
Abstract
Inhibitors of sodium-glucose cotransporters type 2 (SGLT2) reduce hyperglycaemia by decreasing renal glucose threshold and thereby increasing urinary glucose excretion. They are proposed as a novel approach for the management of type 2 diabetes mellitus. They have proven their efficacy in reducing glycated haemoglobin, without inducing hypoglycaemia, as monotherapy or in combination with various other glucose-lowering agents, with the add-on value of promoting some weight loss and lowering arterial blood pressure. As they may be used concomitantly with many other drugs, we review the potential drug–drug interactions (DDIs) regarding the three leaders in the class (dapagliglozin, canagliflozin and empagliflozin). Most of the available studies were performed in healthy volunteers and have assessed the pharmacokinetic interferences with a single administration of the SGLT2 inhibitor. The exposure [assessed by peak plasma concentrations (C max) and area under the concentration-time curve (AUC)] to each SGLT2 inhibitor tested was not significantly influenced by the concomitant administration of other glucose-lowering agents or cardiovascular agents commonly used in patients with type 2 diabetes. Reciprocally, these medications did not influence the pharmacokinetic parameters of dapagliflozin, canagliflozin or empagliflozin. Some modest changes were not considered as clinically relevant. However, drugs that could specifically interfere with the metabolic pathways of SGLT2 inhibitors [rifampicin, inhibitors or inducers of uridine diphosphate-glucuronosyltransferase (UGT)] may result in significant changes in the exposure of SGLT2 inhibitors, as shown for dapagliflozin and canagliflozin. Potential DDIs in patients with type 2 diabetes receiving chronic treatment with an SGLT2 inhibitor deserve further attention, especially in individuals treated with several medications or in more fragile patients with hepatic and/or renal impairment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The incidence of type 2 diabetes mellitus (T2DM) is increasing worldwide. Because this chronic disease leads to numerous medical complications and represents a huge burden for both patients and society, extensive research is still ongoing to develop new pharmacological approaches to tackle T2DM. The oral therapy of T2DM was dominated during decades by only two pharmacological classes, sulphonylureas and biguanides [1, 2]. The risk of potential drug–drug interactions (DDIs) was rather limited at that time, even if DDIs with sulphonylureas (mainly glibenclamide) leading to severe hypoglycaemia have been reported [3]. α-Glucosidase inhibitors were the next available antidiabetic agents and act directly in the gut. Acarbose was shown to slightly reduce the bioavailability of metformin in healthy volunteers [4], although DDIs with α-glucosidase inhibitors are probably not clinically relevant. Numerous other oral glucose-lowering agents are now available, which target either insulin secretion (insulin-secreting agents: besides sulphonylureas, glinides and, more recently, incretin-based therapies called gliptins) or insulin action (insulin sensitizers: besides metformin, thiazolidinediones or glitazones). Thus, numerous patients are currently treated with various combinations of glucose-lowering agents [5, 6]. Furthermore, to reduce the risk of cardiovascular complications, a global approach is recommended so that most patients with T2DM also receive various antihypertensive medications, lipid-lowering drugs and antiplatelet agents [7, 8]. Finally, patients with T2DM are more prone to have cardiovascular diseases [7] or various infections, and thereby may also receive, in the long run or transiently, several other medications, which could potentially interfere with their glucose-lowering agents. Because most T2DM patients receive concomitantly numerous medications, the issue of DDIs is becoming increasingly important in diabetes [9]. DDIs may have clinical implications and expose the diabetic patient to severe complications, as reported with the occurrence of severe hypoglycaemia in patients receiving sulphonylureas (glibenclamide, glimepiride, glipizide), presumably due to cytochrome P450 (CYP) 2C9-mediated DDIs [10].
Since our updated general overview on DDIs of clinical importance with antihyperglycaemic agents published in 2005 [9], several reviews have more specifically focused on one particular class of glucose-lowering agents: thiazolidinediones (rosiglitazone, pioglitazone) [11], glinides (repaglinide, nateglinide) [12], inhibitors of dipeptidyl peptidase-4 (DPP-4; sitagliptin, vildagliptin, saxagliptin, linagliptin, alogliptin) [13] and agonists of the glucagon-like peptide-1 (exenatide, liraglutide, lixisenatide) [14].
