blood. 2010;115(22):4597–604. https://doi.org/10.1182/blood-2010-01-265405.
Ishiwata Y, Nagata M, Tsuge K, Takahashi H, Suzuki S, Imai K, et al. Population pharmacokinetics of intravenous busulfan in Japanese pediatric patients with primary immunodeficiency diseases. J Clin Pharmacol. 2018;58(3):327–31. https://doi.org/10.1002/jcph.1027.
van Hoogdalem MW, Emoto C, Fukuda T, Mizuno T, Mehta PA, Vinks AA. Population pharmacokinetic modelling of busulfan and the influence of body composition in paediatric Fanconi anaemia patients. Br J Clin Pharmacol. 2020;86(5):933–43. https://doi.org/10.1111/bcp.14202.
Johnson L, Orchard PJ, Baker KS, Brundage R, Cao Q, Wang X, et al. Glutathione S-transferase A1 genetic variants reduce busulfan clearance in children undergoing hematopoietic cell transplantation. J Clin Pharmacol. 2008;48(9):1052–62. https://doi.org/10.1177/0091270008321940.
Ansari M, Curtis PH, Uppugunduri CRS, Rezgui MA, Nava T, Mlakar V, et al. GSTA1 diplotypes affect busulfan clearance and toxicity in children undergoing allogeneic hematopoietic stem cell transplantation: a multicenter study. Oncotarget. 2017;8(53):90852–67. https://doi.org/10.18632/oncotarget.20310.
Ten Brink MH, Swen JJ, Bohringer S, Wessels JAM, van der Straaten T, Marijt EWA, et al. Exploratory analysis of 1936 SNPs in ADME genes for association with busulfan clearance in adult hematopoietic stem cell recipients. Pharmacogenet Genom. 2013;23(12):675–83. https://doi.org/10.1097/fpc.0000000000000007.
Lee JW, Kang HJ, Lee SH, Yu KS, Kim NH, Yuk YJ, et al. Highly variable pharmacokinetics of once-daily intravenous busulfan when combined with fludarabine in pediatric patients: phase I clinical study for determination of optimal once-daily busulfan dose using pharmacokinetic modeling. Biol Blood Marrow Transplant. 2012;18(6):944–50. https://doi.org/10.1016/j.bbmt.2011.11.025.
Ansari M, Rezgui MA, Theoret Y, Uppugunduri CR, Mezziani S, Vachon MF, et al. Glutathione S-transferase gene variations influence BU pharmacokinetics and outcome of hematopoietic SCT in pediatric patients. Bone Marrow Transplant. 2013;48(7):939–46. https://doi.org/10.1038/bmt.2012.265.
Ansari M, Lauzon-Joset JF, Vachon MF, Duval M, Theoret Y, Champagne MA, et al. Influence of GST gene polymorphisms on busulfan pharmacokinetics in children. Bone Marrow Transplant. 2010;45(2):261–7. https://doi.org/10.1038/bmt.2009.143.
Nava T, Rezgui MA, Uppugunduri CRS, Curtis PH, Theoret Y, Duval M, et al. GSTA1 genetic variants and conditioning regimen: missing key factors in dosing guidelines of busulfan in pediatric hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2017;23(11):1918–24. https://doi.org/10.1016/j.bbmt.2017.07.022.
McCune JS, Baker KS, Blough DK, Gamis A, Bemer MJ, Kelton-Rehkopf MC, et al. Variation in prescribing patterns and therapeutic drug monitoring of intravenous busulfan in pediatric hematopoietic cell transplant recipients. J Clin Pharmacol. 2013;53(3):264–75. https://doi.org/10.1177/0091270012447196.
Bartelink IH, van Kesteren C, Boelens JJ, Egberts TC, Bierings MB, Cuvelier GD, et al. Predictive performance of a busulfan pharmacokinetic model in children and young adults. Ther Drug Monit. 2012;34(5):574–83. https://doi.org/10.1097/FTD.0b013e31826051bb.
