
Abstract
A proper understanding of the structure and microstructure of additively manufactured (AM) alloys is essential not only to the prediction and assessment of their material properties, but also to the validation and verification of computer models needed to advance AM technologies. To accelerate AM development, as part of the AM-Bench effort, we conducted rigorous synchrotron-based X-ray scattering and diffraction experiments on two types of AM alloys (AM 15-5 stainless steel and AM Inconel 625). Taking advantage of the high penetration of synchrotron hard X-rays, we determined the phases present in these alloys under different build conditions and their statistically meaningful phase fractions using high-resolution X-ray diffraction. Using in situ multi-scale X-ray scattering and diffraction, we quantitatively analyzed the phase evolution and development of major precipitates in these alloys as a function of time during stress relief heat treatments. These results serve to validate AM microstructure models and provide input to higher-level AM processing and property models to predict the material properties and performances.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction and Background
Additive manufacturing (AM) of metals encompasses a class of versatile manufacturing technologies that build upon three-dimensional digital models and enable fabrication of parts with complex, often previously unattainable, shapes and geometries [1]. In its current development and commercialization state, AM represents high value-added manufacturing and draws much attention from industries including defense, aerospace, medicine, and oil and gas.
AM of metals requires repeated heating and cooling to and from the melting temperature of the underlying metallic materials at rates as high as 106 K/s [2]. Fundamentally, this can lead to highly heterogeneous solidification microstructures [3], manifestation of which includes asymmetric crystalline grains, compositional heterogeneities, and unexpected phase landscapes. Such complexity can introduce a monumental challenge in reliably creating certifiable materials with reproducible microstructures and predictable material properties [4, 5]. To overcome this challenge, post-build heat treatment is often required.
AM technologies, from an experimental point of view, have an exceedingly large parameter space. Experimental exploration of such a parameter space is time- and cost-prohibitive. Recent developments in modeling, particularly Integrated Computational Material Engineering (ICME)-based computer models, allow the complex AM process to be described and modeled by considering the materials and their associated materials processing methods at a range of different time and length scales [6,7,8]. These integrated models seek to evaluate material properties on one length and time scale with one or multiple modeling components, while using information provided by other model components on other length and time scales as input. ICME models for AM frequently include CALPHAD-based computational thermodynamic and thermokinetic models for prediction of equilibrium and near-equilibrium phase diagrams, phase field models to simulate microstructure evolution, and finite element models to describe the fabrication process.
While computer models have demonstrated their potential in predicting AM material behavior, their fidelity cannot be established unless these models are validated and verified against rigorous and controlled experiments. To meet this critical need and to enable rapid and reliable development in the emerging AM field, the US National Institute of Standards and Technology (NIST) has worked with a number of national and international stakeholders to develop the Additive Manufacturing Benchmark (AM-Bench) test series [9]. The results reported here address two 2018 AM-Bench challenges:
- 1.
Phase fractions (CHAL-AMB2018-01-PF): Predict the phases and phase fractions, including major precipitates, within the transverse samples from the thick and thin legs of the as-built 15-5 stainless steel (SS) and Inconel 625 (IN625) AM-Bench test artifacts.
- 2.
Phase evolution (CHAL-AMB2018-01-PFRS): Predict the phases and phase fractions, including major precipitates, as a function of time for residual stress anneals of 15-5 SS and IN625, from transverse specimens cut from the thick and thin legs of the AM-Bench test artifacts.
Specifically, these two challenges aim at revealing the atomic structure and microstructure of two classes of AM materials built under two different conditions. Such information is essential in establishing the structure–property–performance relationship of the AM materials at the most fundamental level and needs to be considered in comprehensive ICME-type models to understand how processes produce material structures and give rise to material properties, and to ultimately fulfill their potential in industrial utility.
The results reported here are acquired using synchrotron hard X-rays. The high energy of the synchrotron X-rays leads to low X-ray absorption in metallic materials, hence enabling accurate measurements of bulk material properties in special sample environments, such as furnace and tensile stages. The high flux of synchrotron X-rays enables a temporal resolution that can be as low as sub-second, which allows in situ characterization of structure phase transformation [10], stress relaxation [11], homogenization [12], and precipitation [13, 14]. With these advantages, we present rigorous and statistically meaningful studies using two types of synchrotron-based hard X-ray scattering and diffraction methods, namely ex situ high-resolution X-ray diffraction experiments and in situ combined ultra-small-angle and small-angle X-ray scattering and X-ray diffraction experiments. In this paper, we focus on establishing the needs of the measurements, describing the measurement methodology, and presenting experimental data and results, without providing any thermodynamic modeling results of our own that potentially can be regarded as preconceiving. Our overarching goal is to provide rigorous and statistically meaningful data for the validation of AM models.
Materials and Methods
Materials
IN625 and 15-5 SS parts were printed from virgin IN625 and 15-5 powders by the Measurement Science for Additive Manufacturing program at NIST, Gaithersburg, USA, using an EOSFootnote 1 INT M270 laser sintering powder-bed fusion instrument (EOS GmbH, Munich, Germany) that was modified for quantitative in situ monitoring. The manufacturer-certified chemical compositions of the raw IN625 and 15-5 powders used in this study are listed in Tables 1 and 2, respectively. The compositions reported in these tables were provided by vendor-supplied data sheets and determined following ASTM E1019 and ASTM E2823/#1479. The compositions were also remeasured and confirmed by third-party ASTM E1019 measurements. These tables clearly show that the starting powders used in this study are within the standard composition ranges for IN625 and 15-5 alloys. The measured size distributions of the powders are listed in Table 3.
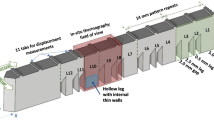
During the build, a Nd:YAG laser was operated at 195 W with an approximately Gaussian-shaped power distribution function at the sample position and an estimated spot size (full width at half maximum) of 50 µm. The scanning speed was 800 mm/s. The hatch distance was 100 µm. The build was conducted in N2 with an approximate oxygen level of 0.5%. The height of each layer is ≈ 20 µm. The AM builds of IN625 and 15-5 follow the same bridge structure geometry that has 12 legs with different sizes. This bridge structure is illustrated in Fig. 1. More details about the build can be found elsewhere.