The existing therapeutic classes of oral antidiabetic drugs are not adequately effective in maintaining long-term glycaemic control in most patients with T2DM, even when used in combination [5], and there remains a medical need for improving pharmacological therapy of T2DM [15]. Inhibitors of sodium-glucose cotransporters type 2 (SGLT2) are new glucose-lowering agents, which specifically target the kidney by blocking the reabsorption of filtered glucose, thus leading to glucosuria [16, 17]. This mechanism of action holds potential promise for patients with T2DM, not only in terms of improvements in glycaemic control, but also considering the potential benefits of weight loss and arterial blood pressure reduction [16, 17]. SGLT2 inhibitors may be used as monotherapy in diet-treated patients or in combination with any other glucose-lowering agent [18]. Pharmacokinetic characteristics of SGLT2 inhibitors show an excellent oral bioavailability, a rather long half-life allowing once-daily administration, a low accumulation index, no active metabolites and a negligible renal clearance (Table 1) [19]. To our knowledge, there is no review analyzing DDIs with SGLT2 inhibitors.
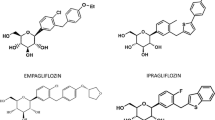
The aim of this review is to provide an extensive analysis of potential DDIs of the recently launched SGLT2 inhibitors—dapagliflozin [20, 21] and canagliflozin [22, 23]—and in late phase of clinical development—empagliflozin [24] (Fig. 1).

Chemical structures of dapagliflozin, canagliflozin and empagliflozin
2 Literature Search
To identify relevant studies, an extensive literature search in MEDLINE was performed from 2008 to October 2013, with the following MESH terms: ‘SGLT2 inhibitor’, ‘canagliflozin’, ‘dapagliflozin’, or ‘empagliflozin’ combined with ‘drug–drug interaction’ or ‘drug interaction’. No language restrictions were imposed. Reference lists of original studies, narrative reviews and previous systematic reviews were also carefully examined.
DDIs can be evaluated in two ways: either the effect of a second drug on the pharmacokinetics of the SGLT2 inhibitor (Table 2) or the effect of the SGLT2 inhibitor on the pharmacokinetics of the second drug (Table 3). Whenever available, data were expressed as the ratio of adjusted geometric mean point estimates (GMR) with 90 % confidence interval (CI). We will successively analyze the DDIs of SGLT2 inhibitors with other glucose-lowering agents, with cardiovascular medications commonly used in patients with T2DM and with various drugs of special interest because they have a narrow therapeutic margin or they may interfere with the metabolic pathways of SGLT2 inhibitors. Most of the data found in the literature were obtained in healthy volunteers after a single administration of the SGLT2 inhibitor. Caution is thus recommended in the interpretation of the available data, which could not be automatically extrapolated to patients with T2DM, especially when comorbidities are present such as chronic kidney disease (CKD) [19, 25] or hepatic impairment [19].
3 Pharmacokinetic/Pharmacodynamic Characteristics of SGLT2 Inhibitors
The main pharmacokinetic characteristics of the three SGLT2 inhibitors (dapagliflozin, canagliflozin, empagliflozin), which were essentially derived from studies in healthy volunteers, are summarized in Table 1. No clinically relevant differences were observed in either compound exposure with respect to age, race, sex, body weight, food or presence of T2DM. Pharmacodynamic changes, assessed by urinary glucose excretion (UGE), are dependent on plasma glucose level and renal function [estimated by glomerular filtration rate (GFR)]. Decreases in UGE were observed due to the lower filtered load (plasma glucose × GFR) in healthy volunteers compared with subjects with T2DM, as well as in patients with moderate to severe CKD compared with patients with normal renal function [19, 25]. In patients with T2DM and normal renal function, UGE increased according to the dose of SGLT2 given. However, for the doses used in clinical practice, no major changes in glucose-lowering efficacy should be observed, even if DDIs result in changing the plasma concentrations of dapagliflozin [20, 21], canagliflozin [22, 23] or empagliflozin [24, 26] by a factor two.