Kawazoe A, Funaki T, Kim S. Population pharmacokinetic analysis of busulfan in Japanese pediatric and adult HCT patients. J Clin Pharmacol. 2018;58(9):1196–204. https://doi.org/10.1002/jcph.1120.
Ansari M, Theoret Y, Rezgui MA, Peters C, Mezziani S, Desjean C, et al. Association between busulfan exposure and outcome in children receiving intravenous busulfan before hematopoietic stem cell transplantation. Ther Drug Monit. 2014;36(1):93–9. https://doi.org/10.1097/FTD.0b013e3182a04fc7.
Benadiba J, Ansari M, Krajinovic M, Vachon M-F, Duval M, Teira P, et al. Pharmacokinetics-adapted Busulfan-based myeloablative conditioning before unrelated umbilical cord blood transplantation for myeloid malignancies in children. PLoS ONE. 2018;13(4):e0193862. https://doi.org/10.1371/journal.pone.0193862.
Philippe M, Neely M, Rushing T, Bertrand Y, Bleyzac N, Goutelle S. Maximal concentration of intravenous busulfan as a determinant of veno-occlusive disease: a pharmacokinetic-pharmacodynamic analysis in 293 hematopoietic stem cell transplanted children. Bone Marrow Transplant. 2018. https://doi.org/10.1038/s41409-018-0281-7.
Kerl K, Diestelhorst C, Bartelink I, Boelens J, Trame MN, Boos J, et al. Evaluation of effects of busulfan and DMA on SOS in pediatric stem cell recipients. Pediatr Blood Cancer. 2014;61(2):306–11. https://doi.org/10.1002/pbc.24827.
Gonzalez-Vicent M, Molina B, Perez A, Diaz MA. Once-daily intravenous busulfan for 47 pediatric patients undergoing autologous hematopoietic stem cell transplantation: a single center study. J Pediatr Hematol Oncol. 2012;34(3):180–3. https://doi.org/10.1097/MPH.0b013e3182431e1b.
Andion M, Molina B, Gonzalez-Vicent M, Alonso L, Hernandez C, Lassaletta A, et al. High-dose busulfan and cyclophosphamide as a conditioning regimen for autologous peripheral blood stem cell transplantation in childhood non-hodgkin lymphoma patients: a long-term follow-up study. J Pediatr Hematol Oncol. 2011;33(3):E89–91. https://doi.org/10.1097/MPH.0b013e3181fd6c79.
Al-Seraihy A, Ayas M, Al-Nounou R, El-Solh H, Al-Ahmari A, Al-Jefri A, et al. Outcome of allogeneic stem cell transplantation with a conditioning regimen of busulfan, cyclophosphamide and low-dose etoposide for children with myelodysplastic syndrome. Hematol Oncol Stem Cell Ther. 2011;4(3):121–5. https://doi.org/10.5144/1658-3876.2011.121.
Schechter T, Perez-Albuerne E, Lin TF, Irwin MS, Essa M, Desai AV, et al. Veno-occlusive disease after high-dose busulfan-melphalan in neuroblastoma. Bone Marrow Transplant. 2020;55(3):531–7. https://doi.org/10.1038/s41409-018-0298-y.
Kim AH, Tse JC, Ikeda A, Moore TB. Evaluating pharmacokinetics and pharmacodynamics of intravenous busulfan in pediatric patients receiving bone marrow transplantation. Pediatr Transplant. 2009;13(8):971–6. https://doi.org/10.1111/j.1399-3046.2008.01098.x.
Huezo-Diaz Curtis P, Uppugunduri CRS, Muthukumaran J, Rezgui MA, Peters C, Bader P, et al. Association of CTH variant with sinusoidal obstruction syndrome in children receiving intravenous busulfan and cyclophosphamide before hematopoietic stem cell transplantation. Pharmacogenom J. 2018;18(1):64–9. https://doi.org/10.1038/tpj.2016.65.
