
Abstract
Background and Objectives
The thrombin inhibitor dabigatran is administered as the prodrug dabigatran etexilate, which is a substrate of esterases and P-glycoprotein (P-gp). Dabigatran is eliminated via renal excretion but is also a substrate of uridine 5ʹ-diphospho (UDP)-glucuronosyltransferases (UGTs). The objective of this study was to build a physiologically based pharmacokinetic (PBPK) model comprising dabigatran etexilate, dabigatran, and dabigatran 1-O-acylglucuronide to describe the pharmacokinetics in healthy adults and renally impaired patients mechanistically.
Methods
Model development and evaluation were carried out using (i) physicochemical and absorption, distribution, metabolism, and excretion (ADME) parameter values of all three analytes; (ii) concentration–time profiles from 13 studies of healthy and renally impaired individuals after varying doses (0.1–300 mg), intravenous (dabigatran) and oral (dabigatran etexilate) administration, and different formulations of dabigatran etexilate (capsule, solution); and (iii) drug–drug interaction studies of dabigatran with the P-gp perpetrators rifampin (inducer) and clarithromycin (inhibitor).
Results
A PBPK model of dabigatran was successfully developed. The predicted area under the plasma concentration–time curve, trough concentration, and half-life values of the assessed clinical studies satisfied the two-fold acceptance criterion. Metabolic clearances of dabigatran etexilate and dabigatran were implemented using data on carboxylesterase 1/2 enzymes and UGT subtype 2B15. In severe renal impairment, the UGT2B15 metabolism and the P-gp transport in the model were reduced to 67% and 65% of the rates in healthy adults.
Conclusion
This is the first implementation of a PBPK model for dabigatran to distinguish between the prodrug, active moiety, and main active metabolite. Following adjustment of the UGT2B15 metabolism and P-gp transport rates, the PBPK model accurately predicts the pharmacokinetics in renally impaired patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A comprehensive physiologically based pharmacokinetic (PBPK) model of dabigatran etexilate, dabigatran, and dabigatran 1-O-acylglucuronide has been developed based on varying dabigatran (etexilate) doses, administration routes (intravenous, oral), and different formulations (capsule, solution) of dabigatran etexilate in healthy and renally impaired individuals. |
PBPK disease modeling allows mechanistic incorporation of hypothesized alterations in absorption, distribution, metabolism, and excretion (ADME) mechanisms such as reduced uridine 5ʹ-diphospho (UDP)-glucuronosyltransferase (UGT) metabolism and P-glycoprotein (P-gp) transport due to renal impairment. |
Independently developed PBPK models of the P-gp inducer rifampin and P-gp inhibitor clarithromycin were linked with the dabigatran PBPK model. The drug–drug interactions, primarily based on the interaction with intestinal P-gp, could be described (rifampin) and predicted (clarithromycin). |
1 Introduction
Dabigatran (Pradaxa®) is a synthetic, non-peptidic, potent, specific, competitive, and reversible inhibitor of thrombin that overcomes the major drawbacks of vitamin K antagonists. It is indicated for the treatment and prophylaxis of deep vein thrombosis (DVT) and pulmonary embolism (PE) in patients treated with a parenteral anticoagulant for 5–10 days or who have undergone hip replacement surgery. Moreover, dabigatran reduces the risks of (i) stroke and systemic embolism in patients with non-valvular atrial fibrillation and (ii) recurrence of DVT and PE in patients who have been previously treated [1].
Dabigatran is administered orally as the pharmacologically inactive prodrug and P-glycoprotein (P-gp) substrate dabigatran etexilate. Carboxylesterases (CESs) have been proposed to be involved in the two-step conversion of dabigatran etexilate to dabigatran in humans: CES1 in the liver and CES2 in the intestine [2, 3]. In healthy male volunteers the mean oral bioavailability of dabigatran is 6–7% [4] and the inter-individual variability in pharmacokinetic parameters has been described with an average inter-individual coefficient of variation (CV) of approximately 30% [5]. Maximum plasma concentrations (Cmax) of dabigatran occur approximately 2–3 h after oral dabigatran etexilate dosing and the disposition is bi-exponential. In renally healthy subjects, the half-life (t½) is 12–17 h and steady state is attained within 3 days of multiple dosing [5, 6]. The Cmax and the area under the plasma concentration–time curve (AUC) are dose proportional after dabigatran doses of 0.1–5.0 mg (single intravenous dose) and dabigatran etexilate doses of 50–400 mg (three times daily orally) [5, 7]. Following intravenous dabigatran administration, about 80% of the dose is eliminated via renal excretion [4]. Dabigatran itself has not been identified as a substrate of any transporter. Its main metabolite is the 1-O-acylglucuronide (dabigatran glucuronide), which in vitro data suggest is formed primarily by the uridine 5ʹ-diphospho (UDP)-glucuronosyltransferase (UGT) 2B15 [8]. Cytochrome P450 enzymes play a negligible role in the pharmacokinetic characteristics of dabigatran, which lowers the likelihood of drug–drug interactions (DDIs) [4]. Dabigatran glucuronide exhibits a similar concentration-dependent pharmacological activity as its parent compound dabigatran [8]. Dabigatran concentrations are measured either as non-conjugated (‘free’) dabigatran or as the total (‘sum’) of non-conjugated dabigatran and dabigatran acylglucuronides, which account for 3% or 20% of the total exposure to dabigatran following intravenous or oral dosing, respectively [4].
As dabigatran is mainly eliminated via urinary excretion, elderly and patients with chronic kidney disease have higher dabigatran plasma concentrations when given the same dose as healthy adults. Clinical studies of dabigatran in patients with different stages of renal impairment showed that the exposure of dabigatran increases in correlation with the severity of renal dysfunction [9,10,11].
Overall, dabigatran shows favorable and predictable linear pharmacokinetics in healthy adults and renally impaired patients. So far, the pharmacokinetic properties of dabigatran have been intensively characterized in empirical population pharmacokinetic modeling analyses considering only ‘sum’ dabigatran concentration data [12,13,14,15]. An attempt to include all three analytes (dabigatran etexilate, dabigatran, and dabigatran glucuronide) separately in a physiologically based pharmacokinetic (PBPK) model has not been undertaken previously. Such a model may incorporate physiological differences between patient populations (healthy, renally impaired, children) that lead to population-specific pharmacokinetic characteristics of the analytes.
1.1 Objectives
The objectives of this study were as follows:
- 1.
To build a whole-body PBPK model of the prodrug dabigatran etexilate, the active moiety dabigatran, and its pharmacologically active metabolite dabigatran glucuronide in healthy adults.
- 2.
To apply this model to describe the pharmacokinetics of dabigatran in renally impaired patients, exploring the potential influence of protein binding, glomerular filtration, and P-gp and UGT2B15 activity due to various degrees of renal impairment.