3.1 Dapagliflozin
Clinical pharmacokinetics and pharmacodynamics of dapagliflozin have been recently extensively reviewed [21]. Orally administered dapagliflozin is rapidly absorbed, generally reaching peak plasma concentrations (t max) within 1–2 h. Dose-proportional systemic exposure to dapagliflozin has been observed over a wide dose range (0.1–500 mg) with an oral bioavailability of 78 %. Dapagliflozin has extensive extravascular distribution, as shown by a mean volume of distribution averaging 118 L (Table 1). Dapagliflozin metabolism occurs predominantly in the liver and kidneys by uridine diphosphate-glucuronosyltransferase (UGT) 1A9 to the major metabolite dapagliflozin 3-O-glucuronide (D3OG; this metabolite is not an SGLT2 inhibitor at clinically relevant exposures). Dapagliflozin is not appreciably cleared by renal excretion (<2 % of dose is recovered in urine as parent), in contrast to its major metabolite that is mainly eliminated via renal excretion. Based on the pharmacokinetic characteristics of dapagliflozin, there is a potential for clinically relevant interactions with inhibitors and inducers of UGT1A9. However, potent inhibitors of UGT1A9 seem to be rare. One study investigated the interaction between dapagliflozin and mefenamic acid, a UGT1A9 inhibitor (see Sect. 6.1).
3.2 Canagliflozin
The pharmacokinetics/pharmacodynamics and metabolism of canagliflozin have also been recently reviewed [22, 23]. After oral administration, canagliflozin is rapidly absorbed in a dose-dependent manner across a dose range of 50–300 mg. The mean absolute oral bioavailability is approximately 65 %. After canagliflozin 100 or 300 mg, median t max values occurred within 1–2 h, with steady-state levels attained after 4–5 days following multiple once-daily doses. Canagliflozin does not exhibit time-dependent pharmacokinetics and, following multiple 100 and 300 mg doses, accumulates in the plasma up to 36 %. The drug is extensively (99 %) bound to plasma proteins, mainly albumin (Table 1). Canagliflozin is mainly metabolized via glucuronidation by UGT1A9 and UGT2B4 to two inactive O-glucuronide metabolites. Such UGT enzymes can be inhibited or induced and have the potential to change canagliflozin systemic exposure. Canagliflozin has only minor (≈7 % in humans) metabolism by CYP3A4. In in vitro studies, canagliflozin did not induce or inhibit most CYP enzyme expression, although it weakly inhibited CYP2B6, CYP2C8, CYP2C9 and CYP3A4 [22]. Whether these effects might be relevant in vivo and have clinical consequences remains unknown.
3.3 Empagliflozin
Single oral doses of empagliflozin were rapidly absorbed, reaching t max after 1.0–2.0 h (Table 1). Increases in empagliflozin exposure were roughly dose-proportional and a dose-dependent increase in UGE was observed for empagliflozin doses up to 100 mg [26]. Pharmacokinetic/pharmacodynamic characteristics of empagliflozin have been recently reviewed in healthy volunteers and patients with T2DM, including patients with CKD or hepatic impairment [24]. However, the description of absorption, distribution, metabolism, and excretion (ADME) characteristics of empagliflozin in humans has not yet been extensively reported [27].
4 Drug–Drug Interactions (DDIs) with other Glucose-Lowering Agents
Because of their mode of action, SGLT2 inhibitors can be combined with any other glucose-lowering agent [18]. Pharmacodynamic investigations and clinical studies have shown complementary efficacy in reducing fasting and postprandial glucose and glycated haemoglobin (HbA1c) levels.