Schematic of the bridge structure and the cuts necessary for X-ray samples with leg indices highlighted. The red lines denote EDM cuts
To prepare the X-ray samples, three different sets of wire electron discharge machining (EDM) cuts were made. The first cut, highlighted as “Cut location 1” in Fig. 1, removes the top of the bridge. The second set of cuts consisted of 8 vertical cuts from the centers of the 5-mm-thick legs, which were made to produce 4 specimens of 0.5 mm thickness. The last cut was a single EDM cut that removed the legs from the base plate. All these cuts were made in the as-built condition, i.e., without a stress relief heat treatment. The X-ray specimens were mechanically thinned and polished to remove surface abnormalities, following a standard metallographic procedure.
Methods
X-ray Diffraction (XRD) Measurements
High-resolution synchrotron XRD experiments were conducted at the dedicated powder XRD beamline 11-BM-B at the Advanced Photon Source (APS), Argonne National Laboratory [15]. This instrument makes use of Si (111) analyzing crystals, leading to a q resolution Δq/q ≈ 2 × 10−4. Here q = 4π sin(θ)/λ, where θ is one half of the scattering angle and λ is the X-ray wavelength.
The monochromatic X-ray energy was 30 keV (wavelength, λ = 0.414554 Å). The X-ray flux was 5 × 1011 photons s−1 mm−2. The beam size was 500 µm × 200 µm. We acquired XRD data in a 2θ (diffraction angle) range of 0.5°–50°, with a step size of 0.0025° and dwell time per step of 0.25 s. This 2θ range translates to a q range of 0.1323–12.811 Å−1. The instrument was calibrated using NIST Standard Reference Material, SRM 660a (LaB6: lanthanum hexaboride) [16]. The unique combination of high X-ray flux, angular resolution, and sensitivity allows the lattice parameters to be accurately determined and weak phases to be detected.
We performed XRD experiments on as-built IN625 and 15-5 SS specimens. The samples are listed in Table 4. The samples were thinned to ≈ 100 µm in thickness and subsequently cut to thin strips with approximate dimensions of 1 mm × 10 mm × 0.1 mm. The strips were loaded in Kapton capillaries and mounted on the standard sample holders of the beamline. During data collection, the samples spun rapidly in the beam (at ≈ 3000 RPM). All measurements were conducted at room temperature (≈ 298 K). The phase identification and analyses were conducted using GSAS-II [17]. To examine the structures at different locations of the samples, we conducted measurements from three non-overlapping positions for each sample. The separation distance between these non-overlapping positions is at least 1 mm.
Ultra-Small-Angle X-ray Scattering and Diffraction Measurements
In situ synchrotron ultra-small-angle X-ray scattering (USAXS), small-angle X-ray scattering (SAXS), and XRD experiments were conducted at the 9-ID-C USAXS facility at the APS [18]. The X-ray energy was 21 keV (λ = 0.5904 Å). The flux density was ≈ 1013 photons s−1 mm−2.
We used combined USAXS and SAXS to monitor the changes in the morphology of the precipitates during the heat treatment. The USAXS measurements were conducted using Bonse-Hart-type crystal optics [19] with a beam size of 0.8 mm × 0.8 mm. USAXS covers a q range from 1 × 10−4 to 0.3 Å−1 and directly provides absolute scattering intensity (scattering cross section) [20, 21]. SAXS measurements were conducted with a single-photon-counting PILATUS 100 K detector (model: 100K-S, Dectris, Baden, Switzerland) in a pinhole small-angle scattering geometry. The SAXS beam size was 0.2 × 0.8 mm, which provides the best possible counting statistics while matching the detector resolution. The SAXS camera was calibrated using a AgBeh calibration standard [22]. The combined accessible q range for USAXS and SAXS is 1 × 10−4–1.5 Å−1, i.e., a continuous size range from ≈ 4 Å to 6 µm, and the combined dynamic range in linear intensity response exceeds 10 orders of magnitude.
To evaluate the changes in the atomic structures in the samples during the heat treatments, we made use of a modified PILATUS 300KW detector to perform area detector-based XRD experiments in a q range between 1.4 and 6.8 Å−1 [18]. We calibrated the q values and sample-to-detector distances using NIST SRM 660a (LaB6) [16]. We used a beam size of 0.2 mm × 0.8 mm for the in situ XRD measurements.
For the in situ experiments, the as-built specimens were mechanically polished to ≈ 50 µm for 15-5 SS and ≈ 40 µm for IN625 in thickness. This difference in thickness is due to different X-ray absorption coefficients of these materials. The in situ heat treatment experiments were conducted using a Linkam 1500 thermal stage (Linkam Scientific Instruments Ltd., Tadworth, UK). The thermal stage was calibrated using a 99.999% purity Pt foil sample. The temperature uncertainty depends on the set temperature and is estimated to be within 1 °C in the temperature range used for this study. We performed the in situ isothermal heat treatment under stress relief conditions on AM 15-5 SS and AM IN625 samples. Details of the in situ samples are listed in Table 5. The heat treatment temperature for AM 15-5 SS was 650 °C. The heat treatment temperature for AM IN625 was 800 °C. This temperature was recommended by Lass et al. [23] as an alternative stress relief heat treatment temperature to the EOS recommended temperature of 870 °C. The heating rate from room temperature to the target temperatures was set at 200 °C per min. For each heat treatment series, we conducted the combined measurements in a repeated sequence of USAXS, SAXS, and XRD, with individual scan times set at 90 s, 30 s, and 60 s, respectively. Including the time required for motor motions, each set of measurements took ≈ 5 min. For each isothermal heat treatment condition, we conducted the in situ experiment for at least 2 h.
We performed detailed SAXS analyses to investigate the morphological evolution of the precipitates and XRD analyses to investigate the changes in the phase components and their phase fractions. SAXS data reduction and analysis were conducted using the small-angle scattering analysis software Indra, Irena, and Nika, developed in the Igor Pro programming environment [24, 25]. For the SAXS analysis, we adopted a model previously developed for precipitates in alloys [13, 14]. XRD data reduction was conducted using Nika [25].
Results
Phase Fraction Analysis
AM 15-5 SS XRD Results
15-5 SS is designed to be a martensitic stainless steel, which can be further strengthened by copper-containing precipitates with a relatively low-temperature heat treatment to provide a greater toughness than 17-4 SS. The as-built AM 15-5 SS, as we will show, is not 100% martensitic. Instead, it contains a small fraction of austenite. The crystal symmetries of the martensitic and austenitic phases are Im-3 m and Fm-3 m, respectively. In other words, for the martensitic phase, the c/a ratio is 1 within our measurement uncertainties.
Rietveld analysis provides information related to the lattice parameters of the martensitic and austenitic phases and their phase fractions. In this analysis, we assumed no microsegregation. A typical analysis result is demonstrated in Fig. 2.