2 Methods
2.1 Model Development and Evaluation
A schematic overview of the PBPK model development is shown in Fig. 1 and the detailed steps of model building are outlined in the following sections. In brief, PBPK modelling was performed in a stepwise procedure. First, a PBPK model of dabigatran alone following intravenous application of dabigatran was developed. Second, this intravenous dabigatran model was expanded by adding the prodrug dabigatran etexilate (oral administration form) and the metabolite dabigatran glucuronide to establish a PBPK model for all three analytes following oral application of dabigatran etexilate. This step includes the incorporation of the P-gp transport of dabigatran etexilate using DDI study data with rifampin and clarithromycin. It was assumed that both CES1 and CES2 can directly cleave the two ester groups of dabigatran etexilate to release dabigatran. Third, the impact of renal impairment was implemented into the PBPK model. Finally, sensitivity analyses were performed to investigate the impact of changes in parameter estimates on the AUC during a dosing interval at steady state (AUCτ,ss) and the trough concentration (Ctrough).

Workflow of the physiologically based pharmacokinetic model development and evaluation from healthy adults to renally impaired patients. ADME absorption, distribution, metabolism, and excretion, DDI drug–drug interaction, GFR glomerular filtration rate, kcat enzymatic rate constant, PBPK physiologically based pharmacokinetic, PK pharmacokinetic, UGT uridine 5ʹ-diphospho (UDP)-glucuronosyltransferase
Experimental datasets from 13 clinical studies were used for PBPK model development in healthy adults and patients with different stages of renal impairment. All studies were reviewed by the relevant regulatory authorities. These studies were split into an internal model development and an external model evaluation dataset with six internal and seven external studies, respectively (Table 1). Physicochemical parameters and information on the absorption, distribution, metabolism, and excretion (ADME) of the three analytes dabigatran etexilate, dabigatran, and dabigatran glucuronide were gathered from the literature or in-house reports (Table 2). Parameter optimizations were carried out using either a Monte-Carlo or Simplex method [16, 17]. For the single process where the KM value had to be optimized (P-gp), a stepwise change of KM with subsequent optimization of the remaining parameters was performed until the best model was found.
Model evaluation was carried out (i) using visual predictive checks; (ii) by comparison of observed and simulated pharmacokinetic parameters and urinary excretion data; and (iii) by prediction of DDIs. Pharmacokinetic parameter comparisons were deemed successful if they satisfied the two-fold acceptance criterion [18, 19] or, for DDI prediction, if they satisfied the acceptance limits proposed by Guest et al. [20]. Mathematical implementation of DDIs, DDI modelling, and evaluation is detailed in the Electronic Supplementary Material (ESM).
Population simulations were performed for all model evaluation steps. Virtual populations were created according to the ranges of age, weight, and height described in the clinical studies (Table 1) [21]. To assess the inter-study variability of pharmacokinetic parameters, ten virtual populations were created for each study using the reported number of study individuals of each study. Dabigatran etexilate doses were administered in a fasted state in all studies used for PBPK model development. Dosing regimens and creatinine clearance (CLCR) values were incorporated into the model as listed in the clinical study reports. The CLCR was used as an estimator of the glomerular filtration rate (GFR) and was calculated using the Cockcroft-Gault equation [22].
Sensitivity analyses were generated in PK-Sim® using a variation range of 1.0 and the maximum number of nine steps [16]. A detailed description of the sensitivity calculation can be found in the ESM.
PBPK simulations and parameter optimizations were carried out in PK-Sim® and MoBi® version 6.0.3; sensitivity analyses and quality check were performed in version 7.2.0 (Bayer Technology Services, Leverkusen, Germany). Statistical analysis of the results and graphics were compiled using MATLAB® (version R2013b, The MathWorks Inc., Natick, MA, USA).
2.1.1 Intravenous Application of Dabigatran
Dabigatran plasma and urine concentrations obtained following intravenous administration of dabigatran were used to establish a PBPK model (intravenous PBPK model) for the active moiety dabigatran, including a description of its distribution, metabolism, and excretion [4, 7]. To describe the distribution phase correctly, the cellular permeability of dabigatran needed to be estimated. Tissue-to-plasma water partition coefficients for dabigatran were calculated using the method of Rodgers and Rowland [23,24,25]. The blood-to-plasma ratio was taken from the literature. The UGT2B15 liver reference concentration was assumed to be 2.05 µmol/L [26, 27]. The contribution of other UGTs was assumed to be negligible. The PK-Sim® gene expression database was used to implement organ-specific expressions of all enzymes and transporters. The plasma and urine concentrations were used to adjust the enzymatic rate constant (kcat) of the UGT2B15 metabolism, which was described by Michaelis–Menten kinetics. Adjustment was necessary as the model was not able to describe the plasma and urine concentrations using the initial value. The turnover number kcat is the maximum metabolic rate (Vmax) divided by the enzyme concentration. The obtained parameter values of dabigatran were fixed for all further model development steps (Table 2). Excretion of unchanged dabigatran into feces accounts for a negligible 1.9% of the dose following intravenous administration and was therefore not considered in the model [4]. The PK-Sim® default passive drug transport from plasma to urine (Tplasma-urine) in the kidneys [28] was altered in MoBi® to allow direct input of individual CLCR values (Eq. 1):
where fu is the unbound fraction of dabigatran, CLCRi is the individual observed creatinine clearance, GFRfraction is the fraction of GFR with which a drug is passively excreted into urine, and Cplasma,kidney is the plasma drug concentration in the kidney. This equation applies equally to dabigatran etexilate and dabigatran glucuronide.