4.1 Dapagliflozin
Pharmacokinetic DDI studies in healthy volunteers showed that dapagliflozin can be coadministered with pioglitazone, metformin, glimepiride or sitagliptin without dose adjustment of either drug [28]. The other glucose-lowering agents did not impact pharmacokinetics of dapagliflozin (only a slight- +8 % -greater exposure to dapagliflozin after coadministration of sitagliptin was observed, without any clinically relevance) [Table 2]. Reciprocally, dapagliflozin did not significantly influence the pharmacokinetic parameters of the other glucose-lowering agents (only a trend for a slightly greater exposure to glimepiride was detected) [Table 3]. The addition of dapagliflozin in patients insufficiently controlled with glimepiride resulted in a significant reduction in HbA1c with only a slightly increased risk of hypoglycaemia [29]. A Japanese study demonstrated that voglibose, an α-glucosidase inhibitor, does not modify the pharmacokinetics of dapagliflozin [30]. Several clinical studies have confirmed that dapagliflozin is effective and safe when combined with other glucose-lowering agents for the management of T2DM [20].
4.2 Canagliflozin
Rather few data are available regarding DDIs with canagliflozin [31], and they are only published as congress abstracts [32]. Metformin slightly increased exposure to canagliflozin [peak plasma concentrations (C max) +5 %; area under the concentration-time curve (AUC) +10 %] (Table 2), whereas canagliflozin did not modify the exposure to metformin (Table 3). However, these changes were rather modest without any clinical relevance [32]. No pharmacokinetic interferences were reported between canagliflozin and glyburide (glibenclamide) [Table 2] [32].
4.3 Empagliflozin
Empagliflozin has been tested in single-dose studies when coadministered with various other glucose-lowering agents, metformin [33], glimepiride [34], sitagliptin [35], linagliptin [36]. Glimepiride slightly decreased exposure to empagliflozin, whereas sitagliptin modestly increased it (Table 2). However, these changes are too small for being considered as clinically relevant. Empagliflozin did not significantly modify exposure to other glucose-lowering agents tested so far (Table 3).
5 DDIs with Cardiovascular Agents
Because of the high cardiovascular risk of patients with T2DM [7], it is important to assess the potential interferences with other cardiovascular drugs commonly prescribed in those patients [37].
5.1 Dapagliflozin
Coadministration of dapagliflozin was evaluated with simvastatin, valsartan, warfarin or digoxin in healthy volunteers [38]. Simvastatin and valsartan did not influence the exposure to dapagliflozin (Table 2). Dapagliflozin 20 mg was associated with modest increases in the exposure to simvastatin (GMR 1.19; 90 % CI 1.01–1.40) and simvastatin acid (GMR 1.30; 1.15–1.47), but these changes were considered as not clinically meaningful. A mild increase in the exposure to R-warfarin and S-warfarin was also detected when warfarin was prescribed with dapagliflozin (Table 3), without any change in the International Normalized Ratio (INR; GMR 1.007; 90 % CI 0.989–1.025) [38]. No other meaningful DDIs were detected, including changes in digoxin levels after dapagliflozin administration [38] (Table 3).
5.2 Canagliflozin
No clinically relevant pharmacokinetic interferences were reported between canagliflozin and simvastatin or hydrochlorothiazide (Tables 2 and 3) [31]. Of potential interest, canagliflozin significantly increased the exposure to digoxin (C max +36 %; AUC +20 %) [Table 3] [39]. Therefore, patients taking canagliflozin with concomitant digoxin should be monitored appropriately regarding plasma digoxin concentrations. In contrast, no significant changes in exposure to warfarin and no change in INR were noticed after canagliflozin administration (Table 3) [39].
5.3 Empagliflozin
DDI studies in healthy volunteers showed no clinically significant interactions between empagliflozin and warfarin [40], simvastatin [41] or other cardiovascular compounds (verapamil, ramipril and digoxin) [42]. The lack of DDIs between verapamil and empagliflozin indicates there is no relevant effect of P-glycoprotein inhibition on the pharmacokinetics of empagliflozin. Another study evaluated empagliflozin combined with diuretics, such as hydrochlorothiazide and torasemide, in patients with T2DM [43]. In the latter study, a slight increase in AUC of empagliflozin was observed with hydrochlorothazide (+7.1 %) and with torasemide (+7.8 %) [Table 2]. These modest changes should not have clinical consequences.