Gokcebay DG, Azik F, Ozbek N, Isik P, Avci Z, Tavil B, et al. Clinical comparison of weight- and age-based strategy of dose administration in children receiving intravenous busulfan for hematopoietic stem cell transplantation. Pediatr Transplant. 2015;19(3):307–15. https://doi.org/10.1111/petr.12430.
Feng XY, Wu YJ, Zhang JR, Li JP, Zhu GH, Fan DF, et al. Busulfan systemic exposure and its relationship with efficacy and safety in hematopoietic stem cell transplantation in children: a meta-analysis. BMC Pediatr. 2020;20(1):176. https://doi.org/10.1186/s12887-020-02028-6.
Hines RN. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther. 2008;118(2):250–67. https://doi.org/10.1016/j.pharmthera.2008.02.005.
Treluyer JM, Gueret G, Cheron G, Sonnier M, Cresteil T. Developmental expression of CYP2C and CYP2C-dependent activities in the human liver: in-vivo/in-vitro correlation and inducibility. Pharmacogenetics. 1997;7(6):441–52. https://doi.org/10.1097/00008571-199712000-00002.
Matalová P, Urbánek K, Anzenbacher P. Specific features of pharmacokinetics in children. Drug Metab Rev. 2016;48(1):70–9. https://doi.org/10.3109/03602532.2015.1135941.
Miners JO, Birkett DJ. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol. 1998;45(6):525–38. https://doi.org/10.1046/j.1365-2125.1998.00721.x.
Almog S, Kurnik D, Shimoni A, Loebstein R, Hassoun E, Gopher A, et al. Linearity and stability of intravenous busulfan pharmacokinetics and the role of glutathione in busulfan elimination. Biol Blood Marrow Transplant. 2011;17(1):117–23. https://doi.org/10.1016/j.bbmt.2010.06.017.
Hassan Z, Hellstrom-Lindberg E, Alsadi S, Edgren M, Hagglund H, Hassan M. The effect of modulation of glutathione cellular content on busulphan-induced cytotoxicity on hematopoietic cells in vitro and in vivo. Bone Marrow Transplant. 2002;30(3):141–7. https://doi.org/10.1038/sj.bmt.1703615.
Sjoo F, Aschan J, Barkholt L, Hassan Z, Ringden O, Hassan M. N-acetyl-L-cysteine does not affect the pharmacokinetics or myelosuppressive effect of busulfan during conditioning prior to allogeneic stem cell transplantation. Bone Marrow Transplant. 2003;32(4):349–54. https://doi.org/10.1038/sj.bmt.1704143.
Vassord C, Lapoumeroulie C, Koumaravelou K, Srivastava A, Krishnamoorthy R. Endothelial cells do not express GSTA1: potential relevance to busulfan-mediated endothelial damage during haematopoietic stem cell transplantation. Eur J Haematol. 2008;80(4):299–302. https://doi.org/10.1111/j.1600-0609.2008.01031.x.
Gulbis AM, Culotta KS, Jones RB, Andersson BS. Busulfan and metronidazole: an often forgotten but significant drug interaction. Ann Pharmacother. 2011;45(7–8):e39. https://doi.org/10.1345/aph.1Q087.y
Myers AL, Kawedia JD, Champlin RE, Kramer MA, Nieto Y, Ghose R, et al. Clarifying busulfan metabolism and drug interactions to support new therapeutic drug monitoring strategies: a comprehensive review. Expert Opin Drug Metab Toxicol. 2017;13(9):901–23. https://doi.org/10.1080/17425255.2017.1360277.
Hassan M, Svensson JO, Nilsson C, Hentschke P, Al-Shurbaji A, Aschan J, et al. Ketobemidone may alter busulfan pharmacokinetics during high-dose therapy. Ther Drug Monit. 2000;22(4):383–5. https://doi.org/10.1097/00007691-200008000-00003.