A typical synchrotron XRD dataset demonstrates that as-built AM 15-5 SS contains two phases with face-centered cubic structure and body-centered cubic structure, respectively. The lattice parameters and phase fractions of these phases are listed in Table 6
Figure 2 clearly shows the presence of two families of peaks that can be described by face-centered cubic (FCC) austenite and body-centered cubic (BCC) martensite/ferrite, which are highlighted by red and blue stick patterns. With these two crystal structures, the entire XRD pattern is well described, as demonstrated by a difference curve shown in green.
We repeated this analysis for all four 15-5 SS samples at three positions in each case. The lattice parameters and phase fractions of martensite/ferrite and austenite phases are listed in Table 6. To aid direct visual comparison, they are also plotted in Fig. 3. In Fig. 3, results acquired on three different positions of the same sample are grouped by color. Figure 3 and Table 6 show that, independent of build history, the as-built AM 15-5 SS consists of mostly (> 98% phase fraction) BCC martensite/ferrite and a small amount of (< 2%) FCC austenite. The lattice parameters, even on the same sample, vary slightly from position to position. This is not surprising, since the thermal history is location specific and far from uniform.

Position-dependent lattice constants and phase fractions of martensite/ferrite and austenite in AM 15-5 SS. The data are also reported in Table 6. The results acquired on three different positions of the same sample are grouped by the same color
We further performed a statistical analysis to calculate the sample-specific mean and standard deviation of the lattice parameters and the phase fractions. They are reported in Table 7. Similar to Fig. 4, the position-averaged lattice parameters and volume fractions are plotted in Fig. 4. There is no significant evidence for suggesting that the differences in the build conditions for the thick and thin legs strongly affect the lattice parameter and phase fractions of austenite and martensite/ferrite.

AM Inconel 625 XRD Results
IN625 is a nickel-based superalloy strengthened by solid solution hardening of Nb and Mo in a Ni–Cr matrix. Crystallographically, wrought IN625 has an FCC structure (crystal symmetry of Fm-3 m). As-built AM IN625 also has an FCC structure, as shown in Fig. 5. Notably, the XRD peaks are asymmetric, as illustrated by the inset of Fig. 5. No secondary phases or precipitates were detected in the as-built materials.

Typical high-resolution XRD pattern reveals that the as-built IN625 is single-phase FCC. The peak shape, however, is asymmetric, as highlighted by the inset. Possible cause of the peak asymmetry includes local elastic strains within the dislocation structures and elemental segregation
We made use of the centers of mass of the diffraction peaks to decide the overall lattice parameters. In this analysis, we first identified the 2θ angle corresponding to the maximum intensity of the nth peak, 2θnmax. We then calculated the center of mass within an angular range of 2θnmax – 0.25° to 2θnmax + 0.25°. Here, 0.25° was chosen such that it is well above the full width at half maximum of the diffraction peaks (≈ 0.03°) yet does not cause overlapping between the integration 2θ range (2θnmax – 0.25° and 2θnmax + 0.25°) of neighboring peaks. The successful phase identification, as illustrated in Fig. 5, allows us to determine the h, k, l values of each peak. Hence, based on the centers of mass, we can determine the lattice parameter corresponding to each peak in a given diffraction pattern. A statistical analysis of these lattice parameters leads to location-specific lattice parameters, which are listed in Table 8 and are plotted in Fig. 6. The results clearly demonstrate that within each sample, the lattice parameter does not strongly depend on the sample location. Furthermore, the different thermal history that leads to a slightly different degree of peak asymmetry in thin and thick legs (inset of Fig. 5) is not strongly reflected in the analyzed lattice parameters following this center of mass analysis.

Location-specific lattice parameters of as-built IN625 as determined by the center of mass positions of the diffraction peaks