6 DDIs with other Medications of Interest
Drugs of special interest should also be tested because they may interfere with a common metabolic pathway with SGLT2 inhibitors (i.e. rifampicin, mefenamic acid) or because they may have a low therapeutic index (contraceptive pill).
6.1 Dapagliflozin
As dapagliflozin is primarily metabolized via the UGT1A9 pathway to its major inactive metabolite D3OG, the potential for DDIs between dapagliflozin and two potential UGT1A9 modulators was evaluated: rifampicin, a pleiotropic drug-metabolizing enzyme inducer, and mefenamic acid, a strong UGT1A9 inhibitor. Significant changes in dapagliflozin exposure were seen with rifampicin (a significant decrease of AUC −22 %), and with mefenamic acid (a significant increase of AUC +51 %) [Table 2]. However, only minor changes in UGE were detected, none of which were considered clinically relevant [44].
6.2 Canagliflozin
No clinically relevant pharmacokinetic interferences were reported between canagliflozin and an oral contraceptive containing ethinylestradiol and levonorgestrel [45] (Tables 2 and 3).
However, GMRs of key pharmacokinetic parameters of canagliflozin were significantly reduced after rifampicin 600 mg once daily for 8 days: C max −28 % and AUC −51 % (Table 2) [31]. This decrease in exposure to canagliflozin may slightly decrease efficacy. Rifampicin is a non-selective inducer of several UGT enzymes, including UGT1A9 and UGT2B4. If an inducer of these UGTs (e.g. rifampicin, phenytoin, phenobarbital, ritonavir) must be coadministered with canagliflozin, advice is given to consider increasing the dose to 300 mg once daily if patients are currently tolerating canagliflozin 100 mg once daily, have an estimated GFR greater than 60 ml/min/1.73 m2, and require additional glycaemic control [31]. Otherwise, alternative glucose-lowering agents should be considered.
Finally, exposure to canagliflozin was modestly but significantly increased by cyclosporine (AUC +23 %) and probenecid (AUC +21 %) [31] (Table 2). Considering the results obtained with ascending doses of canagliflozin in patients with T2DM [46], these modest changes should not have clinically relevant implications.
6.3 Empagliflozin
DDI studies showed no clinically significant interactions between empagliflozin and the oral contraceptive ethinylestradiol/levonorgestrel (Table 3) [47].
7 Clinical Implications
The metabolic pathways of SGLT2 inhibitors expose them to a low risk of clinically relevant pharmacokinetic DDIs. This is important because most patients with T2DM are treated with many medications, not only for controlling blood glucose but also for managing other risk factors (including hypertension and dyslipidaemia) and treating various comorbidities (coronary artery disease, congestive heart failure, etc).
Several studies have demonstrated that the effects of SGLT2 inhibitors on UGE is reduced when GFR is decreasing [19, 25]. Specific pharmacokinetic studies have shown heterogeneous results in patients with various degrees of CKD [19]: according to reported changes in systemic exposure, the daily dose of dapagliflozin [48] or empagliflozin [49] should not be reduced in patients with moderate CKD, whereas the maximum dose recommended for canagliflozin is 100 mg instead of 300 mg [50, 51]. Even if canagliflozin has been shown to be efficacious and safe in diabetic patients with mild to moderate CKD [52], all drugs that may interfere with renal function, by decreasing GFR (for instance, non-steroidal anti-inflammatory drugs), may exert pharmacodynamic (rather than pharmacokinetic) DDIs and reduce the glucose-lowering effects of SGLT2 inhibitors in fragile patients. In clinical practice, renal function should be regularly monitored in diabetic patients treated with SGLT2 inhibitors, especially in patients with mild/moderate CKD, and all agents that may interfere with kidney function should be used with caution [25]. Finally, dose adjustment and special caution may be recommended in patients taking loop diuretics, especially in elderly people, if there are concerns or symptoms of volume-related side effects [23]. The observation that there were only negligible pharmacokinetic interactions between canagliflozin and hydrochlorothiazide [31], or between empagliflozin and hydrochlorothiazide or torasemide, a loop diuretic [43], in patients with T2DM does not mean that a clinically relevant pharmacodynamic interaction could be excluded, at least in some more fragile patients.
