
Abstract
Alzheimer’s disease (AD) is a distinctive medical condition characterized by loss of memory, orientation, and cognitive impairments, which is an exceptionally universal form of neurodegenerative disease. The statistical data suggested that it is the 3rd major cause of death in older persons. Butyrylcholinesterase (BChE) and acetylcholinesterase (AChE) inhibitors play a vital role in the treatment of AD. Coumarins, natural derivatives, are reported as cholinesterase inhibitors and emerges as a promising scaffold for design of ligands targeting enzymes and pathological alterations related to AD. In this regard, the 3D QSAR pharmacophore models were developed for coumarin scaffold containing BChE and AChE inhibitors. Several 3D QSAR pharmacophore models were developed with FAST, BEST, and CEASER methods, and finally, statistically robust models (based on correlation coefficient, cost value, and RMSE value) were selected for further analysis for both targets. The important features ((HBA 1, HBA 2, HY, RA (BChE) HBA 1, HBA 2, HY, PI, (AChE)) were identified for good inhibitory activity of coumarin derivatives. Finally, the selected models were applied to various database compounds to find potential BChE and AChE inhibitors, and we found 13 for BChE and 1 potent compound for AChE with an estimated activity of IC50 < 10 µM. Further, the Lipinski filters, and ADMET analysis supports the selected compounds to become a drug candidate. These selected BChE and AChE inhibitors can be used in the treatment of AD.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is one of the typical medical and social problems which is most widely prevalent in older people. Clinically, it is distinguished by increasing memory and orientation loss and also other cognitive impairments such as poor impulse control and decision-making, apraxia, and communication difficulties. It is a highly general form of neurodegenerative disease. According to the Alzheimer’s Association, 13% of individuals over 65 in growing countries have AD (Alzheimer’s Association 2021; Folch et al. 2016). It is currently projected that AD may rank as the 3rd primary cause of mortality in the United States, just behind heart disease and cancer, as a significant cause of death for the elderly (NIH National Institute of Aging 2021). Previous studies convey that this disease is also caused by genetics, environmental factors, and lifestyle changes (NIH National Institute of Aging 2021; Wang et al. 2021). At the time of the COVID-19 pandemic, researchers discovered that approximately 56% of patients had cognitive deterioration (Healthline 2021; Alzheimer’s Association 2021). Several biological targets against AD have been identified recently, including acetylcholinesterase (AChE), butyrylcholinesterase (BChE), N-methyl-d-aspartate (NMDA) receptor, tau protein, amyloid-beta plaques, and others. Among all these targets, the cholinergic pathways has long been the most accepted pathogenesis in the drug discovery of AD (Ferreira-Vieira et al. 2016; Francis et al. 1999; Hampel et al. 2018).
According to the cholinergic pathways, degeneration of cholinergic neurons and a deficiency of the neurotransmitter acetylcholine (Ach) are the reasons for the loss of memory and decline of cognitive functions (Contestabile 2011; Francis 2005; Francis et al. 1999; Hampel et al. 2018; Wilkinson et al. 2004). Cholinesterase catalyzes the breakdown of choline; hence inhibiting it can be an effective treatment for AD. Two cholinesterase enzymes are responsible for the hydrolysis of Ach in the central nervous system they are: butyrylcholinesterase (BChE) and acetylcholinesterase (AChE) (Atatreh et al. 2019; Coyle et al. 1983). In advanced AD, the level of AChE in the brain gradually decrease from its average value, whereas the level of BChE increase or remaine unchanged, indicating that BChE plays a crucial role in hydrolyzing Ach in the late stages of the disease, whereas AChE only plays a supplementary role (Nordberg et al. 2013; Yang et al. 2019a). Thus, considering the numerous functions of BChE and AChE in different AD process the discovery of BChE and AChE inhibitors provide a feasible therapeutic approach to prevent AD.