For completeness, we performed a statistical analysis of the three sets of results acquired on each sample. These results are listed in Table 9 and Fig. 7. Again, thermal history does not appear to strongly affect the lattice parameters of the matrix phase of as-built IN625.

AM 15-5 SS In Situ USAXS/SAXS/XRD Results
We performed combined USAXS/SAXS/XRD measurements to both AM 15-5 SS and IN625 samples to provide an across-length-scale examination of the microstructure evolution. The results are reported in this subsection.
Figures 8 and 9 show representative in situ XRD and SAXS data of AM 15-5 SS under isothermal heat treatment at 650 °C.

Typical in situ XRD data of AM 15-5 acquired during isothermal heat treatment at 650 °C. The data acquisition time is color coded following the colored arrow in the figure. The inset shows the intensity variation in two peaks (left: austenite {111}; right: martensite/ferrite {011}) during the isothermal heat treatment

a Typical in situ SAXS data of AM 15-5 acquired during isothermal heat treatment at 650 °C. The data acquisition time is color coded following the colored arrow in the figure. b An illustration of the SAXS model. Experimental uncertainty of the USAXS data is within the extent of the plotting symbol
Figure 8 clearly shows that the preexisting phases (martensite/ferrite and austenite) remain in the alloy during the heat treatment, with no new, crystallographically detectable phases. It is worth noting that the Pilatus XRD detector is a highly sensitive single-photon-counting device. The lack of new XRD peaks strongly indicates no appreciable amount of other crystalline phases, such as carbides, formed during this heat treatment. The inset shows that the intensities of austenite peaks continuously grow and the intensities of the martensite/ferrite peaks continuously decrease, indicating that at least some martensite/ferrite was transformed to austenite during this heat treatment.
Figure 9a shows that during the heat treatment, a monotonic increase in the scattering intensity is observed. We constructed a model, as illustrated in Fig. 9b, where the observed scattering intensity is modeled as a sum of a preexisting scattering baseline and contributions from two types of scattering inhomogeneities. More details of this type of scattering model can be found elsewhere [13, 14, 26, 27]. Using this model, it is readily seen that two scattering inhomogeneities, well separated in length scales, contribute to the increase in the overall scattering intensity. We attribute one component as precipitate scattering, as it is known that at this temperature, nanoscopic Cu-based precipitates form to harden the alloy. Here, we assume that the precipitates are spherical in shape, as shown by transmission electron microscope (TEM) [28]. The origin of second component may involve changing of size and shape of the crystalline domains in the size range of 100 nm. Its determination requires further investigation. However, in situ XRD suggests that martensite/ferrite is being transformed to austenite during this heat treatment, and scattering contrast between austenite and martensite/ferrite, albeit small, exists. Because of these reasons, we tentatively attribute this second scattering component to domain scattering from austenite.
The analysis of the in situ XRD data provides the evolution of the phase fraction, whereas the analysis of the in situ SAXS data provides information related to the size and shape evolution of the precipitates and the domains. We present the detailed results of the four AM 15-5 SS samples as follows.
Figure 10 shows results acquired using sample AMB2018-01-15_5-CBM-B3-P1_L1. It shows that the phase fractions of both austenite and martensite/ferrite change monotonically as a function of time during heat treatment at 650 °C. The fraction of austenite increased from ≈ 1.1% in the as-built condition to ≈ 4.4% after 2 h of heat treatment at 650 °C, representing a fourfold increase. The fraction of martensite/ferrite decreased accordingly. As expected, the mean size of the precipitates, as shown in Fig. 10c, also increased monotonically, reaching ≈ 15 nm at the end of the heat treatment.