Yasar U, Annas A, Svensson JO, Lazorova L, Artursson P, Al-Shurbaji A. Ketobemidone is a substrate for cytochrome P4502C9 and 3A4, but not for P-glycoprotein. Xenobiotica. 2005;35(8):785–96. https://doi.org/10.1080/00498250500183181.
Uppugunduri CRS, Rezgui MA, Diaz PH, Tyagi AK, Rousseau J, Daali Y, et al. The association of cytochrome P450 genetic polymorphisms with sulfolane formation and the efficacy of a busulfan-based conditioning regimen in pediatric patients undergoing hematopoietic stem cell transplantation. Pharmacogenom J. 2014;14(3):263–71. https://doi.org/10.1038/tpj.2013.38.
Palmer J, McCune JS, Perales MA, Marks D, Bubalo J, Mohty M, et al. Personalizing busulfan-based conditioning: considerations from the American society for blood and marrow transplantation practice guidelines committee. Biol Blood Marrow Transplant. 2016;22(11):1915–25. https://doi.org/10.1016/j.bbmt.2016.07.013.
Strouse C, Zhang Y, Zhang M-J, DiGilio A, Pasquini M, Horowitz MM, et al. Risk score for the development of veno-occlusive disease after allogeneic hematopoietic cell transplant. Biol Blood Marrow Transplant. 2018;24(10):2072–80. https://doi.org/10.1016/j.bbmt.2018.06.013.
McCune JS, Quinones CM, Ritchie J, Carpenter PA, van Maarseveen E, Yeh RF, et al. Harmonization of busulfan plasma exposure unit (BPEU): a community-initiated consensus statement. Biol Blood Marrow Transplant. 2019;25(9):1890–7. https://doi.org/10.1016/j.bbmt.2019.05.021.
Booth BP, Rahman A, Dagher R, Griebel D, Lennon S, Fuller D, et al. Population pharmacokinetic-based dosing of intravenous busulfan in pediatric patients. J Clin Pharmacol. 2007;47(1):101–11. https://doi.org/10.1177/0091270006295789.
Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44(10):1051–65. https://doi.org/10.2165/00003088-200544100-00004.
Okamoto Y, Nagatoshi Y, Kosaka Y, Kikuchi A, Kato S, Kigasawa H, et al. Prospective pharmacokinetic study of intravenous busulfan in hematopoietic stem cell transplantation in 25 children. Pediatr Transplant. 2014;18(3):294–301. https://doi.org/10.1111/petr.12236.
Brooks KM, Jarosinski P, Hughes T, Kang E, Shah NN, Gall JBL, et al. Test dose pharmacokinetics in pediatric patients receiving once-daily IV busulfan conditioning for hematopoietic stem cell transplant: a reliable approach? J Clin Pharmacol. 2017. https://doi.org/10.1002/jcph.1049.
Kishimoto K, Hasegawa D, Irie K, Okada A, Nakamura S, Tamura A, et al. Pharmacokinetic analysis for model-supported therapeutic drug monitoring of busulfan in Japanese pediatric hematopoietic stem cell transplantation recipients. Pediatr Transplant. 2020;24(4):e13696. https://doi.org/10.1111/petr.13696.
Le Gall JB, Milone MC, Waxman IM, Shaw LM, Harrison L, Duffy D, et al. The pharmacokinetics and safety of twice daily i.v. BU during conditioning in pediatric allo-SCT recipients. Bone Marrow Transplant. 2013;48(1):19–25. https://doi.org/10.1038/bmt.2012.105.
Mehta PA, Emoto C, Fukuda T, Seyboth B, Teusink-Cross A, Davies SM, et al. Busulfan pharmacokinetics and precision dosing: are patients with Fanconi anemia different? Biol Blood Marrow Transplant. 2019;25(12):2416–21. https://doi.org/10.1016/j.bbmt.2019.07.014.
Nath CE, Earl JW, Pati N, Stephen K, Shaw PJ. Variability in the pharmacokinetics of intravenous busulphan given as a single daily dose to paediatric blood or marrow transplant recipients. Br J Clin Pharmacol. 2008;66(1):50–9. https://doi.org/10.1111/j.1365-2125.2008.03166.x.