2.1.2 Oral Application of Dabigatran Etexilate
The developed intravenous PBPK model of dabigatran was expanded by including its prodrug dabigatran etexilate (oral administration form) and the major metabolite of dabigatran, namely dabigatran glucuronide, which is formed by UGT2B15 metabolism (oral PBPK model). Clinical studies assessing the pharmacokinetics of the etexilate and the glucuronide individually following intravenous administration of these compounds were not available. Tissue-to-plasma water partition coefficients for dabigatran etexilate and dabigatran glucuronide were also calculated using the method of Rodgers and Rowland [23,24,25]. Parameters for dabigatran etexilate and dabigatran glucuronide were optimized to improve the description of free dabigatran and total dabigatran plasma concentrations following oral administrations of dabigatran etexilate with and without rifampin coadministration [29,30,31]. In MoBi®, total dabigatran concentrations were calculated as the sum of dabigatran and dabigatran glucuronide using Eq. 2:
where DAB(SUM) denotes total dabigatran, DAB the unconjugated dabigatran, DAB-GLUC the dabigatran glucuronide, and MW(DAB) the molecular weight of dabigatran of 471.51 g/mol. The reference concentrations for CES1, CES2, and P-gp were set to 54.93, 19.81, and 1 µmol/L, respectively [27, 32,33,34]. For CES1 it has been found that the single nucleotide polymorphism (SNP) rs2244613 in the CES1 gene is associated with a decrease of the Ctrough values of total dabigatran by 15% and 28% for one or two minor alleles, respectively [35]. For the purpose of simulations, it has been assumed that two minor alleles of CES1 (liver) would equal a loss of function of the CES1 enzyme, setting the CES1 kcat value to 0 in the model so that dabigatran etexilate is solely metabolized to dabigatran by CES2 (intestine). The CES1/2 metabolism and the P-gp transport were implemented using Michaelis–Menten kinetics. Some physicochemical and ADME parameters needed for PBPK modeling of dabigatran glucuronide have not been reported. Due to large structural similarities between dabigatran and dabigatran glucuronide, the unavailable parameter values for fu and the acid dissociation constant (pKa) were assumed to be equal to the dabigatran values. Further, the glucuronide was assumed to be solely renally cleared by GFR and tubular secretion (modelled by allowing the apparent GFRfraction to be > 1 in Eq. 1).
2.1.3 Oral Application of Dabigatran Etexilate in Renal Impairment
The oral PBPK model of dabigatran etexilate, dabigatran, and dabigatran glucuronide was subsequently expanded to predict plasma concentration–time profiles of the analytes in patients with different stages of renal impairment. The considered key physiological parameters were GFR, metabolic enzymes, and albumin concentration change with the degree of renal impairment [36]. The CLCR value was used in the simulations as stated in the study reports. The fu values of dabigatran etexilate, dabigatran, and dabigatran glucuronide in plasma for different stages of renal impairment (fui), based on the concentration of albumin in the plasma ([Pi]), were estimated using Eq. 3:
where [P] is the average concentration of albumin in plasma and fu is the fraction unbound in the healthy population [36, 37]. The intestinal P-gp activity was reduced to 65% for patients with severe renal impairment (CLCR ≤ 30 mL/min), in accordance with literature reports in rats [38,39,40]. With CLCR, fui, and P-gp contributions set to values as already described, the potential contribution of UGT2B15 kcat was estimated for patients with severe renal impairment based on a clinical study in patients with different stages of renal disease [9].
3 Results
A comprehensive PBPK model for the prediction of dabigatran concentrations after different intravenous doses of dabigatran or oral doses (capsule or solution) of dabigatran etexilate has been developed. The predicted AUC, Ctrough, and t½ values of the internal and external study datasets satisfy the two-fold acceptance criterion as shown in Fig. 2 and ESM Table 1. The final model parameters are presented in Table 2.

Observed versus predicted pharmacokinetic parameters of total dabigatran (left: linear scale, right: log scale): a AUC, bCtrough, and c half-life. Squares represent intravenous studies in healthy subjects; triangles represent oral studies in healthy subjects; circles represent oral studies in renally impaired patients. Blue markers show the internal dataset studies; red markers show the external dataset studies; and the size of the markers correlates to the number of individuals per study. AUC area under the plasma concentration–time curve, Ctrough trough concentration
3.1 Intravenous Application of Dabigatran
The model describes intravenous concentration–time profiles in the internal data set well (Fig. 3a), with only 3% over-prediction of Cmax (observed Cmax: 234 ng/mL, predicted Cmax: 241 ng/mL) and t½ (observed t½: 8.04 h, predicted t½: 8.29 h). After intravenous administration of 5.0 mg, a slight over-prediction of dabigatran concentrations is visible between 6 and 24 h. The urinary fraction excreted of dabigatran after 72 h is well-described (Fig. 3b). The observed mean fraction excreted in urine is 76.7% with minimum and maximum values of 57.6% and 90.6%. The predicted mean fraction excreted in comparison is 70.3% with minimum and maximum values of 47.5% and 88.5%. External dataset profiles after intravenous doses of 0.1, 1.0, and 5.0 mg are equally well-predicted (ESM Fig. 1). The t½ of the 0.1 mg dose group (4.31) is under-predicted (3.41) by 21%, which might be due to the relatively small number of measured dabigatran plasma concentrations in the terminal elimination phase.

Individually observed (dots) and population simulated (lines) plasma concentration–time profiles for dabigatran etexilate (c [29]), dabigatran (a [7], d [29]), and total dabigatran (e). b The fraction of dose excreted in urine following a 30 min intravenous infusion of 5.0 mg [7]. f All analytes after dabigatran etexilate 110 mg (twice daily) in a typical individual. Simulated data are represented as median (black lines), 90% prediction interval (grey shaded areas), and minimum and maximum values (dotted lines). bid twice daily, conc. concentration, SUM total
3.2 Oral Application of Dabigatran Etexilate
The PBPK model shows good descriptive and predictive performance for all internal and external clinical study simulations (Fig. 3c–e and ESM Fig. 2). The geometric mean AUCτ,ss values are slightly under-predicted by 2% and 10% for 110 and 150 mg twice daily, respectively, and the geometric mean minimum concentration (Cmin) in a dosing interval at steady state is slightly over-predicted by 16% and 4% for 110 and 150 mg twice daily, respectively. The deviations of predicted from observed geometric mean Ctrough values range from – 4% to + 19%, with one outlier of + 33%. The predicted and observed fractions of dose excreted in urine as unchanged dabigatran, following oral administration of dabigatran etexilate, are comparable, with 5% predicted and 4% observed [4]. To capture the lower Cmax after administration of capsule compared to solution formulations (ESM Fig. 3), the capsule dissolution is described using a Weibull function [41]. The time to set 50% of dabigatran etexilate free has been estimated to be 1.1 min (standard error (SE) = 0.2%) and the dissolution shape factor has been estimated at 0.26 (SE = 0.003%), which means that the capsule needs 1.6 h to set 90% of the dose free for absorption. The reported absolute bioavailability of dabigatran of 6.0% (range 3.0–7.0%) is closely reproduced with the oral PBPK model, which predicts a bioavailability of 7.1% [1, 4]. The model is furthermore able to describe the fraction of total dabigatran in plasma that the glucuronide accounts for (20% observed, 23% predicted) [4]. A decreased CES1 function (due to two minor CES1 alleles) was found to be associated with 28% lower total dabigatran Ctrough values in vivo, in which case the model predicts a 27% decrease (inactive CES1). To increase the confidence in the model, the Ctrough and the 2 h post-dose plasma concentration at steady state (C2,ss) of the RE-LY (Randomized Evaluation of Long-term Anticoagulant Therapy) trial (n = 9522 patients) have been simulated, assuming no co-medications for the simulations, and compared to observed values (ESM Table 2). The median Ctrough is slightly over-predicted by 6.4% for the 110 and 150 mg treatments. The C2,ss is slightly under-predicted by – 7.9% for the 110 mg twice daily treatment and over-predicted by + 8.8% for the 150 mg twice-daily treatment. A simulation of the concentration–time profiles of the three analytes and total dabigatran after oral dosing (twice daily) is schematically shown in Fig. 3f.