Computational tools are currently used in the drug discovery process to identify and develop a potential lead/hit (Arya and Coumar 2021). Nowadays, computational techniques are becoming popular in the academic and pharmaceutical industries (Sliwoski et al. 2014). Pharmacophore modeling is one of the most common methods, which deals with the common chemical functions and maintain a similar spatial arrangement, resulting in biological activity on targets. A pharmacophore model can be constructed based on ligands or structures. Pharmacophore approaches have been broadly used in virtual screening, de novo design, and other applications such as lead optimization and multitarget drug design (Khedkar et al. 2007). The objective of virtual screening is to identify molecules (hits) with physicochemical characteristics that are similar to those substances from which the models are developed. Some of the hits may be comparable to known active compounds, and some may be completely new scaffolds (Yang 2010; Sun 2008). Moreover, pharmacophore modeling is used to facilitate and promote the efficiency of the drug discovery process. In addition, this approach is a low-cost and rapid alternative to high-throughput screening (Lu et al. 2011).
Coumarin is an important class of natural compounds and has been reported to have various biological activities such as anti-viral, anti-tumor, anti-cancer, anti-inflammatory, anti-bacterial, anti-fungal, anti-alzheimer, etc. (Menezes and Diederich 2019; Abdelmohsen et al. 2021; Buran et al. 2021; Pibuel et al. 2021; Yang et al. 2022; Yun et al. 2020). Notably, earlier research has shown that coumarin can reduce cholinesterase activity. The possible chemical substitution in the basic nucleus of coumarin structure makes them interesting molecules in drug discovery (Abu-Aisheh et al. 2019). In this regard, we have considered several coumarin derivatives reported in various literatures (Piazzi et al. 2008; Yang et al. 2019b; Heo et al. 2020; Hu et al. 2019; Pisani et al. 2017; Hirbod et al. 2017) as model data and subjected to pharmacophore modeling. Further, the models were used for virtual screening to find potential BChE and AChE inhibitors for the treatment of AD.
Materials and methods
Preparation of datasets
At the beginning of this research work, a total of 136 coumarin derivatives with BChE and AChE inhibitory activity were gathered from different literatures (Piazzi et al. 2008; Yang et al. 2019b; Heo et al. 2020; Hu et al. 2019; Pisani et al. 2017; Hirbod et al. 2017). The dataset collection was done considering different parameters such as donepezil as standard drug, Ellman’s method (Ellman et al. 1961) for the biological evaluation etc. Further, the enzymes used in collected dataset reported the diffrernt source of enzymes (BChE and AChE) like human BChE, human AChE, eeAChE, eqBChE, etc. These all data were gathered in this study because they (collected literatures) used the extract of enzymes and hence the extracted enzymes were pure BChE and AChE. Further, it is reported the eeAChE and eqBChE are highly homologous and cheaper surrogate for human enzymes (Rullo et al. 2019). Hence there is no or less chance of error in results of in silico study. In this way all the data were gathered for this study. Further, the collected dataset were subjected to data curation (deletion of qualitative data and influential compounds for modeling) to refine the dataset. Finally, 58 and 100 compounds for BChE and AChE, respectively, were used for further analysis. The structures of the collected dataset were drawn using ChemDraw Ultra 12.0. Finally, ligand preparation was done by a different process, such as addition of hydrogen and energy minimization of ligands. The 2D structures of total collected compounds (n = 136) with IC50 value were reported in the supplementary file (Table S1).