8 Conclusion
All available studies investigating potential DDIs between SGLT2 inhibitors (dapagliflozin, canagliflozin and empagliflozin) and other drugs commonly used in patients with T2DM provide reassuring results with no clinically relevant pharmacokinetic interferences detected when combined with other glucose-lowering agents or cardiovascular drugs. However, these data should be interpreted with some caution because they were obtained in healthy volunteers and in most instances after a single acute administration of the SGLT2 inhibitor. Nevertheless, large phase III clinical trials lasting up to 1–2 years showed no clinically relevant safety issues when the SGLT2 inhibitors are prescribed with other glucose-lowering agents and several cardiovascular drugs. However, when agents selectively acting on pathways playing a role in the metabolism of SGLT2 inhibitors (UGT inducers or inhibitors) are prescribed, larger changes in exposure to dapagliflozin and canagliflozin may occur, which might have clinical consequences. Further studies are required to investigate more extensively potential DDIs in patients with T2DM chronically treated with an SGLT2 inhibitor.
References
Scheen AJ, Lefebvre PJ. Oral antidiabetic agents: a guide to selection. Drugs. 1998;55(2):225–36.
Krentz AJ, Bailey CJ. Oral antidiabetic agents: current role in type 2 diabetes mellitus. Drugs. 2005;65(3):385–411.
Scheen AJ, Lefèbvre PJ. Antihyperglycemic agents: drug interactions of clinical importance. Drug Saf. 1995;12(1):32–45.
Scheen AJ, Demagalhaes A, Salvatore T, et al. Reduction of the acute bioavailability of metformin by the alpha-glucosidase inhibitor acarbose in normal man. Eur J Clin Investig. 1994;24:50–4.
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2012;55(6):1577–96.
Bennett WL, Maruthur NM, Singh S, et al. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011;154(9):602–13.
Ryden L, Grant PJ, Anker SD, et al. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: The Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD). Eur Heart J. 2013;34(39):3035–87.
Lafeber M, Grobbee DE, Spiering W, et al. The combined use of aspirin, a statin, and blood pressure-lowering agents (polypill components) in clinical practice in patients with vascular diseases or type 2 diabetes mellitus. Eur J Prev Cardiol. 2013;20(5):771–8.
Scheen AJ. Drug interactions of clinical importance with antihyperglycaemic agents: an update. Drug Saf. 2005;28(7):601–31.
Tirkkonen T, Heikkila P, Huupponen R, et al. Potential CYP2C9-mediated drug–drug interactions in hospitalized type 2 diabetes mellitus patients treated with the sulphonylureas glibenclamide, glimepiride or glipizide. J Intern Med. 2010;268(4):359–66.
Scheen AJ. Pharmacokinetic interactions with thiazolidinediones. Clin Pharmacokinet. 2007;46(1):1–12.
Scheen AJ. Drug–drug and food-drug pharmacokinetic interactions with new insulinotropic agents repaglinide and nateglinide. Clin Pharmacokinet. 2007;46(2):93–108.
Scheen AJ. Dipeptidylpeptidase-4 inhibitors (gliptins): focus on drug–drug interactions. Clin Pharmacokinet. 2010;49(9):573–88.
Hurren KM, Pinelli NR. Drug–drug interactions with glucagon-like peptide-1 receptor agonists. Ann Pharmacother. 2012;46(5):710–7.
Tahrani AA, Bailey CJ, Del Prato S, et al. Management of type 2 diabetes: new and future developments in treatment. Lancet. 2011;378(9786):182–97.
Bailey CJ. Renal glucose reabsorption inhibitors to treat diabetes. Trends Pharmacol Sci. 2011;32(2):63–71.
Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32(4):515–31.
Vasilakou D, Karagiannis T, Athanasiadou E, et al. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med. 2013;159(4):262–74.
Scheen AJ. Evaluating SGLT2 inhibitors for type 2 diabetes: pharmacokinetic and toxicological considerations. Expert Opin Drug Metab Toxicol. doi:10.1517/17425255.2014.873788.