3.3 Oral Application of Dabigatran Etexilate in Renal Impairment
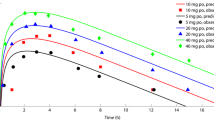
The final parameter values for the prediction of renally impaired patients are given in Table 3. Applying calculated values for fu and reported values for P-gp activity and CLCR, only the UGT2B15 kcat needed to be estimated for severe renal impairment. The development dataset’s concentration–time profiles and the fractions of dose excreted in urine after a single dose of dabigatran etexilate 150 mg are very well captured (Fig. 4). While the exposure (AUC and Cmax) increases significantly with decreasing renal function, the fraction of dose excreted unchanged in urine stays constant within different groups of renal impairment at approximately 2.4% (CV = 50%), ranging from 0.6% to 4.9% within individuals. The external dataset, a multiple-dose study of 75 mg twice daily in a patient population with severely impaired renal function, is successfully predicted (Fig. 5a, b). The predicted total dabigatran values of AUCτ,ss (2355 ng·h/mL), Ctrough (176 ng/mL), and Cmax (214 ng/mL) are in close agreement with the observed values of AUCτ,ss (2140 ng·h/mL), Ctrough (187 ng/mL), and Cmax (207 ng/mL) (ESM Table 1). GFR, UGT2B15, and P-gp impact (i) the shape of the concentration–time profiles and (ii) pharmacokinetic parameters such as AUC, Ctrough, Cmax, and t½ differently (Fig. 5c). For AUC and Ctrough, the parameter ranking from highest to smallest impact is GFR > P-gp > UGT2B15, for Cmax it is P-gp > GFR > UGT2B15, and for t½ it is GFR > UGT2B15 > P-gp.

Individually observed (dots) and population simulated (lines) plasma concentration–time profiles for total dabigatran (left) and fractions of dose excreted in urine as dabigatran (right) after oral administration of 150 mg [9]. a Healthy renal function, b mild renal impairment, c moderate renal impairment, and d severe renal impairment. Simulated data are represented as median (black lines), geometric mean (dashed-dotted lines), 90% prediction interval (grey shaded areas), and minimum and maximum values (dotted lines). CLCR creatinine clearance, conc. concentration, sd single dose, SUM total

a Individually observed (dots) and population simulated (lines) plasma concentration–time profiles for total dabigatran after twice-daily oral administration of 75 mg [10]. Simulated data are represented as median (black lines), 90% prediction interval (grey shaded areas), and minimum and maximum values (dotted lines). b Unconjugated dabigatran: solid line (predicted) and circles (observed), total dabigatran: dashed line (predicted) and triangles (observed). c Impact of parameter alterations in the renal impairment model on single- (150 mg, left) and multiple-dose (75 mg, right) dabigatran concentration–time profiles for a typical individual. bid twice daily, CLCR creatinine clearance, conc. concentration, GFR glomerular filtration rate, kcat enzymatic rate constant, P-gp P-glycoprotein, sd single dose, SUM total, UGT uridine 5ʹ-diphospho (UDP)-glucuronosyltransferase
3.4 Drug–Drug Interaction Simulations
To assess the impact of the estimated P-gp KM and kcat values and to test the applicability of the model, a DDI description with rifampin and a prediction with clarithromycin have been performed. The predicted DDI AUCratios (dabigatran AUCwith perpetrator/dabigatran AUCcontrol) following induction (rifampin, Fig. 6a) and during competitive inhibition (clarithromycin, Fig. 6b, c) of P-gp are in good agreement with the observed values and meet the DDI acceptance criterion (Fig. 6d). The predicted DDI AUCratios are 0.42 (rifampin) and 1.7 (clarithromycin) compared to the observed DDI AUCratios of 0.33 (rifampin) and 1.6 (clarithromycin). Thus, the ratios of predicted divided by observed DDI AUCratios (predicted DDI AUCratio/observed DDI AUCratio) for rifampin and clarithromycin are 1.27 and 1.06, respectively.

Drug–drug interaction predictions. a Geometric mean observed (filled circles) and simulated (line) plasma concentration–time profiles of unconjugated dabigatran following oral administration of dabigatran etexilate 150 mg and rifampin 600 mg (dosing indicated by black triangles) [31]. b Left: individually observed (dots) and population simulated (lines) plasma concentration–time profiles of unconjugated dabigatran following oral administration of dabigatran etexilate 300 mg. Right: unconjugated dabigatran following oral administration of dabigatran etexilate 300 mg with a concomitant dose of clarithromycin 500 mg [63]. Simulated data are represented as median (black lines), 90% prediction interval (grey shaded areas), and minimum and maximum values (dotted lines). c Mean unconjugated dabigatran following oral administration of dabigatran etexilate 300 mg with (prediction: dashed line, observed: triangles) and without (prediction: solid line, observed: circles) a concomitant dose of clarithromycin 500 mg. d Observed versus predicted AUCratios with the curved dotted acceptance limits according to Guest et al. [20], and the straight dashed lines showing the two-fold acceptance limits. The triangle represents the dabigatran-rifampin DDI; the circle represents the dabigatran-clarithromycin DDI. bid twice daily, conc. concentration, DDI drug–drug interaction, qd once daily, sd single dose
3.5 Parameter Sensitivity Analysis
The parameter sensitivity analysis (ESM Fig. 4) shows that, among the evaluated parameters, P-gp kcat and the intestinal permeability of dabigatran etexilate have the highest impact on the predicted AUCτ,ss of dabigatran. The UGT2B15 kcat has the highest impact on the predicted Ctrough of dabigatran. The CES1 kcat, the cellular permeabilities, and the GFR fraction of dabigatran glucuronide have low or zero impact on AUCτ,ss and Ctrough.
4 Discussion
For the first time a comprehensive PBPK model of dabigatran etexilate, dabigatran, and dabigatran glucuronide has been developed that is able to accurately predict the concentration–time profiles of all compounds after different intravenous or oral doses (capsule or solution). Important ADME mechanisms have been implemented, enabling application of the model for healthy individuals as well as patients with different stages of renal impairment. The developed oral renal impairment model attempts to differentiate and describe the potential impact of changes in GFR, fu, P-gp, and UGT2B15 in different stages of renal disease. Furthermore, the final model has been applied for the simulation of DDIs with two different perpetrators, showing excellent performance as a P-gp victim drug.