a Phase fraction of austenite as a function of time during heat treatment at 650 °C. b Phase fraction of martensite/ferrite as a function of time during heat treatment at 650 °C. c The mean size of the precipitate as a function of time during heat treatment at 650 °C. The sample is AMB2018-01-15_5-CBM-B3-P1_L1
Similarly, Figs. 11, 12, and 13 show the evolution of phase fractions of austenite and martensite/ferrite and the nominal size of the precipitates as a function of time for samples AMB2018-01-15_5-CBM-B3-P1_L2, AMB2018-01-15_5-CBM-B3-P1_L7, and AMB2018-01-15_5-CBM-B3-P1_L8, respectively. The same general trend as AMB2018-01-15_5-CBM-B3-P1_L1 was observed; namely, austenite phase fraction increases monotonically as time increases while martensite/ferrite phase fraction decreases. The size of the precipitates also increases monotonically. The phase fraction of martensite/ferrite, phase fraction of ferrite, and the nominal size of the precipitates at the end of the heat treatment is reported in Table 10. Overall, at the end of the heat treatment, austenite phase fraction ranges between 4.4 and 7.6%, and the size of the precipitates ranges from 12.6 to 20.2 nm. Based on these results, we did not identify a strong dependence between these parameters and the build conditions.

a Phase fraction of austenite as a function of time during heat treatment at 650 °C. b Phase fraction of martensite/ferrite as a function of time during heat treatment at 650 °C. c The mean size of the precipitate as a function of time during heat treatment at 650 °C. The sample is AMB2018-01-15_5-CBM-B3-P1_L2

a Phase fraction of austenite as a function of time during heat treatment at 650 °C. b Phase fraction of martensite/ferrite as a function of time during heat treatment at 650 °C. c The mean size of the precipitate as a function of time during heat treatment at 650 °C. The sample is AMB2018-01-15_5-CBM-B3-P1_L7

a Phase fraction of austenite as a function of time during heat treatment at 650 °C. b Phase fraction of martensite/ferrite as a function of time during heat treatment at 650 °C. c The mean size of the precipitate as a function of time during heat treatment at 650 °C. AMB2018-01-15_5-CBM-B3-P1_L8
AM IN625 In Situ USAXS/SAXS/XRD Results
Figures 14 and 15 show representative in situ XRD and SAXS data of AM IN625 under isothermal heat treatment at 800 °C.

a Typical in situ XRD data of AM IN625 acquired during isothermal heat treatment at 800 °C. The data acquisition time is color coded following the colored arrow in the figure. The inset shows the monotonic increase in peaks belong to an orthorhombic δ phase as time increases. b A comparison between the XRD pattern of IN625 acquired at 40 °C, after a 2 h heat treatment at 800 °C and stick patterns calculated for an FCC matrix (lattice parameter 3.60 Å) and an orthorhombic δ phase (lattice parameters a = 5.09 Å, b = 4.48 Å, and c = 4.09 Å)