Plosker GL. Dapagliflozin: a review of its use in type 2 diabetes mellitus. Drugs. 2012;72(17):2289–312.
Kasichayanula S, Liu X, Lacreta F, et al. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin Pharmacokinet. 2014;53(1):17–27.
Elkinson S, Scott LJ. Canagliflozin: first global approval. Drugs. 2013;73(9):979–88.
Lamos EM, Younk LM, Davis SN. Canagliflozin, an inhibitor of sodium-glucose cotransporter 2, for the treatment of type 2 diabetes mellitus. Expert Opin Drug Metab Toxicol. 2013;9(6):763–75.
Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet. doi:10.1007/s40262-013-0126-x.
Scheen AJ. Pharmacokinetic considerations for the treatment of diabetes in patients with chronic kidney disease. Expert Opin Drug Metab Toxicol. 2013;9(5):529–50.
Seman L, Macha S, Nehmiz G, et al. Empagliflozin (BI 10773), a potent and selective SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Drug Dev. 2013;2(2):152–61.
Grempler R, Thomas L, Eckhardt M, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14(1):83–90.
Kasichayanula S, Liu X, Shyu WC, et al. Lack of pharmacokinetic interaction between dapagliflozin, a novel sodium-glucose transporter 2 inhibitor, and metformin, pioglitazone, glimepiride or sitagliptin in healthy subjects. Diabetes Obes Metab. 2011;13(1):47–54.
Strojek K, Yoon KH, Hruba V, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13(10):928–38.
Imamura A, Kusunoki M, Ueda S, et al. Impact of voglibose on the pharmacokinetics of dapagliflozin in Japanese patients with type 2 diabetes. Diabetes Ther. 2013;4(1):41–9.
Janssen Pharmaceuticals Inc. Invokana™ (canagliflozin) tablets, for oral use: US prescribing information. 2013. http://www.janssenmd.com/pdf/invokana/PI-INVOKANA.pdf. Accessed 11 Oct 2013.
Devineni D, Sarich TC, Wexler D, et al. Effects of canagliflozin on the pharmacokinetics (PK) and pharmacodynamics (PD) of metformin and glyburide [abstract no. 2268-PO 2011]. Presented at the American Diabetes Association (ADA) 71st Scientific Sessions; San Diego, CA; 24–28 June 2011.
Macha S, Dieterich S, Mattheus M, et al. Pharmacokinetics of empagliflozin, a sodium glucose cotransporter-2 (SGLT2) inhibitor, and metformin following co-administration in healthy volunteers. Int J Clin Pharmacol Ther. 2013;51(2):132–40.
Macha S, Mattheus M, Pinnetti S, et al. Pharmacokinetics of empagliflozin, a sodium glucose cotransporter 2 inhibitor, and glimepiride following co-administration in healthy volunteers: a randomised, open-label, crossover study. Diab Res Clin Metab. 2012;1:1–7.
Brand T, Macha S, Mattheus M, et al. Pharmacokinetics of empagliflozin, a sodium glucose cotransporter-2 (SGLT-2) inhibitor, coadministered with sitagliptin in healthy volunteers. Adv Ther. 2012;29(10):889–99.
Friedrich C, Metzmann K, Rose P, et al. A randomized, open-label, crossover study to evaluate the pharmacokinetics of empagliflozin and linagliptin after coadministration in healthy male volunteers. Clin Ther. 2013;35(1):A33–42.
Scheen AJ. Cytochrome P450-mediated cardiovascular drug interactions. Expert Opin Drug Metab Toxicol. 2011;7(9):1065–82.
Kasichayanula S, Chang M, Liu X, et al. Lack of pharmacokinetic interactions between dapagliflozin and simvastatin, valsartan, warfarin, or digoxin. Adv Ther. 2012;29(2):163–77.
Devineni D, et al. Lack of clinically meaningful interaction between canagliflozin, a sodium glucose co-transporter 2 inhibitor, and digoxin or warfarin in healthy subjects [poster]. Presented at the 2012 Annual Meeting of the American College of Clinical Pharmacology (ACCP), San Diego, CA: 23–25 September 2012.