Although our goal was to fix as many parameters to literature values as possible, some values needed to be estimated to establish this complex model. Comparison of estimated and literature values in Table 2 shows differences in the cellular permeabilities of all three modeled compounds, in the intestinal permeability of dabigatran etexilate, and in the kcat and KM values of P-gp. The estimated cellular permeabilities are higher than the values calculated within PK-Sim®. This could indicate that the literature lipophilicity values used in the model are too low to accurately describe the lipid membrane affinity of the three compounds. A second possibility might be that there are unknown transport processes missing in the model, especially for the dabigatran glucuronide. However, dabigatran etexilate is described as a selective probe drug for P-gp inhibition, unaffected by other transport processes [42]. The lower estimated intestinal permeability of dabigatran etexilate might reflect differences between the Caco-2 assay used to determine the literature value [2] and the human in vivo situation. The literature P-gp reference concentration (see Sect. 2) and KM value in Table 2 are predicted values [33] without experimental evidence. As the P-gp kcat in the model equals the P-gp Vmax normalized by the P-gp reference concentration, misspecification of the reference concentration (or of Vmax) is compensated by optimization of the kcat, but probably creates the observed discrepancy between estimated and literature P-gp kcat values in Table 2.
The presented model has a wider application and validation range than a recently published PBPK model for dabigatran [33] that was based on a limited dataset for model development. Furthermore, the model’s capability to capture the fraction of administered dabigatran etexilate dose excreted in urine as dabigatran is not demonstrated. The glucuronide, the CES2 metabolism, and the differentiation between formulations are not included. Also, no information is given on the performance of population simulation. Finally, the DDI predictions are static and not dynamic. The model presented here overcomes these limitations: a larger number of clinical studies were used with clearly defined internal and external datasets, the fraction of dose excreted in urine after intravenous and oral administration is well-predicted, the glucuronide, the CES2 metabolism in the intestine, and a differentiation between capsule and solution formulation are included and can be simulated with the model. Furthermore, model performance was assessed in virtual populations and DDI studies were predicted dynamically.
The presented modelling approach also has limitations. To reduce the uncertainty of the parameter estimates, concentration–time profiles after intravenous administration of dabigatran etexilate and dabigatran glucuronide would be needed. For the glucuronide, measurements of fu and pKa would be helpful. It has been assumed that the fu value of the glucuronide was equal to the value of the parent compound dabigatran, which is reasonable comparing other compounds and their glucuronides [43,44,45,46]. For the glucuronide modelling it has further been assumed that its excretion rate to urine could exceed the GFR. As with glucuronides of other drugs, biliary clearance, hydrolysis, or isomerization might also contribute to its elimination. Unfortunately, there was not enough information on all processes possibly involved for a more mechanistic implementation. Since dabigatran pharmacokinetics are influenced by kidney function it was decided that the glucuronide GFRfraction should be > 1. The fitted renal clearance of the glucuronide is 6.36-fold higher than the GFR. This is a reasonable value as, in general, the renal clearance of glucuronides exceeds that of their parent compounds (e.g., acetaminophen 15.8-fold, furosemide 4.3-fold, ketoprofen 2.5-fold, zomepirac 11.3-fold) [43, 47,48,49,50,51,52,53].
In the renal impairment model it has been assumed that intestinal P-gp activity is reduced to 65% in severe renal impairment [38, 39]. This value is based on measurements in rats with end-stage renal disease, which might differ from human P-gp activity alterations in renal dysfunction. It has been further assumed that the metabolic clearance via UGT2B15 is reduced due to renal impairment. For other drugs it has been reported that uremic toxins accumulating during renal impairment can inhibit UGT metabolism at concentrations found in vivo [40, 54,55,56]. Another mechanism potentially responsible for reduced metabolism by UGT is the ‘futile cycle’ of glucuronides, allowing glucuronides at increased concentrations to be hydrolyzed back to the parent compound [47, 57]. Concerning the fu values of dabigatran in the renally impaired populations, it is important to point out that the calculated values are not relevantly different, which is in agreement with measured fu values in renal impairment. Apart from the implemented alterations, there are other physiological parameters that could potentially impact the pharmacokinetics due to renal dysfunction, such as hematocrit, gastric pH, gastric emptying time, and renal blood flow [58,59,60]. However, the extent and exact mechanisms of these changes is not fully understood, and since none of these parameters significantly increased the AUC or t½ of dabigatran, the corresponding parameter values were not varied in the model [61, 62].
The parameter sensitivity analysis shows that the oral PBPK model is sensitive to the P-gp kcat value. As both, the P-gp kcat and KM values were estimated, successful prediction of the DDIs with well-established P-gp perpetrator models of rifampin and clarithromycin was important to increase the confidence in the correct implementation of the P-gp transport.
In general, future work should strive for a better mechanistic understanding and modelling of the pharmacokinetics in renally impaired patients. GFR and plasma albumin changes during the different stages of chronic kidney disease are well-documented, but it is unclear how the activities of renal enzymes and transporters depend on the patient’s renal function. Furthermore, the identity of all uremic toxins that accumulate during renal impairment is unknown, as is their qualitative and quantitative impact on drug-metabolizing enzymes and transporters. Therefore, comprehensive research is needed to gain new insights into the physiological changes that arise due to chronic kidney dysfunction and to transfer this knowledge into PBPK models. Future studies of dabigatran could use the presented PBPK model in systems pharmacology models of the human blood coagulation network or in the field of DDI simulations, where the model can be applied to predict the impact of investigational drugs on dabigatran via P-gp inhibition or induction. Finally, the model could be expanded for the prediction of dabigatran in pediatric individuals.
5 Conclusion
We developed a comprehensive PBPK model of dabigatran etexilate, dabigatran, and dabigatran glucuronide that is able to describe and predict concentration–time profiles following a broad range of dabigatran (etexilate) doses, administration routes, and formulations in healthy and renally impaired individuals. The model was applied in DDI simulations with the P-gp inducer rifampin and the P-gp inhibitor clarithromycin. The model hypothesizes that, in addition to a reduced GFR, the P-gp transport of dabigatran etexilate and the UGT2B15 metabolism of dabigatran are inhibited due to (severe) renal dysfunction.
References
Boehringer Ingelheim Pharmaceuticals Inc. Full prescribing information: Pradaxa, 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022512s024lbl.pdf. Accessed 29 Oct 2018.
Ishiguro N, Kishimoto W, Volz A, Ludwig-Schwellinger E, Ebner T, Schaefer O. Impact of endogenous esterase activity on in vitro p-glycoprotein profiling of dabigatran etexilate in caco-2 monolayers. Drug Metab Dispos. 2013;42:250–6. https://doi.org/10.1124/dmd.113.053561.
Laizure SC, Parker RB, Herring VL, Hu Z-Y. Identification of carboxylesterase-dependent dabigatran etexilate hydrolysis. Drug Metab Dispos. 2013;42:201–6. https://doi.org/10.1124/dmd.113.054353.