Nishikawa T, Yamaguchi H, Ikawa K. Influence of GST polymorphisms on busulfan pharmacokinetics in Japanese children [erratum]. Pediatr Int. 2019;61(8):841. https://doi.org/10.1111/ped.13985.
Wall DA, Chan KW, Nieder ML, Hayashi RJ, Yeager AM, Kadota R, et al. Safety, efficacy, and pharmacokinetics of intravenous busulfan in children undergoing allogeneic hematopoietic stem cell transplantation. Pediatr Blood Cancer. 2010;54(2):291–8. https://doi.org/10.1002/pbc.22227.
Behfar M, Koochakzadeh L, Yazdanian N, Salajegheh P, Rostami T, Khodayari-Namini N, et al. Outcome of allogeneic hematopoietic stem cell transplantation on diamond-Blackfan anemia using busulfan-based myeloablative regimen. Turk J Pediatr. 2019;61(3):407–12. https://doi.org/10.24953/turkjped.2019.03.013.
Beier R, Albert MH, Bader P, Borkhardt A, Creutzig U, Eyrich M, et al. Allo-SCT using BU, CY and melphalan for children with AML in second CR. Bone Marrow Transplant. 2013;48(5):651–6. https://doi.org/10.1038/bmt.2012.204.
Chiesa R, Cappelli B, Crocchiolo R, Frugnoli I, Biral E, Noe A, et al. Unpredictability of intravenous busulfan pharmacokinetics in children undergoing hematopoietic stem cell transplantation for advanced beta thalassemia: limited toxicity with a dose-adjustment policy. Biol Blood Marrow Transplant. 2010;16(5):622–8. https://doi.org/10.1016/j.bbmt.2009.11.024.
Gungor T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383(9915):436–48. https://doi.org/10.1016/s0140-6736(13)62069-3.
Hafez HA, Abdallah A, Hammad M, Hamdy N, Yassin D, Salem S, et al. Outcomes of allogenic hematopoietic cell transplantation for childhood chronic myeloid leukemia: single-center experience. Pediatr Transplant. 2020;24(4):e13664. https://doi.org/10.1111/petr.13664.
Harris AC, Boelens JJ, Ahn KW, Fei M, Abraham A, Artz A, et al. Comparison of pediatric allogeneic transplant outcomes using myeloablative busulfan with cyclophosphamide or fludarabine. Blood Adv. 2018;2(11):1198–206. https://doi.org/10.1182/bloodadvances.2018016956.
Ishida H, Kato M, Kudo K, Taga T, Tomizawa D, Miyamura T, et al. Comparison of outcomes for pediatric patients with acute myeloid leukemia in remission and undergoing allogeneic hematopoietic cell transplantation with myeloablative conditioning regimens based on either intravenous busulfan or total body irradiation: a report from the Japanese society for hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2015;21(12):2141–7. https://doi.org/10.1016/j.bbmt.2015.08.011.
Kato M, Takahashi Y, Tomizawa D, Okamoto Y, Inagaki J, Koh K, et al. Comparison of intravenous with oral busulfan in allogeneic hematopoietic stem cell transplantation with myeloablative conditioning regimens for pediatric acute leukemia. Biol Blood Marrow Transplant. 2013;19(12):1690–4. https://doi.org/10.1016/j.bbmt.2013.09.012.
Katsanis E, Sapp LN, Pelayo-Katsanis L, Whitney K, Zeng Y, Kopp LM. Alternative donor hematopoietic cell transplantation conditioned with myeloablative busulfan, fludarabine, and melphalan is well tolerated and effective against high-risk myeloid malignancies. J Pediatr Hematol Oncol. 2016;38(8):E315–8. https://doi.org/10.1097/mph.0000000000000621.