a Typical in situ SAXS data of AM IN625 acquired during isothermal heat treatment at 800 °C. The data acquisition time is color coded following the colored arrow in the figure. As time increases, we observed a monotonic increase in the scattering intensity across the observed q range. b An illustration of the SAXS model. The overall fit, depicted by the blue solid line, is the sum of the scattering baseline and two scattering levels related to the diameter and thickness of the δ phase precipitates. More details about this model can be found elsewhere [14]
Figure 14 demonstrates the changes in the phases present in the sample as a function of time. In the as-built condition, only FCC matrix peaks are observed, consistent with the high-resolution XRD results reported in the previous section. However, within 5 min after the sample reached 800 °C, a family of orthorhombic peaks emerged. This is highlighted by the inset of Fig. 14a, where two peaks identified as δ {012} and δ {211} monotonically grew as a function of time. The entire XRD pattern acquired at heat treatment conditions can be satisfactorily accounted for by the matrix and δ phase peaks, as demonstrated by the example shown in Fig. 14b. This result is consistent with a previous study of phase evolution of AM IN625 under several different heat treatment conditions [14], where δ phase was identified as the precipitating phase and their growth kinetics was determined.
From the microstructure point of view, Fig. 15 shows that during the heat treatment, as time increases, a monotonic increase in the entire range of scattering intensity is observed. In the scattering sense, this means that scattering inhomogeneities with dimensions within the instrumental detection limits are developed through the heat treatment. The SAXS model is illustrated in Fig. 15b. Based on the XRD results shown in Fig. 14, we attributed the increase in the scattering intensity to the precipitation of δ phase. Also, δ phase is known to be platelet in shape [23]. Hence, two characteristic dimensions exist. We account for this by constructing a scattering model with two scattering levels, highlighted by the solid black and orange lines in Fig. 15b. We found that this model describes the scattering data well for all the in situ datasets. More details about this model can be found in [14].
The analysis of the in situ XRD data provides the evolution of the relative phase fraction of the δ phase. Because AM IN625 is known to be textured, to convert the relative phase fraction (integrated peak intensity) to absolute phase fraction, a synchrotron XRD analysis of the heat-treated IN625 under an identical heat treatment condition is required. This can be achieved following a previously established analysis protocol [29, 30]. The analysis of the in situ SAXS data provides information related to the size and shape evolution of the precipitates.
The detailed results acquired from four AM IN625 samples, namely AMB2018-01-625-CBM-B1-P4_L1, AMB2018-01-625-CBM-B1-P1_L2, AMB2018-01-625-CBM-B1-P1_L7, and AMB2018-01-625-CBM-B1-P1_L8, are shown in Figs. 16, 17, 18, and 19, respectively. Each figure consists of three panels. The first panel, (a), shows the integrated peak intensity of δ {211} reflection. This reflection has the strongest diffraction intensity. The XRD pattern of δ phase precipitates shows very limited, if not none, texturing effect. Hence, the integrated intensity of δ phase peaks is proportional to the volume fraction of δ phase [29]. In all four samples, we found that the total volume of δ phase increases monotonically as a function of time, consistent with the previous results [14, 29, 30].

a Relative phase fraction (in the form of integrated peak intensity) of δ phase as a function of time during heat treatment at 800 °C. b Mean thickness of δ phase platelet as a function of time during heat treatment at 800 °C. c Mean diameter of δ phase platelet as a function of time during heat treatment at 800 °C for sample AMB2018-01-625-CBM-B1-P4_L1

a Relative phase fraction (in the form of integrated peak intensity) of δ phase as a function of time during heat treatment at 800 °C. b Mean thickness of δ phase platelet as a function of time during heat treatment at 800 °C. c Mean diameter of δ phase platelet as a function of time during heat treatment at 800 °C of sample AMB2018-01-625-CBM-B1-P1_L2

a Relative phase fraction (in the form of integrated peak intensity) of δ phase as a function of time during heat treatment at 800 °C. b Mean thickness of δ phase platelet as a function of time during heat treatment at 800 °C. c Mean diameter of δ phase platelet as a function of time during heat treatment at 800 °C of AMB2018-01-625-CBM-B1-P4_L7