Blech S, Ebner T, Ludwig-Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos. 2008;36:386–99. https://doi.org/10.1124/dmd.107.019083.
Stangier J, Rathgen K, Stähle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol. 2007;64:292–303. https://doi.org/10.1111/j.1365-2125.2007.02899.x.
Stangier J, Stähle H, Rathgen K, Fuhr R. Pharmacokinetics and pharmacodynamics of the direct oral thrombin inhibitor dabigatran in healthy elderly subjects. Clin Pharmacokinet. 2008;47:47–59. https://doi.org/10.2165/00003088-200847010-00005.
Boehringer Ingelheim Clinical Study Synopsis for Public Disclosure - BI Trial No. 1160.5. Tolerability of single rising doses of 0.1 mg, 1 mg, and 5 mg BIBR 953 ZW IV (placebo-controlled in each dose group; substudy 1) and absolute and relative bioavailability of 100 mg BIBR 1048 tablet and of solution and of 1 mg or 5 mg BIBR 953 ZW IV, 2001. https://trials.boehringer-ingelheim.com/public/trial_results_documents/1160/1160.5_U01-1807.pdf. Accessed 29 Oct 2018.
Ebner T, Wagner K, Wienen W. Dabigatran acylglucuronide, the major human metabolite of dabigatran: In vitro formation, stability, and pharmacological activity. Drug Metab Dispos. 2010;38:1567–75. https://doi.org/10.1124/dmd.110.033696.
Stangier J, Rathgen K, Stähle H, Mazur D. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate: an open-label, parallel-group, single-centre study. Clin Pharmacokinet. 2010;49:259–68. https://doi.org/10.2165/11318170-000000000-00000.
Kooiman J, Van Der Hulle T, Maas H, Wiebe S, Formella S, Clemens A, et al. Pharmacokinetics and pharmacodynamics of dabigatran 75 mg b.i.d. in patients with severe chronic kidney disease. J Am Coll Cardiol 2016;67:2442–4. https://doi.org/10.1016/j.jacc.2016.03.516.
Prins MH, Lensing AWA, Bauersachs R, van Bellen B, Bounameaux H, Brighton TA, et al. Oral rivaroxaban versus standard therapy for the treatment of symptomatic venous thromboembolism: a pooled analysis of the EINSTEIN-DVT and PE randomized studies. Thromb J. 2013;11:21. https://doi.org/10.1186/1477-9560-11-21.
Trocóniz IF, Tillmann C, Liesenfeld K-H, Schäfer H-G, Stangier J. Population pharmacokinetic analysis of the new oral thrombin inhibitor dabigatran etexilate (BIBR 1048) in patients undergoing primary elective total hip replacement surgery. J Clin Pharmacol. 2007;47:371–82. https://doi.org/10.1177/0091270006297228.
Liesenfeld K-H, Lehr T, Dansirikul C, Reilly PA, Connolly SJ, Ezekowitz MD, et al. Population pharmacokinetic analysis of the oral thrombin inhibitor dabigatran etexilate in patients with non-valvular atrial fibrillation from the RE-LY trial. J Thromb Haemost. 2011;9:2168–75. https://doi.org/10.1111/j.1538-7836.2011.04498.x.
Dansirikul C, Lehr T, Liesenfeld K-H, Haertter S, Staab a. A combined pharmacometric analysis of dabigatran etexilate in healthy volunteers and patients with atrial fibrillation or undergoing orthopaedic surgery. Thromb Haemost 2012;107:775–85. https://doi.org/10.1160/TH11-09-0656.
Liesenfeld K-H, Staab A, Härtter S, Formella S, Clemens A, Lehr T. Pharmacometric characterization of dabigatran hemodialysis. Clin Pharmacokinet. 2013;52:453–62. https://doi.org/10.1007/s40262-013-0049-6.
Open Systems Pharmacology Suite manual, version 7.4. 2018. https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/Open-Systems-Pharmacology-Suite.pdf. Accessed 22 Feb 2019.
Nelder JA, Mead R. A simplex method for function minimization. Comput J. 1965;7:308–13.
Abduljalil K, Cain T, Humphries H, Rostami-Hodjegan A. Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: an analysis based on in vivo observations. Drug Metab Dispos. 2014;42:1478–84. https://doi.org/10.1124/dmd.114.058099.
Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97:247–62. https://doi.org/10.1002/cpt.37.
Guest EJ, Aarons L, Houston JB, Rostami-Hodjegan A, Galetin A. Critique of the two-fold measure of prediction success for ratios: application for the assessment of drug-drug interactions. Drug Metab Dispos. 2011;39:170–3. https://doi.org/10.1124/dmd.110.036103.
Willmann S, Höhn K, Edginton A, Sevestre M, Solodenko J, Weiss W, et al. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34:401–31. https://doi.org/10.1007/s10928-007-9053-5.
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41.
Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci. 2005;94:1259–76. https://doi.org/10.1002/jps.20322.
Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95:1238–57. https://doi.org/10.1002/jps.20502.
Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24:918–33. https://doi.org/10.1007/s11095-006-9210-3.
Achour B, Russell MR, Barber J, Rostami-Hodjegan A. Simultaneous quantification of the abundance of several cytochrome P450 and uridine 5’-diphospho-glucuronosyltransferase enzymes in human liver microsomes using multiplexed targeted proteomics. Drug Metab Dispos. 2014;42:500–10. https://doi.org/10.1124/dmd.113.055632.
Wilson ZE, Rostami-Hodjegan A, Burn JL, Tooley A, Boyle J, Ellis SW, et al. Inter-individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. Br J Clin Pharmacol. 2003;56:433–40.
Claassen K, Willmann S, Eissing T, Preusser T, Block M. A detailed physiologically based model to simulate the pharmacokinetics and hormonal pharmacodynamics of enalapril on the circulating endocrine renin-angiotensin-aldosterone system. Front Physiol. 2013;4:4. https://doi.org/10.3389/fphys.2013.00004.
Boehringer Ingelheim clinical study synopsis for public disclosure - BI trial no. 1160.61. Pharmacokinetics, safety and pharmacodynamics after multiple oral doses of dabigatran etexilate capsule (110 mg and 150 mg b.i.d., 7 days) in healthy Japanese and Caucasian male subjects (open label study), 2006. https://trials.boehringer-ingelheim.com/public/trial_results_documents/1160/1160.61_U06-3420.pdf. Accessed 29 Oct 2018.
Huang F, Wiebe S, Jungnik A, Gropper S, Brueckmann M, Hohl K, et al. Assessment of the relative bioavailability of dabigatran etexilate as pellets on food and as granules resolved in reconstitution solution, compared with dabigatran etexilate as hard capsules [poster no. 008]. Clin Pharmacokinet Pharmacodyn 2017;6 Suppl. 1. https://doi.org/10.1002/cpdd.385.