3D QSAR pharmacophore model development
To develop a statistically robust pharmacophore model, the original dataset was divided into training and test set compounds in the ratio of (50–70% training set and 50–30% test set). The training set compounds were used for the model development, and the developed models were applied to test set compounds to validate the developed models. After dividing the dataset into training and test set compounds, the feature mapping was done to find the possible features responsible for BChE and AChE inhibitory activities of the coumarin derivatives (Jiang and Gao 2018). In this regard, the two most active compounds were selected for the feature mapping (Ambure and Roy 2014), and based on these two compounds, the five most probable features were identified, which include hydrogen-bond donor (HBD), hydrogen-bond acceptor (HBA), hydrophobic (HY), ring aromatic (RA), positive ionizable features. After feature mapping, for both BChE and AChE inhibitors, the uncertainty value of 1.5 was set in the training set, which states that the biological activity of the training set is in the range of 1.5 times more than or less than the reported value (Ambure and Roy 2014; Lu et al. 2011; Ma et al. 2014). Then, considering these features, the pharmacophore models were developed by using the FAST, BEST, and CEASER algorithms. Initially, 10 hypotheses were generated by each algorithm. The results of the FAST, BEST, and CEASER algorithms were reported in supplementary (Table S2). Among the 10 hypotheses, the best models were selected based on different internal and external validation parameters such as total cost value, null cost value, fixed cost value, cost error, features, feature weight, maximum fit value, correlation (r) value, cross-validation correlation (q), root mean square error (RMSE), mean absolute error value (MAE), accuracy, precision, sensitivity, specificity, F-measure and precision (Fawcett 2006), the area under the curve-receiver operating characteristics (AUC-ROC) (Fawcett 2006), Matthews correlation coefficient (MCC) (Matthews 1975), and geometric mean (G-means). The details of the different parameters are given below:
Cost analysis
It is an essential parameter in evaluating a good pharmacophore model. Three cost values were kept in mind to calculate cost analysis, i.e., fixed cost, null cost, and total cost. Δ Cost (Null cost-Total cost) If the difference is in the range of (i) > 60–90%, it shows 90% statistical significance (ii) ~ 30–40%, it shows 70–80% statistical significance (iii) < 20%, it shows 40–50% statistical significance (Jiang and Gao 2018; Ma et al. 2014).
Fischer’s randomization test
It is a statistical validation test used to evaluate the significance level of generated models. Fischer's randomization test is used to assess if there is a substantial relationship between chemical structures and biological activity. The desired confidence level for Fischer's randomization test are 90%, 95%, 98%, and 99%, and the selected confidence level in this study was set to 95%, where 19 random spreadsheets (random hypothesis) were generated (Chhabria et al. 2012).
The other validation parameters were reported in Table 1. From dataset division to pharmacophore model development, all steps were completed in BIOVIA Discovery Studio Client 4.1 software on a windows operating system.
Pharmacophore-based virtual screening
The final step of this research work was the pharmacophore-based virtual screening to get potent BChE and AChE inhibitors, the developed pharmacophore models were utilized for the virtual screening (Chhabria et al. 2012). A total of 2428 compounds were retrieved from different database, which upon the data curation, i.e. deletion of duplicate and structural error compounds, 1884 molecules were used for virtual screening (Fourches et al. 2010; Ambure et al. 2019). Finally, the ligand preparation was done for 1884 curated molecules. After the ligand preparation, the selected pharmacophore models were applied to these curated 1884 compounds for the virtual screening using the ligand pharmacophore mapping tool in Discovery Studio Client 4.1. Finally, the active compounds were selected and subjected to ADMET characteristics and the Lipinski filters (Lipinski 2000) for drug-likeness property analysis. The whole methodology is represented in Fig. 1

Flow diagram of the entire work
Heat map ligand profiler
The heat map of the Ligand profiler is displayed using different colors. The various colors represent the degree of matching. The red one with the maximum fit value means good matching; the blue one with the minimum fit value shows imperfect matching (Jiang and Gao 2018).
Results and discussion
Development of 3D QSAR pharmacophore model
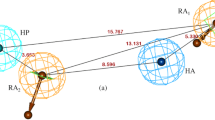
3D QSAR pharmacophore models were developed by using all three confirmation methods available in the software, i.e., FAST, BEST, and CEASER. The BEST method produced superior results based on various validation parameter values for the BChE and AChE datasets. Moving further with this method, ten quantitative hypotheses were generated and ranked based on the "total cost" values. Results obtained from the BEST method were considered for further analysis. The BEST method identified the four pharmacophore features: HBA, HBA, HY, and RA for the BChE. The significant statistical parameter values such as total cost, correlation coefficient (r), pharmacophore features, maximum fit value, cross-validation correlation, and root-mean-square deviation (RMSD) for each selected hypothesis from FAST, BEST, and CEASER methods have been enumerated in Table 2. The statistical parameters for the remaining hypotheses from FAST, BEST, and CEASER methods were reported in Table S2. The Hypo 5 of the BEST method (for BChE), shown in Table 2, was categorized best among all the 10 hypotheses. It showed the lowest total cost (130.85), the highest cost difference (54.55), the lowest RMSD (0.61), the lowest error (111.50), and the best correlation coefficient (0.80). The other cost values, such as fixed cost and null cost, were 85.02 and 185.40 bits. The total cost value of hypo 5 was relatively close to the fixed cost and was lower than the null cost. This evidence suggests that Hypo 5, which accounts for three pharmacophore features, first HBA, second HY, and third RA, has a high level of predictability. The structural features, feature distance, and angles of the Hypo 5 pharmacophore model were shown in Fig. 2a. The features of Hypo 5 were mapped completely with the most potent compound (compound no. 108) having a maximum fit value of 4.85 of the training set, whereas one of the hydrogen bond acceptor feature of Hypo 5 was not mapped with the least active compound (compound no. 13), having a maximum fit value of 3.10 and was illustrated in Fig. 3a, b respectively. According to the parameters mentioned above, in BChE, Hypo 3 of the FAST algorithm and Hypo 1 of the CEASER algorithm were selected as good hypotheses. The structural features of the selected models were represented in the supplementary Fig. S1.