a Relative phase fraction (in the form of integrated peak intensity) of δ phase as a function of time during heat treatment at 800 °C. b Mean thickness of δ phase platelet as a function of time during heat treatment at 800 °C. c Mean diameter of δ phase platelet as a function of time during heat treatment at 800 °C of sample AMB2018-01-625-CBM-B1-P4_L8
The second panel, (b), of Figs. 16, 17, 18, and 19 shows the time-dependent evolution of the nominal thickness of δ phase precipitates. In all cases, we observed an initial sharp increase in this dimension during the first half an hour of the heat treatment; then, its increase gradually slows down and almost reaches a plateau. The nominal diameter of the δ phase precipitates, as shown by the third panel, (c), of Figs. 16, 17, 18, and 19, also shows a rapid increase in the initial stage of the heat treatment, followed by a continuous growth in later stage of the isothermal heat treatment, again, consistent with the previous results [14].
Summary
Following AM-Bench challenges CHAL-AMB2018-01-PF and CHAL-AMB2018-01-PFRS, we conducted rigorous measurements of the phase, phase fraction, and phase evolution using synchrotron X-rays measurements. Our major conclusions include:
XRD analysis shows that the as-built 15-5 SS consists of most (> 98% phase fraction) BCC martensite/ferrite and a small amount of (< 2%) FCC austenite. The lattice parameters and phase fractions of the martensite/ferrite and austenite do not demonstrate strong dependence on the build condition.
An isothermal heat treatment of 15-5 SS at 650 °C leads a partial transformation from martensite/ferrite to austenite. No other crystalline phases were detected. The size of Cu-based precipitates increases monotonically as time increases. At the end of the heat treatment, austenite phase fraction ranges between 4.4% and 7.6%, and the size of the precipitates ranges from 12.6 nm to 20.2 nm. Based on these results, we did not identify a strong dependence between these parameters and the build conditions.
The as-built IN625 shows a single-phase FCC structure with no detectable secondary phases. However, the XRD peaks are highly asymmetric, indicating structure inhomogeneity. The lattice parameters of the matrix phase do not show a strong dependence on the thermal history.
Isothermal heat treatment of IN625 leads to an almost immediate formation of δ phase precipitates. The total volume of δ phase increases monotonically as a function of time. The long dimension (diameter) of the platelet-shaped δ phase precipitates increases continuously through the heat treatment while the short dimension (thickness) reaches a plateau after an initial rapid increase.
Availability and Software Used
All data reported in this paper are freely available upon request.
X-ray diffraction data were analyzed using GSAS-II, developed by Robert Von Dreele and Brian Toby at the Advanced Photon Source, Argonne National Laboratory. GSAS-II is available for free use. More details about this software can be found here: https://subversion.xray.aps.anl.gov/trac/pyGSAS.
Small-angle X-ray scattering data reduction and analysis were conducted using Igor Pro based Indra, Irena, and Nika, developed by Jan Ilavsky at the Advanced Photon Source, Argonne National Laboratory. Igor Pro is a commercial software. Indra, Irena, and Nika are available for free use. More details about these small-angle scattering reduction and analysis packages can be found here: https://usaxs.xray.aps.anl.gov/software/irena.
Figures are generated using Igor Pro, Origin, and MATLAB.
Notes
Certain commercial equipment, instruments, software, or materials are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the Department of Commerce or the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified is necessarily the best available for the purpose.
References
Herzog D, Seyda V, Wycisk E, Emmelmann C (2016) Additive manufacturing of metals. Acta Mater 117:371–392
Keller T, Lindwall G, Ghosh S, Ma L, Lane BM, Zhang F, Kattner UR, Lass EA, Heigel JC, Idell Y (2017) Application of finite element, phase-field, and calphad-based methods to additive manufacturing of Ni-based superalloys. Acta Mater 139:244–253
Kok Y, Tan XP, Wang P, Nai M, Loh NH, Liu E, Tor SB (2018) Anisotropy and heterogeneity of microstructure and mechanical properties in metal additive manufacturing: a critical review. Mater Des 139:565–586
King WE, Anderson AT, Ferencz R, Hodge N, Kamath C, Khairallah SA, Rubenchik AM (2015) Laser powder bed fusion additive manufacturing of metals; physics, computational, and materials challenges. Appl Phys Rev 2(4):041304
Gao W, Zhang Y, Ramanujan D, Ramani K, Chen Y, Williams CB, Wang CC, Shin YC, Zhang S, Zavattieri PD (2015) The status, challenges, and future of additive manufacturing in engineering. Comput Aided Des 69:65–89
Seifi M, Salem A, Beuth J, Harrysson O, Lewandowski JJ (2016) Overview of materials qualification needs for metal additive manufacturing. JOM 68(3):747–764
Megahed M, Mindt H-W, N’Dri N, Duan H, Desmaison O (2016) Metal additive-manufacturing process and residual stress modeling. Integr Mater Manuf Innov 5(1):4