Härtter S, Koenen-Bergmann M, Sharma A, Nehmiz G, Lemke U, Timmer W, et al. Decrease in the oral bioavailability of dabigatran etexilate after co-medication with rifampicin. Br J Clin Pharmacol. 2012;74:490–500. https://doi.org/10.1111/j.1365-2125.2012.04218.x.
Boberg M, Vrana M, Mehrotra A, Pearce RE, Gaedigk A, Bhatt DK, et al. Age-dependent absolute abundance of hepatic carboxylesterases (CES1 and CES2) by LC-MS/MS proteomics: application to PBPK modeling of oseltamivir in vivo pharmacokinetics in infants. Drug Metab Dispos. 2017;45:216–23. https://doi.org/10.1124/dmd.116.072652.
Zhao Y, Hu Z-Y. Physiologically based pharmacokinetic modelling and in vivo [I]/K(i) accurately predict P-glycoprotein-mediated drug-drug interactions with dabigatran etexilate. Br J Pharmacol. 2014;171:1043–53. https://doi.org/10.1111/bph.12533.
Meyer M, Schneckener S, Ludewig B, Kuepfer L, Lippert J. Using expression data for quantification of active processes in physiologically based pharmacokinetic modeling. Drug Metab Dispos. 2012;40:892–901. https://doi.org/10.1124/dmd.111.043174.
Paré G, Eriksson N, Lehr T, Connolly S, Eikelboom J, Ezekowitz MD, et al. Genetic determinants of dabigatran plasma levels and their relation to bleeding. Circulation. 2013;127:1404–12. https://doi.org/10.1161/CIRCULATIONAHA.112.001233.
Rowland Yeo K, Aarabi M, Jamei M, Rostami-Hodjegan A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev Clin Pharmacol. 2011;4:261–74. https://doi.org/10.1586/ecp.10.143.
McNamara PJ, Alcorn J. Protein binding predictions in infants. AAPS PharmSci. 2002;4:19–26. https://doi.org/10.1208/ps040104.
Veau C, Leroy C, Banide H, Auchère D, Tardivel S, Farinotti R, et al. Effect of chronic renal failure on the expression and function of rat intestinal P-glycoprotein in drug excretion. Nephrol Dial Transplant. 2001;16:1607–14.
Naud J, Michaud J, Boisvert C, Desbiens K, Leblond F a, Mitchell A, et al. Down-regulation of intestinal drug transporters in chronic renal failure in rats. J Pharmacol Exp Ther 2006;320:978–85. https://doi.org/10.1124/jpet.106.112631.
Nolin TD, Naud J, Leblond F a, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther 2008;83:898–903. https://doi.org/10.1038/clpt.2008.59.
Langenbucher F. Letters to the Editor: Linearization of dissolution rate curves by the Weibull distribution. J Pharm Pharmacol. 1972;24:979–81. https://doi.org/10.1111/j.2042-7158.1972.tb08930.x.
Chu X, Liao M, Shen H, Yoshida K, Zur AA, Arya V, et al. Clinical probes and endogenous biomarkers as substrates for transporter drug-drug interaction evaluation: perspectives from the international transporter consortium. Clin Pharmacol Ther. 2018;104:836–64. https://doi.org/10.1002/cpt.1216.
Morris ME, Levyx G. Renal clearance and serum protein binding of acetaminophen and its major conjugates in humans. J Pharm Sci. 2018;73:1038–41. https://doi.org/10.1002/jps.2600730806.
Milne R, Nation R, Somogyi A, Bochner F, Griggs W. The influence of renal function on the renal clearance of morphine and its glucuronide metabolites in intensive-care patients. Br J Clin Pharmacol. 1992;34:53–9. https://doi.org/10.1111/j.1365-2125.1992.tb04107.x.
Varma MVS, Lin J, Bi Y, Kimoto E, Rodrigues AD. Quantitative rationalization of gemfibrozil drug interactions: consideration of transporters-enzyme interplay and the role of circulating metabolite gemfibrozil 1-O-β-glucuronide. Drug Metab Dispos. 2015;43:1108–18. https://doi.org/10.1124/dmd.115.064303.
Sallustio BC, Fairchild BA, Pannall PR. Interaction of human serum albumin with the electrophilic metabolite 1-O-gemfibrozil-beta-D-glucuronide. Drug Metab Dispos. 1997;25:55–60.
Shipkova M, Armstrong VW, Oellerich M, Wieland E. Acyl glucuronide drug metabolites: toxicological and analytical implications. Ther Drug Monit. 2003;25:1–16. https://doi.org/10.1097/00007691-200302000-00001.
Regan S, Maggs J, Hammond T, Lambert C, Williams D, Park B. Acyl glucuronides: the good, the bad and the ugly. Biopharm Drug Dispos. 2010;31:367–95. https://doi.org/10.1002/bdd.720.
Skeith KJ, Dasgupta M, Lange R, Jamali F. The influence of renal function on the pharmacokinetics of unchanged and acyl-glucuroconjugated ketoprofen enantiomers after 50 and 100 mg racemic ketoprofen. Br J Clin Pharmacol. 1996;42:163–9.
Spahn H, Spahn I, Benet LZ. Probenecid-induced changes in the clearance of carprofen enantiomers: a preliminary study. Clin Pharmacol Ther. 1989;45:500–5. https://doi.org/10.1038/clpt.1989.64.
Hyneck M, Smith P, Munafo A, McDonagh A, Benet L. Disposition and irreversible plasma protein binding of tolmetin in humans. Clin Pharmacol Ther. 1988;44:107–14. https://doi.org/10.1038/clpt.1988.120.
Reimers A, Helde G, Bråthen G, Brodtkorb E. Lamotrigine and its N2-glucuronide during pregnancy: the significance of renal clearance and estradiol. Epilepsy Res. 2011;94:198–205. https://doi.org/10.1016/j.eplepsyres.2011.02.002.
Pauli-Magnus C, Hofmann U, Mikus G, Kuhlmann U, Mettang T. Pharmacokinetics of morphine and its glucuronides following intravenous administration of morphine in patients undergoing continuous ambulatory peritoneal dialysis. Nephrol Dial Transplant. 1999;14:903–9. https://doi.org/10.1093/ndt/14.4.903.
Yeung CK, Shen DD, Thummel KE, Himmelfarb J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int. 2018;85:522–8. https://doi.org/10.1038/ki.2013.399.
Mutsaers HAM, Wilmer MJG, Reijnders D, Jansen J, van den Broek PHH, Forkink M, et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim Biophys Acta. 2013;1832:142–50. https://doi.org/10.1016/j.bbadis.2012.09.006.