The structural features of Hypo 5 of BChE (a) and Hypo 9 of AChE inhibitor (b)

Most and Least active compounds mapped with developed models of BChE a most active (compound no 108) and b least active (compound no. 13)) and AChE c most active (compound no 81) and d least active (compound no. 10))
Similarly, for AChE inhibitors, 10 hypotheses were generated and validated using various statistical parameters reported in Table S2. Among the 10 hypotheses of the BEST algorithm, Hypo 9 was selected for further analysis as it has the lowest total cost value (384.39), the highest cost difference (321.83), the lowest RMSE (2.30), and the best correlation coefficient (0.80) as compared to all hypotheses reported in Table 2. The fixed cost was 189.91, and the null cost was 706.22 bits. The features generated by the Hypo 9 model were HBA1, HBA2, one HY, and one PI, which were given in Fig. 2b. The training set was perfectly mapped with all four features of Hypo 9 after mapping with the most potent compound (compound no. 81), with a maximum fit value of 4.00 (Fig. 3c). While mapping with the least active compound (compound no. 10), a maximum fit value of 1.75 was observed, and the feature PI was not mapped with the least active compound, as shown in Fig. 3d. On the basis of validation parameters, Hypo 1 in the FAST algorithm and Hypo 6 in the CEASER algorithm were chosen as good, and the selected models Hypo 1 in FAST and Hypo 6 in CEASER, were shown in Fig. S1.
Validation of pharmacophore models
Cost analysis
A "cost difference" is the difference between a null cost and a total cost. The importance and significance of cost values were already mentioned in materials and methods section. The fixed cost represents the simplest model, and it fits with all the predicted data ideally in the training test compounds, while the null cost has no features with the high-cost value, and the experimental activities are usually spread around their mean value (Chadha and Silakari 2017). According to these parameters, the values of Hypo 5 and Hypo 9 of BChE and AChE could be selected as the best hypotheses with a greater cost difference of 54.55 and 321.83, respectively, as reported in Table 2. Thus, this data indicates that the probability of correlation was more than 90%. Figure 4a, b shows the cost analysis results for Hypo 5 of BChE and Hypo 9 of AChE inhibitors. Figure S2 depicts the cost analysis results of FAST and CEASER for BChE and AChE inhibitors.

Cost analysis of BEST for Hypo 5 of the BChE inhibitors (a) and Hypo 9 of the AChE inhibitors (b)
Fischer’s randomization test
A Fischer randomization test was performed to validate the developed pharmacophore model’s significance. For all the developed models, the confidence level was assigned to 95%, and 19 random spreadsheets were generated, which suggests the statistical robustness of Hypo 5 in BChE and Hypo 9 in AChE. The fisher correlation of BEST Hypo 5 in BChE inhibitors and Hypo 9 in AChE inhibitors were shown in Fig. 5a, b. The result suggests that the models were not generated by chance. Also, Fischer’s correlation of FAST and CEASER algorithms for BChE and AChE was represented in Fig. S3.