Law J, Cowan MJ, Dvorak CC, Musick L, Long-Boyle JR, Baxter-Lowe LA, et al. Busulfan, fludarabine, and alemtuzumab as a reduced toxicity regimen for children with malignant and nonmalignant diseases improves engraftment and graft-versus-host disease without delaying immune reconstitution. Biol Blood Marrow Transplant. 2012;18(11):1656–63. https://doi.org/10.1016/j.bbmt.2012.05.006.
Maheshwari S, Kassim A, Yeh RF, Domm J, Calder C, Evans M, et al. Targeted Busulfan therapy with a steady-state concentration of 600–700 ng/mL in patients with sickle cell disease receiving HLA-identical sibling bone marrow transplant. Bone Marrow Transplant. 2014;49(3):366–9. https://doi.org/10.1038/bmt.2013.188.
Oshrine B, Adams L, Nguyen ATH, Amankwah E, Shyr D, Hale G, et al. Comparison of melphalan- And busulfan-based myeloablative conditioning in children undergoing allogeneic transplantation for acute myeloid leukemia or myelodysplasia. Pediatr Transplant. 2020. https://doi.org/10.1111/petr.13672.
Sisler IY, Koehler E, Koyama T, Domm JA, Ryan R, Levine JE, et al. Impact of conditioning regimen in allogeneic hematopoetic stem cell transplantation for children with acute myelogenous leukemia beyond first complete remission: a pediatric blood and marrow transplant consortium (PBMTC) study. Biol Blood Marrow Transplant. 2009;15(12):1620–7. https://doi.org/10.1016/j.bbmt.2009.08.014.
Talleur AC, Triplett BM, Federico S, Mamcarz E, Janssen W, Wu J, et al. Consolidation therapy for newly diagnosed pediatric patients with high-risk neuroblastoma using busulfan/melphalan, autologous hematopoietic cell transplantation, anti-GD2 antibody, granulocyte-macrophage colony-stimulating factor, interleukin-2, and haploidentical natural killer cells. Biol Blood Marrow Transplant. 2017;23(11):1910–7. https://doi.org/10.1016/j.bbmt.2017.07.011.
Tesfaye H, Branova R, Klapkova E, Prusa R, Janeckova D, Riha P, et al. The importance of therapeutic drug monitoring (TDM) for parenteral busulfan dosing in conditioning regimen for hematopoietic stem cell transplantation (HSCT) in children. Ann Transplant. 2014;19:214–24. https://doi.org/10.12659/aot.889933.
Uemura S, Mori T, Ishiko S, Takafuji S, Nino N, Yamamoto N, et al. Retrospective analysis of high-dose chemotherapy followed by autologous stem cell transplantation for high-risk pediatric osteosarcoma. Pediatr Hematol Oncol. 2020;37(4):337–43. https://doi.org/10.1080/08880018.2020.1729906.
Wang JM, Luan Z, Jiang H, Fang JP, Qin MQ, Lee V, et al. Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis-A ten year report from the China children transplant group. Biol Blood Marrow Transplant. 2016;22(11):2104–8. https://doi.org/10.1016/j.bbmt.2016.08.015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used to assist in the preparation of this review.
Conflict of interest
Rachael Lawson, Christine E. Staatz, Christopher J. Fraser and Stefanie Hennig have no conflicts of interest that are relevant to the contents of this review.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
Code availability
Not applicable.
Author contributions
All authors contributed to the study conception and design. Material preparation and data collection was performed by Rachael Lawson. Data analysis was performed by Rachael Lawson, Stefanie Hennig, Christine Staatz and Chris Fraser. The first draft of the manuscript was written by Rachael Lawson and all authors commented on previous versions of the manuscript. All authors read and approved the final version of the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lawson, R., Staatz, C.E., Fraser, C.J. et al. Review of the Pharmacokinetics and Pharmacodynamics of Intravenous Busulfan in Paediatric Patients. Clin Pharmacokinet 60, 17–51 (2021). https://doi.org/10.1007/s40262-020-00947-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00947-2