Barnes KJ, Rowland A, Polasek TM, Miners JO. Inhibition of human drug-metabolising cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in vitro by uremic toxins. Eur J Clin Pharmacol. 2014;70:1097–106. https://doi.org/10.1007/s00228-014-1709-7.
Sallustio BC, Purdie YJ, Birkett DJ, Meffin PJ. Effect of renal dysfunction on the individual components of the acyl-glucuronide futile cycle. J Pharmacol Exp Ther. 1989;251:288–94.
Khatir DS, Pedersen M, Jespersen B, Buus NH. Evaluation of renal blood flow and oxygenation in CKD using magnetic resonance imaging. Am J Kidney Dis. 2018;66:402–11. https://doi.org/10.1053/j.ajkd.2014.11.022.
Lam YWF, Banerji S, Hatfield C, Talbert RL. Principles of drug administration in renal insufficiency. Clin Pharmacokinet. 1997;32:30–57. https://doi.org/10.2165/00003088-199732010-00002.
Hsu C, McCulloch CE, Curhan GC. Iron status and hemoglobin level in chronic renal insufficiency. J Am Soc Nephrol. 2002;13:2783–6.
Strid H, Simrén M, Stotzer P, Abrahamsson H, Björnsson E. Delay in gastric emptying in patients with chronic renal failure. Scand J Gastroenterol. 2004;39:516–20. https://doi.org/10.1080/00365520410004505.
Watanabe H, Hiraishi H, Ishida M, Kazama JJ, Terano A. Pathophysiology of gastric acid secretion in patients with chronic renal failure: influence of Helicobacter pylori infection. J Intern Med. 2003;254:439–46. https://doi.org/10.1046/j.1365-2796.2003.01210.x.
Delavenne X, Ollier E, Basset T, Bertoletti L, Accassat S, Garcin A, et al. A semi-mechanistic absorption model to evaluate drug-drug interaction with dabigatran: application with clarithromycin. Br J Clin Pharmacol. 2013;76:107–13. https://doi.org/10.1111/bcp.12055.
Stangier J, Rathgen K, Stähle H, Reseski K, Körnicke T, Roth W. Coadministration of dabigatran etexilate and atorvastatin: assessment of potential impact on pharmacokinetics and pharmacodynamics. Am J Cardiovasc Drugs. 2009;9:59–68. https://doi.org/10.2165/00129784-200909010-00006.
Stangier J, Stähle H, Rathgen K, Roth W, Reseski K, Körnicke T. Pharmacokinetics and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor, with coadministration of digoxin. J Clin Pharmacol. 2012;52:243–50. https://doi.org/10.1177/0091270010393342.
Boehringer Ingelheim Clinical Study Synopsis for Public Disclosure - BI Trial No. 1160.81. Safety, tolerability and pharmacokinetics study after single and multiple oral doses of dabigatran etexilate capsule (110 mg, 150 mg b.i.d., 7 days) in healthy Chinese subjects (open label study), 2010. https://trials.boehringer-ingelheim.com/public/trial_results_documents/1160/1160.81_U10-3381-01.pdf. Accessed 29 Oct 2018.
Martin J, Esmaeili H, Manuel RC, Petrini M, Wiebe S, Maas H. A prospective, open label study to evaluate the pharmacokinetics of dabigatran in patients with non-valvular atrial fibrillation and severe renal impairment who are receiving dabigatran etexilate (75 mg twice daily). FASEB J 2017;31:674.9-674.9. https://doi.org/10.1096/fasebj.31.1_supplement.674.9.
Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139–51. https://doi.org/10.1056/NEJMoa0905561.
Boehringer Ingelheim Pharmaceuticals Inc. Dabigatran Etexilate Capsules (75 mg, 110 mg, and 150 mg). Environmental Assessment, 2010. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022512orig1s000ea.pdf. Accessed 29 Oct 2018.
U.S. Food and Drug Administration (FDA). Approval package for application number 22-512/S-011 (PRADAXA), 2012. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/022512Orig1s011_replace.pdf. Accessed 29 Oct 2018.
Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. https://doi.org/10.1016/0006-2952(73)90196-2.
Kishimoto W, Ishiguro N, Ludwig-Schwellinger E, Ebner T, Schaefer O. In vitro predictability of drug-drug interaction likelihood of P-glycoprotein-mediated efflux of dabigatran etexilate based on [I]2/IC50 threshold. Drug Metab Dispos. 2014;42:257–63. https://doi.org/10.1124/dmd.113.053769.
Hanke N, Frechen S, Moj D, Britz H, Eissing T, Wendl T, et al. PBPK models for CYP3A4 and P-gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil and digoxin. CPT Pharmacometrics Syst Pharmacol 2018. https://doi.org/10.1002/psp4.12343.
Moj D, Hanke N, Britz H, Frechen S, Kanacher T, Wendl T, et al. Clarithromycin, midazolam, and digoxin: application of PBPK modeling to gain new insights into drug-drug interactions and co-medication regimens. AAPS J. 2017;19:298–312. https://doi.org/10.1208/s12248-016-0009-9.
Reilly PA, Lehr T, Haertter S, Connolly SJ, Yusuf S, Eikelboom JW, et al. The effect of dabigatran plasma concentrations and patient characteristics on the frequency of ischemic stroke and major bleeding in atrial fibrillation patients: the RE-LY Trial (Randomized Evaluation of Long-Term Anticoagulation Therapy). J Am Coll Cardiol. 2014;63:321–8. https://doi.org/10.1016/j.jacc.2013.07.104.
ChemAxon. Marvin JS. https://www.chemaxon.com/products/marvin/marvin-js/. Accessed 18 Jan 2017
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was partly funded by Boehringer Ingelheim Pharma GmbH & Co. KG.
Conflict of interest
Daniel Moj worked as consultant for Boehringer Ingelheim Pharma GmbH & Co. KG. Hugo Maas and José David Gómez-Mantilla are employees of Boehringer Ingelheim Pharma GmbH & Co. KG. André Schaeftlein gave lectures for physicians hosted by Boehringer Ingelheim Pharma GmbH & Co. KG and received a research grant from Boehringer Ingelheim Pharma GmbH & Co. KG. Nina Hanke reported no conflict of interest. Thorsten Lehr received research grants from Boehringer Ingelheim Pharma GmbH & Co. KG.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the studies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moj, D., Maas, H., Schaeftlein, A. et al. A Comprehensive Whole-Body Physiologically Based Pharmacokinetic Model of Dabigatran Etexilate, Dabigatran and Dabigatran Glucuronide in Healthy Adults and Renally Impaired Patients. Clin Pharmacokinet 58, 1577–1593 (2019). https://doi.org/10.1007/s40262-019-00776-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00776-y