Fischer correlation of the BEST algorithm for Hypo 5 of the BChE inhibitors (a) and Hypo 9 of the AChE inhibitors (b)
Training and test set validation
The training and test datasets were used to verify the best pharmacophore model. Hypo 5 and Hypo 9 of the known BChE and AChE inhibitors were found best after the screening of different validation parameters. Based on experimental activity (IC50 μM), the training and test sets were classified into three activity scales: highly active = + + + , IC50 < 1 μM; moderately active = + + , IC50 ≤ 1 μM; low active or inactive = + , IC50 ≥ 10 μM. The experimental Vs. predicted activity of training and test set in BChE and AChE inhibitors of FAST, BEST, and CEASER was highlighted in Table S3. In the training set, the strongest correlation (r) of 0.80 was observed between the experimental and estimated activity of Hypo 5 of BChE (Fig. 6a). Table S3 revealed that two highly active compounds and five inactive compounds based on experimental value were predicted as moderately active and two moderately active compounds as inactive by the developed model based on BEST method (Hypo 5). While in the case of test set compounds, one moderately active compound based on experimental value was predicted as inactive (Table S3). The correlation of the experimental Vs. predicted activity by Hypo 5 for training and test set were represented in Fig. 6a, b, respectively.

Log experimental Vs. log predicted activity (IC50) by Hypo 5 of BChE in the training set compounds (a), test compounds (b), Hypo 9 of AChE in the training set compounds (c), and test set compounds (d)
Similarly, for AChE inhibitors, as reported in Table S3, the training set of AChE inhibitors showed nine highly active compounds and three inactive compounds based on experimental value were predicted as moderately active, and ten moderately active compounds were predicted as inactive by the developed model based on the BEST method (Hypo 9). In the test set compound, two highly active and five inactive compounds based on experimental value were predicted as moderately active and one moderately active compound as inactive. The correlation of the experimental Vs. predicted activity by Hypo 9 for training and test set were represented in Fig. 6c, d, respectively.
Based on these outcomes, Hypo 5 and Hypo 9 were not only capable of appropriately predict the training sets inhibitory activity, but it was also appropriate for the test set or unknown compounds. Thus, both the hypothesis of BChE and AChE has been proven to be reliable pharmacophore models for identifying potential BChE and AChE inhibitors through database screening. The correlation of the experimental and predicted activity for BChE and AChE based on selected models of FAST and CEASER methods were represented in Fig. S4 and Fig. S5.
Some other validation parameters were also applied to check the robustness and predictive activity of the selected models, and the obtained results for the BEST method were reported in Table 3. The ROC curves of the selected models (Best method: Hypo 5 for BChE and Hypo 9 for AChE) were given in Fig. 7. The results of these parameters for FAST and CEASER methods were reported in supporting information (Table S4 and Figs. S6, S7).

The ROC curve of the training set (a), test set (b) of BChE, and ROC curve of the training set (c), test set (d) of AChE
Pharmacophore-based virtual screening
The validated hypo 5 and hypo 9 of the BEST method for BChE and AChE were used as 3D structural queries to find new lead compounds from different databases. Using the protocol Ligand Pharmacophore Mapping, 1884 finally selected ligands were subjected for virtual screening with Hypo 5 of BChE and Hypo 9 of AChE. After screening, 90 compounds for BChE and 6 compopunds for AChE were obtained. Further, the compounds with an estimated activity of less than 10 µM were selected as the potent BChE inhibitors. As a result, 15 compounds (estimated IC50 = 1.13–9.08 µM) for BChE and one compound (estimated IC50 = 6.90 µM) for AChE were identified as potent inhibitor. Finally, Lipinski filters and ADMET analysis were done for these identified potent BChE and AChE inhibitors. Upon the ADMET and Lipinski filters 13 compounds for BChE and 1 compound for AChE were obtained with good ADMET and drug-likeness properties (Table S5). The 2D structures of all 13 hits for BChE and 1 hit for AChE with their IDs and estimated activity were reported in Table S6. The fitting of identified thirteen potent compounds with the pharmacophore model was represented in Fig. 8, which indicates that all the identified compounds were fitted properly with selected pharmacophore model. The most active compounds for BChE and AChE mapped with pharmacophore models were represented in Fig. 9a, b. These hit compounds could be promisingly used as a better alternatives to the currently used pharmaceuticals for the treatment or management of AD.