Macha S, Rose P, Mattheus M, et al. Lack of drug–drug interaction between empagliflozin, a sodium glucose cotransporter 2 inhibitor, and warfarin in healthy volunteers. Diabetes Obes Metab. 2013;24(15):316–23.
Macha S, Lang B, Pinnetti S, et al. Lack of pharmacokinetic interaction between the sodium glucose cotransporter-2 (SGLT-2) inhibitor empagliflozin and simvastatin in healthy volunteers [abstract no. PCS-33-7]. J Diabetes Investig 2012; 3 Suppl 1: 228.
Macha S, Sennewald R, Rose P, et al. Lack of clinically relevant drug–drug interaction between empagliflozin, a sodium glucose cotransporter 2 inhibitor, and verapamil, ramipril, or digoxin in healthy volunteers. Clin Ther. 2013;35(3):226–35.
Giessmann T, Heise T, Macha S, et al. Lack of interaction between the sodium glucose cotransporter-2 inhibitor empagliflozin and hydrochlorothiazide or torasemide in patients with T2DM [abstract no. 2440-PO]. Diabetes 2012;61 Suppl:A614.
Kasichayanula S, Liu X, Griffen SC, et al. Effects of rifampin and mefenamic acid on the pharmacokinetics and pharmacodynamics of dapagliflozin. Diabetes Obes Metab. 2013;15(3):280–3.
Skee D, Shalayda K, Vandebosch A, et al. The effects of multiple doses of canagliflozin on the pharmacokinetics and safety of single doses of an oral contraceptive containing ethinyl estradiol and levonorgestrel [poster]. Presented at the 111th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics (ASCPT), Atlanta, GA; 17–20 March 2010.
Devineni D, Curtin CR, Polidori D, et al. Pharmacokinetics and pharmacodynamics of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in subjects with type 2 diabetes mellitus. J Clin Pharmacol. 2013;53(6):601–10.
Macha S, Mattheus M, Pinnetti S, et al. Effect of empagliflozin on the steady-state pharmacokinetics of ethinylestradiol and levonorgestrel in healthy female volunteers. Clin Drug Investig. 2013;20(33):351–7.
Kasichayanula S, Liu X, Pe Benito M, et al. The influence of kidney function on dapagliflozin exposure, metabolism and pharmacodynamics in healthy subjects and in patients with type 2 diabetes mellitus. Br J Clin Pharmacol 2013;76(3):432–44.
Macha S, Mattheus M, Halabi A, et al. Pharmacokinetics, pharmacodynamics and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in subjects with renal impairment. Diabetes Obes Metab (Epub 16 Jul 2013).
Devineni D, Marbury T, Curtin C, et al. Effects of renal function on canagliflozin (CANA) pharmacokinetics (PK) and pharmacodynamics (PD) in non-diabetic subjects [abstract no. PUB295]. JASN Abstract Supplement of the American Society of Nephrology (ASN) Kidney Week, San Diego California; 30 October–4 November 2012.
Food and Drug Administration. Center for Drug Evaluation and Research report. Canagliflozin (Invokana). http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204042Orig1s000ClinPharmR.pdf. Accessed 09 Jan 2014.
Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab. 2013;15(5):463–73.
Funding and conflicts of interest
No sources of funding were used to assist in the preparation of this manuscript. No conflicts of interest are directly relevant to the content of this manuscript.
A.J. Scheen has received lecture/advisor fees from AstraZeneca/BMS, Boehringer Ingelheim, Eli Lilly, GlaxoSmithKline, Merck Sharp & Dohme, Novartis, NovoNordisk, and Sanofi-Aventis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Scheen, A.J. Drug–Drug Interactions with Sodium-Glucose Cotransporters Type 2 (SGLT2) Inhibitors, New Oral Glucose-Lowering Agents for the Management of Type 2 Diabetes Mellitus. Clin Pharmacokinet 53, 295–304 (2014). https://doi.org/10.1007/s40262-013-0128-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-013-0128-8