Jared BH, Aguilo MA, Beghini LL, Boyce BL, Clark BW, Cook A, Kaehr BJ, Robbins J (2017) Additive manufacturing: toward holistic design. Scr Mater 135:141–147
Levine L, Lane BM, Heigel JC, Migler K, Stoudt MR, Phan T, Ricker R, Strantza M, Hill M, Zhang F, Seppala J, Garboczi E, Bain E, Cole D, Allen AJ, Fox J, Campbell CE (2019) Outcomes and conclusions from the 2018 AM-Bench measurements, challenge problems, modeling submissions, and conference. Integr Mater Manuf Innov (under review)
Wang X-L, Almer J, Liu C, Wang Y, Zhao J, Stoica A, Haeffner D, Wang W (2003) In situ synchrotron study of phase transformation behaviors in bulk metallic glass by simultaneous diffraction and small angle scattering. Phys Rev Lett 91(26):265501
Dubois J, Thilly L, Renault P, Lecouturier F, Di Michiel M (2010) Thermal stability of nanocomposite metals: in situ observation of anomalous residual stress relaxation during annealing under synchrotron radiation. Acta Mater 58(19):6504–6512
Zhang F, Levine LE, Allen AJ, Campbell CE, Lass EA, Cheruvathur S, Stoudt MR, Williams ME, Idell Y (2017) Homogenization kinetics of a nickel-based superalloy produced by powder bed fusion laser sintering. Scr Mater 131:98–102
Zhang F, Levine LE, Allen AJ, Campbell CE, Creuziger AA, Kazantseva N, Ilavsky J (2016) In situ structural characterization of ageing kinetics in aluminum alloy 2024 across angstrom-to-micrometer length scales. Acta Mater 111:385–398
Zhang F, Levine LE, Allen AJ, Stoudt MR, Lindwall G, Lass EA, Williams ME, Idell Y, Campbell CE (2018) Effect of heat treatment on the microstructural evolution of a nickel-based superalloy additive-manufactured by laser powder bed fusion. Acta Mater 152:200–214
Lee PL, Shu D, Ramanathan M, Preissner C, Wang J, Beno MA, Von Dreele RB, Ribaud L, Kurtz C, Antao SM (2008) A twelve-analyzer detector system for high-resolution powder diffraction. J Synchrotron Radiat 15(5):427–432
Black DR, Windover D, Henins A, Filliben J, Cline JP (2011) Certification of standard reference material 660B. Powder Diffr 26(2):155–158
Toby BH, Von Dreele RB (2013) GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J Appl Crystallogr 46(2):544–549
Ilavsky J, Zhang F, Andrews RN, Kuzmenko I, Jemian PR, Levine LE, Allen AJ (2018) Development of combined microstructure and structure characterization facility for in situ and operando studies at the advanced photon source. J Appl Crystallogr 51(3):867–882
Ilavsky J, Jemian PR, Allen AJ, Zhang F, Levine LE, Long GG (2009) Ultra-small-angle X-ray scattering at the advanced photon source. J Appl Crystallogr 42(3):469–479
Allen AJ, Zhang F, Kline RJ, Guthrie WF, Ilavsky J (2017) Nist standard reference material 3600: absolute intensity calibration standard for small-angle X-ray scattering. J Appl Crystallogr 50(2):462–474
Zhang F, Ilavsky J, Long GG, Quintana JP, Allen AJ, Jemian PR (2010) Glassy carbon as an absolute intensity calibration standard for small-angle scattering. Metall Mater Trans 41(5):1151–1158
Gilles R, Keiderling U, Wiedenmann A (1998) Silver behenate powder as a possible low-angle calibration standard for small-angle neutron scattering. J Appl Crystallogr 31(6):957–959
Lass EA, Stoudt MR, Williams ME, Katz MB, Levine LE, Phan TQ, Gnaeupel-Herold TH, Ng DS (2017) Formation of the Ni3Nb δ-phase in stress-relieved inconel 625 produced via laser powder-bed fusion additive manufacturing. Metall Mater Trans A 48(11):5547–5558
Ilavsky J, Jemian PR (2009) Irena: tool suite for modeling and analysis of small-angle scattering. J Appl Crystallogr 42(2):347–353
Ilavsky J (2012) Nika: software for two-dimensional data reduction. J Appl Crystallogr 45(2):324–328
Idell Y, Levine LE, Allen AJ, Zhang F, Campbell CE, Olson G, Gong J, Snyder D, Deutchman H (2016) Unexpected δ-phase formation in additive-manufactured Ni-based superalloy. JOM 68(3):950–959
Petrović ZS, Milić J, Zhang F, Ilavsky J (2017) Fast-responding bio-based shape memory thermoplastic polyurethanes. Polymer 121:26–37
Bajguirani HH (2002) The effect of ageing upon the microstructure and mechanical properties of type 15-5 PH stainless steel. Mater Sci Eng, A 338(1–2):142–159
Lindwall G, Campbell C, Lass E, Zhang F, Stoudt M, Allen A, Levine L (2019) Simulation of TTT curves for additively manufactured Inconel 625. Metall Mater Trans A 50(1):457–467
Stoudt MR, Lass E, Ng DS, Williams ME, Zhang F, Campbell CE, Lindwall G, Levine LE (2018) The influence of annealing temperature and time on the formation of δ-phase in additively-manufactured Inconel 625. Metall Mater Trans A 49(7):3028–3037
Acknowledgements
Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357. This research was partially supported by the Exascale Computing Project (17-SC-20-SC), a collaborative effort of the US Department of Energy Office of Science and the National Nuclear Security Administration.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, F., Levine, L.E., Allen, A.J. et al. Phase Fraction and Evolution of Additively Manufactured (AM) 15-5 Stainless Steel and Inconel 625 AM-Bench Artifacts. Integr Mater Manuf Innov 8, 362–377 (2019). https://doi.org/10.1007/s40192-019-00148-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40192-019-00148-1