The fitting of the 13 hits with the pharmacophore model of BChE along with the most potent compound of the BChE

Most active compounds mapped with pharmacophore features (a BChE and b AChE)
Heat map ligand profiler
The pharmacophore model was further validated with the protocol "Ligand Profiler". The ligand profiler results were displayed as a heat map, a two-dimensional table in which the x-axis represents the 3D QSAR pharmacophore models hypotheses, and the y-axis means compounds. Figure 10a shows the heat map generated by the ligand profiler for the best 15 compounds obtained by virtual screening of BChE, and Fig. 10b shows the heat map generated by the ligand profiler for the one compound obtained by virtual screening of AChE. The color difference indicated the fit value; the color scale was represented in Fig. 10a, b. In the color scale, red suggests the best matching of ligands and blue color with a low fit, while in between blue and moderate red fitting. The selected compounds (15 for BChE and 1 for AChE) for the ligand profiler were good to moderately fitted on hypothesis 5 for BChE and hypothesis 9 for AChE which also supports the selected hypothesis were robust enough and this results suggests that the model can be useful for the discovery of new BChE and AChE inhibitors.

Ligand Profiler Heat map for best hits of a BChE inhibitor and b AChE inhibitor obtained after virtual screening
Overview and conclusion
The rate of new AD cases is continuously increasing and need to slow or to stop with the help of effective drug treatment. BChE and AChE are the two primary promising targets mainly involved in AD, and inhibitors of these two targets are the choice for anti-alzehimer drug development. Coumarins, natural derivatives, are reported as cholinesterase inhibitors and emerges as a promising scaffold for design of ligands targeting enzymes and pathological alterations related to AD with minor structural alterations or finding of similar compounds through virtual screening. With this aim, statistically robust, significant, and predictive 3D QSAR pharmacophore models were developed for BChE and AChE structural data with three different confirmation methods. The best features extracted were HBA, HBA, HY, RA (BChE) and HBA, HBA, HY, and PI (AChE). Overall, the BEST method generated the best quality model and was further utilized for identifying potent BChE and AChE inhibitors from different database (DrugBank, ChEMBL drug candidate clinics, Zinc fragments, ChEMBL approveddrugs, MiniMaybridge, ChemBridge, Bioactive ligandexproBioactive, and ChEMBL complete database). The screening result yields thirteen new BChE inhibitors with IC50 of less than 10 µM and one potent AChE inhibitor with IC50 of 6.90 µM. Besides potency, these compounds have a good ADMET (drug-like) profile. They can be utilized as a drug of choice for the treatment of Alzheimer’s diseases or certain neurodegenerative diseases upon some experimental investigations. The study is useful for filling current needs and may optimize future discovery of novel anti-alzheimer's agents. Additionally, BChE and AChE have crucial roles in some other neurodegenerative diseases, and such advantages can be utilized to design polypharmacological or multi-targeted drug candidates.
References
Abdelmohsen UR, Albohy A, Abdulrazik BS, Bayoumi SAL, Malak LG, Khallaf ISA et al (2021) Natural coumarins as potential anti-SARS-CoV-2 agents supported by docking analysis. RSC Adv 11(28):16970–16979
Abu-Aisheh MN, Al-Aboudi A, Mustafa MS, El-Abadelah MM, Ali SY, Ul-Haq Z et al (2019) Coumarin derivatives as acetyl- and butyrylcholinestrase inhibitors: an in vitro, molecular docking, and molecular dynamics simulations study. Heliyon 5(4):e01552
Alzheimer’s Association (2021) Alzhiemer’s disease facts and figures. Alzheimers Dement 11(3):332–384
Alzheimer’s disease facts and figures. Alzheimers. Dement. [Internet]. 2021;17Available from: http://www.ncbi.nlm.nih.gov/pubmed/33756057
Ambure P, Roy K (2014) Advances in quantitative structure-activity relationship models of anti-Alzheimer’s agents. Expert Opin Drug Discov 9(6):697–723
Ambure P, Bhat J, Puzyn T, Roy K (2019) Identifying natural compounds as multi-target-directed ligands against Alzheimer’s disease: an in silico approach. J Biomol Struct Dyn 37(5):1282–1306
Arya H, Coumar MS (2021) Lead identification and optimization. The design and development of novel drugs and vaccines. Elsevier, pp 31–63
Atatreh N, Al Rawashdah S, Al Neyadi SS, Abuhamdah SM, Ghattas MA (2019) Discovery of new butyrylcholinesterase inhibitors via structure-based virtual screening. J Enzyme Inhib Med Chem 34(1):1373–1379
Buran K, Reis R, Sipahi H, ÖnenBayram FE (2021) Piperazine and piperidine-substituted 7-hydroxy coumarins for the development of anti-inflammatory agents. Arch Pharm (Weinheim) 354(7):e2000354
Chadha N, Silakari O (2017) Pharmacophore based design of some multi-targeted compounds targeted against pathways of diabetic complications. J Mol Graph Model. Elsevier Inc 76:412–418
Chhabria MT, Brahmkshatriya PS, Mahajan BM, Darji UB, Shah GB (2012) Discovery of novel acyl coenzyme a: cholesterol acyltransferase inhibitors: pharmacophore-based virtual screening, synthesis and pharmacology. Chem Biol Drug Des 80(1):106–113
Contestabile A (2011) The history of the cholinergic hypothesis. Behav Brain Res 221(2):334–340
Coyle JT, Price DL, DeLong MR (1983) Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 219(4589):1184–1190
Ellman GL, Courtney KD, Andres V, Featherstone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7(2):88–95
Fawcett T (2006) An introduction to ROC analysis. Pattern Recognit Lett 27(8):861–874
Ferreira-Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM (2016) Alzheimer’s disease: targeting the cholinergic system. Curr Neuropharmacol 14(1):101–115
Folch J, Petrov D, Ettcheto M, Abad S, Sánchez-López E, García ML et al (2016) Current research therapeutic strategies for Alzheimer’s disease treatment. Neural Plast 2016:8501693
Fourches D, Muratov E, Tropsha A (2010) Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J Chem Inf Model 50(7):1189–1204
Francis PT (2005) The interplay of neurotransmitters in Alzheimer’s disease. CNS Spectr 10(S18):6–9
Francis PT, Palmer AM, Snape M, Wilcock GK (1999) The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry 66(2):137–147
Hampel H, Mesulam M-M, Cuello AC, Farlow MR, Giacobini E, Grossberg GT et al (2018) The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141(7):1917–1933
Healthline (2021) COVID-19, Alzheimer’s Disease, and memory loss: what we know
Heo JH, Eom BH, Ryu HW, Kang MG, Park JE, Kim DY et al (2020) Acetylcholinesterase and butyrylcholinesterase inhibitory activities of khellactone coumarin derivatives isolated from Peucedanum japonicum Thurnberg. Sci Rep. Nature Publishing Group UK 10(1):1–11
Hu YH, Yang J, Zhang Y, Liu KC, Liu T, Sun J et al (2019) Synthesis and biological evaluation of 3–(4-aminophenyl)-coumarin derivatives as potential anti-Alzheimer’s disease agents. J Enzyme Inhib Med Chem. Taylor & Francis 34(1):1083–1092
Jiang Y, Gao H (2018) Pharmacophore-based drug design for potential AChE inhibitors from Traditional Chinese Medicine Database. Bioorg Chem 76:400–414
Khedkar SA, Malde AK, Coutinho EC, Srivastava S (2007) Pharmacophore modeling in drug discovery and development: an overview. Med Chem 3(2):187–197
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44(1):235–249
Lu S-H, Wu JW, Liu H-L, Zhao J-H, Liu K-T, Chuang C-K et al (2011) The discovery of potential acetylcholinesterase inhibitors: a combination of pharmacophore modeling, virtual screening, and molecular docking studies. J Biomed Sci 18:8
Ma Y, Jin Y-Y, Wang Y-L, Wang R-L, Lu X-H, Kong D-X et al (2014) The discovery of a novel and selective inhibitor of PTP1B over TCPTP: 3D QSAR pharmacophore modeling, virtual screening, synthesis, and biological evaluation. Chem Biol Drug Des 83(6):697–709
Matthews BW (1975) Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta - Protein Struct 405(2):442–451
Menezes JC, Diederich M (2019) Translational role of natural coumarins and their derivatives as anticancer agents. Future Med Chem 11(9):1057–1082
NIH National Institute of Aging (2021) Alzheimer’s Disease Fact Sheet
Nordberg A, Ballard C, Bullock R, Darreh-Shori T, Somogyi M (2013) A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim Care Companion CNS Disord. https://doi.org/10.4088/PCC.12r01412
Piazzi L, Cavalli A, Colizzi F, Belluti F, Bartolini M, Mancini F et al (2008) Multi-target-directed coumarin derivatives: hAChE and BACE1 inhibitors as potential anti-Alzheimer compounds. Bioorganic Med Chem Lett 18(1):423–426
Pibuel MA, Díaz M, Molinari Y, Poodts D, Silvestroff L, Lompardía SL et al (2021) 4-Methylumbelliferone as a potent and selective antitumor drug on a glioblastoma model. Glycobiology 31(1):29–43
Pisani L, Catto M, De Palma A, Farina R, Cellamare S, Altomare CD (2017) Discovery of Potent dual binding site acetylcholinesterase inhibitors via homo- and heterodimerization of coumarin-based moieties. ChemMedChem 12(16):1349–1358
Rullo M, Catto M, Carrieri A, de Candia M, Altomare CD, Pisani L (2019) Chasing ChEs-MAO B Multi-Targeting 4-Aminomethyl-7-Benzyloxy-2H-Chromen-2-ones. Molecules 24(24):4507
Sliwoski G, Kothiwale S, Meiler J, Lowe EW (2014) Computational methods in drug discovery. Pharmacol Rev 66(1):334–395
Sun H (2008) Pharmacophore-based virtual screening. Curr Med Chem 15(10):1018–1024
Wang H, Yang F, Zhang S, Xin R, Sun Y (2021) Genetic and environmental factors in Alzheimer’s and Parkinson’s diseases and promising therapeutic intervention via fecal microbiota transplantation. Npj Park Dis 7(1):70
Wilkinson DG, Francis PT, Schwam E, Payne-Parrish J (2004) Cholinesterase inhibitors used in the treatment of Alzheimer’s disease: the relationship between pharmacological effects and clinical efficacy. Drugs Aging 21(7):453–478
Yang S-Y (2010) Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 15(11–12):444–450
Yang H, Du C, Li Q, Chen T, Lu X, Li Q et al (2019a) Discovery, molecular dynamic simulation and biological evaluation of structurally diverse cholinesterase inhibitors with new scaffold through shape-based pharmacophore virtual screening. Bioorg Chem 92:103294
Yang J, Zhang P, Hu Y, Liu T, Sun J, Wang X (2019b) Synthesis and biological evaluation of 3-arylcoumarins as potential anti-Alzheimer’s disease agents. J Enzyme Inhib Med Chem 34(1):651–656
Yang X-C, Zhang P-L, Kumar KV, Li S, Geng R-X, Zhou C-H (2022) Discovery of unique thiazolidinone-conjugated coumarins as novel broad spectrum antibacterial agents. Eur J Med Chem 232:114192
Yun Y, Yang J, Miao Y, Wang X, Sun J (2020) Synthesis and biological evaluation of 4-arylcoumarins as potential anti-Alzheimer’s disease agents. Bioorg Med Chem Lett 30(4):126900
Acknowledgements
Financial assistance from the SCIENCE & ENGINEERING RESEARCH BOARD (SERB) DST, Govt. of India, New Delhi (File no. EMR/2017/004497) is gratefully acknowledged by. Partha Pratim Roy. The authors are also thankful to The Department of Pharmacy, Guru Ghasidas Vishwavidyalaya, Bilaspur, Chhattisgarh, India, 495009, for providing the research facilities.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lakra, N., Matore, B.W., Banjare, P. et al. Pharmacophore based virtual screening of cholinesterase inhibitors: search of new potential drug candidates as antialzheimer agents. In Silico Pharmacol. 10, 18 (2022). https://doi.org/10.1007/s40203-022-00133-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40203-022-00133-1